HAL Id: dumas-03129869
https://dumas.ccsd.cnrs.fr/dumas-03129869
Submitted on 3 Feb 2021HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License
Émilie Chotard
To cite this version:
Émilie Chotard. Maladie à dépôts de pyrophosphate de calcium : étude d’une cohorte de 57 patients avec syndrome de Gitelman. Médecine humaine et pathologie. 2019. �dumas-03129869�
AVERTISSEMENT
Cette thèse d’exercice est le fruit d’un travail approuvé par le jury de soutenance et réalisé dans le but d’obtenir le diplôme d’Etat de docteur en médecine. Ce document est mis à disposition de l’ensemble de la communauté universitaire élargie.
Il est soumis à la propriété intellectuelle de l’auteur. Ceci implique une obligation de citation et de référencement lors de l’utilisation de ce document.
D’autre part, toute contrefaçon, plagiat, reproduction illicite encourt toute poursuite pénale.
Code de la Propriété Intellectuelle. Articles L 122.4
Année 2019
N° 126
THÈSE
POUR LE DIPLÔME D’ÉTAT
DE
DOCTEUR EN MÉDECINE
Maladie à dépôts de pyrophosphate de calcium :
étude d’une cohorte de 57 patients avec syndrome de Gitelman
Présentée et soutenue publiquement
le 20 septembre 2019
Par
Émilie CHOTARD
Née le 3 mai 1990 à Nantes (44)
Co-dirigée par M. Le Professeur Hang-Korng Ea, PU-PH, et
Mme Le Docteur Anne Blanchard, PH
Jury :
M. Le Professeur Pascal Richette, PU-PH ……….. Président M. Le Professeur Emmanuel Letavernier, PU-PH
2
Remerciements ... 4
Abréviations ... 6
Introduction ... 7
Partie I ... 7
1) Maladie à dépôts de pyrophosphate de calcium ... 7
2) Etiologies secondaires de la maladie à dépôts de pyrophosphate de calcium ... 12
3) Homéostasie du magnésium dans l’organisme ... 15
4) Complications rhumatologiques de l’hypomagnésémie ... 21
Partie II ... 23
1) Définition du syndrome de Gitelman et épidémiologie ... 23
2) Historique de la description du syndrome de Gitelman ... 23
3) Physiopathologie du syndrome de Gitelman ... 23
4) Diagnostic et présentation clinique ... 24
5) Pénétrance, formes sévères et complications ... 25
6) Tableau biologique ... 26
7) Diagnostic génétique ... 26
8) Diagnostics différentiels ... 27
9) Prise en charge thérapeutique ... 27
Objectifs ... 29 Matériels et méthodes ... 30 Population ... 30 Données recueillies ... 30 Dépistage de la maladie à dépôts de PPC ... 30 Relecture radiologique ... 31 Analyse statistique ... 31 Résultats ... 32
Caractéristiques de la population d’inclusion... 32
Mise en évidence des dépôts de chondrocalcinose ... 33
Localisation des dépôts de chondrocalcinose ... 34
Chondrocalcinose diffuse et facteurs de risque ... 39
Arthropathie et chondrocalcinose disséminée ... 41
3 Thérapeutiques utilisées ... 42 Discussion... 43 Conclusion ... 48 Annexes ... 49 Références ... 50
4
Remerciements
Je remercie tout d’abord Korng qui m’a offert l’opportunité de collaborer sur ce sujet de thèse. Merci Korng, pour tes éclairages scientifiques, tes encouragements pour les présentations orales, tes remarques toujours données avec bienveillance. J’espère pouvoir continuer à travailler ensemble sur de futurs projets.
Je remercie le Dr Blanchard pour sa collaboration qui nous a permis de suivre la cohorte de l’hôpital Européen Georges Pompidou et pour son aide pour la relecture de cette thèse. Je remercie le Pr Richette d’avoir accepté d’être président de mon jury de thèse et pour l’opportunité offerte de travailler à nouveau en rhumatologie à Lariboisière.
Je remercie le Pr Letavernier d’avoir accepté d’être membre de mon jury de thèse.
Je remercie le Pr Bousson pour sa disponibilité pour la relecture des examens radiologiques des patients de cette cohorte.
