Localization of the pre-squalene segment of the isoprenoid biosynthetic pathway in mammalian peroxisomes
18
0
0
Texte intégral
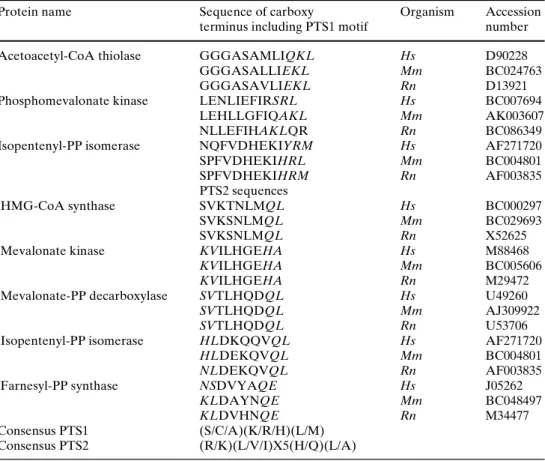
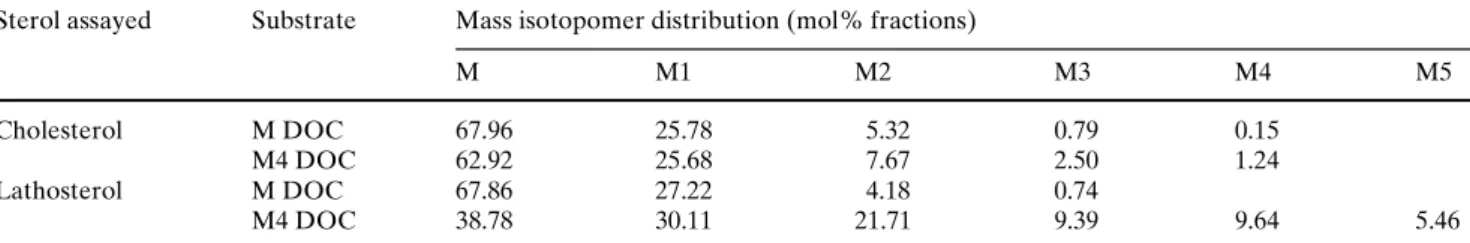
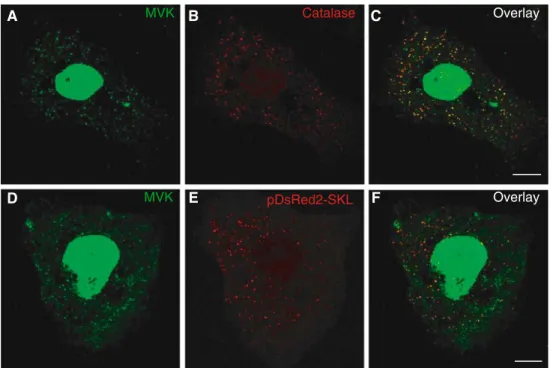
Figure
Documents relatifs