Je remercie le Dr Vargas-Poussou pour son aide concernant la caractérisation génétique des patients de cette cohorte.
Je remercie le Dr Gailly pour son travail d’échographiste auprès des patients de la cohorte. Je remercie le Dr Ostertag pour l’aide à la réalisation des statistiques de cette étude. Je remercie Mylène pour son aide bibliographique au cours de la rédaction de cette thèse. Je remercie toute l’équipe du service de rhumatologie de Lariboisière avec qui j’ai eu la chance de travailler et que je retrouverai prochainement avec plaisir pour le clinicat.
Je remercie les passionnés de l’échographie, en particulier Sébastien Ottaviani et Jimmy : merci de m’avoir enseigné vos bases et techniques, toujours dans la bonne humeur !
Je remercie mes co-internes et chefs de clinique croisés aux cours de ces quatre années d’internat : Marion, Margherita, Maud, Quentin B., Juliette L., Camille G., Etienne, Florence B., Antoine, Camille B., Stéphanie et Anne-Laure, Aude, Sacha, Esteban, Juliette P., Quentin K., Isabelle B., Minh, Vanina, Isabelle S., Florence A., Raphaël. Merci pour ces agréables semestres passés à vos côtés !
Je remercie mes collègues de master 2 avec qui j’ai passé une super année 2017-2018 au laboratoire : Camille, Ji-Yun, Sarah, Maud et Alan.
Je remercie mes amis de toujours, mes amis du lycée, Camille et Virginie de la faculté de médecine de Nantes, Pauline mon ex-coloc, avec qui j’ai partagé les joies et difficultés de l’internat. Grâce à vous, j’ai aussi pu garder un pied dans le monde tout aussi passionnant de la « non-médecine » !
5 Je remercie ma famille et plus particulièrement mes parents, mes frères et mes cousins pour leur présence, leur affection et leur confiance qui m’ont permis de m’épanouir d’abord dans ma vie personnelle mais aussi dans ma vie professionnelle.
Je remercie également ma belle-famille pour son intérêt bienveillant lors de mes années d’internat.
Enfin je remercie mon mari pour son soutien sans faille au cours de ces 7 dernières années, merci d’être toujours à mes côtés.
6
Abréviations
ADN acide désoxyribonucléique
AINS anti-inflammatoire non stéroïdien AMP adénosine monophosphate
ANKH homologue humain de l'ankylose progressive ARN acide ribonucléique
ATP adénosine triphosphate BCP phosphate de calcium basique CCA chondrocalcinose articulaire Cl-CKB chloride channel Kb
CST coefficient de saturation de la transferrine
ENPP1 ectonucléotide pyrophosphatase/phosphodiestérase 1 IL-1β interleukine-1 β
IPP inhibiteur de la pompe à protons NCC Na/Cl cotransporteur
ORKID filière orphan kidney disease
PC-1 ecto-enzyme plasma cell glycoprotein 1 Pi phosphate inorganique
PPC pyrophosphate de calcium PPi pyrophosphate inorganique
PTH parathormone
PAL phosphatase alcaline
ROMK renal outer medullary potassium channel SG syndrome de Gitelman
TLR2 toll-like receptor 2
TNAP tissue non specific alkaline phosphatase
7
Introduction
Partie I
1) Maladie à dépôts de pyrophosphate de calcium
Définition
La maladie à dépôts de pyrophosphate de calcium (PPC) est une arthropathie microcristalline engendrée par la formation de cristaux de pyrophosphate de calcium dans les tissus articulaires et périarticulaires.
Historique
Les cristaux de PPC ont été identifiés pour la première fois au microscope à lumière polarisée en 1961 et 1962 par McCarty chez des patients dont la présentation clinique avait été qualifiée de « pseudo goutte » en raison de sa ressemblance avec une crise de goutte1,2. La même description clinique avait été faite en 1963 par des tchéquoslovaques et qualifiée de chondrocalcinose3.
Epidémiologie
La forme primitive liée à l’âge (ou chondrocalcinose idiopathique) est de loin la forme la plus courante de la maladie : sa fréquence est estimée entre 1 et 2% entre 60 et 70 ans, entre 6 et 8% de 70 à 80 ans et entre 20 et 30% après 80 ans4,5. L’origine « primitive » est communément admise lorsque l’âge de survenue est supérieur ou égal à 60 ans6. L’incidence de la chondrocalcinose varie selon la technique de dépistage utilisée (histologie sur cadavres ou radiographies) et selon l’âge des patients. La maladie touche autant les femmes que les hommes.
Terminologie
La maladie à dépôts de PPC est la terminologie actuellement admise qui regroupe les différentes formes cliniques de la maladie7. Ainsi, le terme de pseudo-goutte est aujourd’hui obsolète et correspond aux arthrites aiguës à PPC. La chondrocalcinose est définie par la présence de calcifications radiographiques des cartilages : elle est le plus souvent due à la maladie à PPC, cependant ces calcifications ne correspondent pas toujours à des dépôts de PPC mais peuvent aussi être constituées de phosphate de calcium comme l’hydroxyapatite carbonatée.
8
Physiopathologie
La formation de cristaux de PPC est favorisée par un excès local de pyrophosphate inorganique (PPi) extracellulaire et de calcium libre8. La régulation du taux de PPi est assurée par 3 protéines principales : l’ecto-enzyme plasma cell glycoprotein 1 (PC-1) qui hydrolyse l’ATP extracellulaire en PPi et adénosine monophosphate (AMP), le transporteur de PPi ankylosis
human (ANKH) qui permet l’excrétion extracellulaire du PPi et travaille en coordination avec
le co-transporteur sodium/phosphate Pit-19, et enfin la phosphatase alcaline (TNAP) qui métabolise le PPi extracellulaire en orthophosphate. L’AMP est hydrolysé par l’enzyme CD73 en phosphate inorganique (Pi) et adénosine qui à son tour inhibe l’activité de TNAP10 (figure 1).
Figure 1 : Régulation des pyrophosphate (PPi) et phosphate (Pi) inorganiques et formation des cristaux de pyrophosphate de calcium (PPC) ; ATP : adénosine triphosphate ; AMP : adénosine monophosphate ; PC-1 : plasma cell glycoprotein 1 ; ANKH : homologue humain de l'ankylose progressive ; TNAP : Tissue non specific alkaline phosphatase (inspiré de A. Abhishek, M. Doherty, Clin Exp Rheumatol 2016)
La réaction inflammatoire aiguë survenant dans les arthrites à PPC peut être assimilée aux processus survenant dans les accès goutteux aigus. Les cristaux de PPC activent des systèmes de l’immunité innée, notamment via les TLR-211, le complexe de l’inflammasome et l’interleukine (IL)-1β12,13 dans les synoviocytes macrophagiques engendrant une cascade de
9 production de cytokines pro-inflammatoires responsables des symptômes aigus. Les cristaux de PPC pourraient également interagir avec les fibroblastes synoviaux et favoriser le relargage de métalloprotéinases14 pro-inflammatoires.
Formes cliniques
La maladie à dépôts de PPC peut revêtir des formes cliniques variées qui ont été clairement définies dans les dernières recommandations EULAR 20117 :
La forme asymptomatique est très probablement la plus fréquente : elle correspond à la chondrocalcinose, par définition radiologique, sans symptômes articulaires associés.
L’arthrite aiguë à dépôts de PPC (anciennement pseudo-goutte) est quant à elle une arthrite aiguë, à début brutal. Sa survenue peut être favorisée par un événement intercurrent (chirurgie, sepsis, traumatisme local, perfusion de bisphosphonate15, parathyroïdectomie16). Il s’agit le plus souvent d’une mono-arthrite, touchant préférentiellement le genou, ou bien d’une oligo-arthrite. Les symptômes cliniques peuvent être associés à un syndrome inflammatoire biologique. L’évolution naturelle est favorable et les crises sont habituellement spontanément résolutives. Cependant, les récidives sont fréquentes.
L’arthropathie chronique à PPC est plus rare. Elle se caractérise par des synovites chroniques volontiers bilatérales et symétriques pouvant mimer une polyarthrite rhumatoïde ou bien une pseudo-polyarthrite rhizomélique lorsque les atteintes sont proximales17.
Enfin la maladie à PPC peut prendre l’aspect d’une arthropathie dégénérative chronique. Dans ce contexte, les atteintes peuvent se distinguer de l’arthrose classique par une distribution articulaire inhabituelle (poignet, coude, gléno- humérale) ou une localisation intra-articulaire
inhabituelle (atteinte radiocarpienne, scapho-trapézienne sans atteinte
trapézométacarpienne18, fémoro-patellaire, talo-calcanéo-naviculaire). Dans ce cas, les géodes sous-chondrales sont souvent plus nombreuses et de grande taille19 en comparaison avec l’arthrose classique. Enfin, la destruction articulaire observée dans l’arthropathie dégénérative à cristaux de PPC pourrait être plus sévère et progresser plus rapidement que dans l’arthrose idiopathique. Toutefois, concernant spécifiquement la gonarthrose, plusieurs études ont suggéré l’absence d’effet voire un effet structural « protecteur » des cristaux de PPC sur l’évolution de l’arthrose. Ainsi, une étude longitudinale utilisant l’IRM pour mesurer l’épaisseur des cartilages n’a pas retrouvé d’association entre la présence de cristaux de PPC
10 au genou et la perte cartilagineuse en comparaison avec des genoux sans chondrocalcinose20. Une autre étude s’intéressant aux patients bénéficiant d’une arthroplastie totale de genou a aussi constaté que les patients avec chondrocalcinose étaient en moyenne plus âgés au moment de la chirurgie que les patients sans chondrocalcinose21.
Présentation radiologique
Les cristaux de PPC se déposent à la surface et dans des cartilages articulaires : les articulations les plus souvent touchées sont les poignets, les genoux, les métacarpo-phalangiennes, les acromio-claviculaires mais aussi les coudes et les coxo-fémorales.
Les cristaux de PPC sont également décrits au niveau des capsules, de la synoviale, des tendons ou des ligaments. La présentation radiologique classique présentée ci-dessous a été décrite à partir des formes idiopathiques de maladie à dépôts de PPC.
Concernant les fibrocartilages, on retrouve des cristaux dans les ménisques, dans le ligament triangulaire du carpe, dans la symphyse pubienne, dans le labrum acétabulaire et glénoïdien ou encore dans l’annulus fibrosus discal22.
Les dépôts synoviaux sont préférentiellement retrouvés aux poignets, aux genoux, dans les métacarpo-phalangiennes ou métatarso-phalangiennes22.
Les capsules articulaires les plus souvent touchées sont celles des coudes et des métatarso-phalangiennes.
Concernant les tendons, les bourses et les ligaments, les cristaux de PPC se déposent fréquemment au niveau achilléen, dans les enthèses tricipitales et quadricipitales, dans les tendons supra-spinatus ou encore dans la bourse sous acromiale.
Enfin, les dépôts de PPC ont été décrits dans les tissus mous péri articulaires notamment au niveau des coudes, des poignets et du bassin constituant parfois des « tophus » de PPC22,23. Des dépôts vasculaires ont également été décrits chez quelques patients apparentés présentant des mutations génétiques codant pour certaines enzymes impliquées dans le métabolisme du pyrophosphate10.
Diagnostic
Le diagnostic de certitude est possible grâce à la mise en évidence de cristaux de PPC dans un liquide articulaire ou plus rarement sur une biopsie synoviale par une étude au microscope à
11 lumière polarisée. Les cristaux sont reconnaissables par leur aspect court, parallélépipède, de localisation souvent intracellulaire et par leur faible biréfringence en lumière polarisée24. Le diagnostic peut également être évoqué devant des calcifications des cartilages méniscaux, de la symphyse pubienne ou du ligament triangulaire du carpe sur des radiographies standard : dans ce cas on parle plutôt de chondrocalcinose articulaire. L’échographie est également un outil de plus en plus utilisé pour mettre en évidence des calcifications de type chondrocalcinose avec une sensibilité supérieure à la radiographie25,26: Les calcifications prennent l’aspect d’une fine bande hyperéchogène à la surface des cartilages ou bien de spots hyperéchogènes avec cône d’ombre postérieur dans les tendons, ligaments, capsules ou autres tissus péri articulaires. Le scanner est également un outil permettant d’identifier des dépôts calciques : l’avènement du scanner low-dose corps entier et du scanner double énergie renforce l’arsenal radiologique et peut permettre de faire le diagnostic de maladie à dépôts de PPC au niveau d’articulations plus complexes à étudier telles que la cheville ou encore le rachis cervical. Ainsi, une étude récente suggère que le scanner double énergie est capable de différencier les dépôts de PPC méniscaux des dépôts d’hydroxyapatite de l’os sous-chondral ou trabéculaire in vivo27.
Prise en charge thérapeutique
Les traitements disponibles sont essentiellement symptomatiques et ont fait l’objet de recommandations européennes en 201128. Ils sont le plus souvent extrapolés de la prise en charge des arthropathies goutteuses.
Pour l’arthrite aiguë à PPC, le repos et le glaçage sont recommandés avec éventuellement une ponction évacuatrice de l’articulation symptomatique (à visée antalgique mais aussi diagnostique) qui peut être associée à une injection intra-articulaire de corticoïdes. La prise d’anti-inflammatoires non stéroïdiens (AINS) ou de colchicine per os est également une possibilité mais les effets secondaires et leur marge thérapeutique étroite peuvent être un frein à leur utilisation chez les patients âgés avec des comorbidités (notamment rénales ou cardiovasculaires).
La prise de corticoïdes oraux ou par voie parentérale en cure courte peut être une alternative lorsqu’une injection intra-articulaire est impossible ou que les AINS et la colchicine sont contre indiqués29,30.
12 Les inhibiteurs de l’IL-1β sont également une option lorsque les traitements suscités ne peuvent pas être employés et l’anakinra a ainsi été utilisé avec succès dans plusieurs cas rapportés dans la littérature31–33.
De façon intéressante, la colchicine à faible dose pourrait être efficace dans le traitement préventif des arthrites récurrentes à PPC34. L’hydroxy-chloroquine a également été suggérée comme efficace dans les formes de polyarthrites chroniques à dépôts de PPC dans une étude prospective de 6 mois réalisée en double aveugle35. Quant au méthotrexate, alors qu’il semblait efficace dans une étude observationelle36, il n’a finalement pas montré son efficacité dans une étude randomisée contre placebo37. Les synoviorthèses isotopiques restent une alternative thérapeutique dans les formes chroniques et dans les hémarthroses récidivantes38. Enfin, dans les formes secondaires liées à l’hypomagnésémie, la supplémentation en Mg2+ pourrait réduire les accès aigus à PPC 39 voire diminuer les dépôts chondrocalcinosiques 40 mais seuls des cas isolés ont été publiés. Le traitement d’une hyperparathyroïdie ou de l’hémochromatose ne semble pas influencer le cours de l’arthropathie microcristalline. Le traitement de l’arthrose liée à la maladie à PPC ne diffère pas de celui de l’arthrose primitive. Les symptômes de gonarthrose associée à la chondrocalcinose pourraient cependant être améliorés par la colchicine prise à faible dose au long cours41. Enfin la chondrocalcinose asymptomatique ne requiert aucun traitement.
2) Etiologies secondaires de la maladie à dépôts de pyrophosphate de calcium
Hypomagnésémie
Une étude rétrospective chez plus de 25 000 vétérans américains a souligné une association entre maladie à dépôts de PPC et hypomagnésémie avec un OR de 1.2342. Une seconde étude de prévalence incluant plus de 1700 patients a également retrouvé une association positive entre chondrocalcinose au genou et utilisation de diurétiques avec un odd ratio ajusté supérieur à 2. Cette association a été attribuée à l’hypomagnésémie induite par les diurétiques, cependant il n’a pas été mis en évidence de relation effet-dose dans cette étude5. A l’inverse, une étude cas témoins de petit effectif n’a pas permis de mettre en évidence une diminution significative de la magnésémie chez des patients atteints de chondrocalcinose idiopathique en comparaison aux contrôles43.
13 Le mécanisme physiopathologique de la formation des dépôts de PPC en lien avec l’hypomagnésémie est détaillé dans la suite du manuscrit (pages 22-23).
Hémochromatose
L’hémochromatose est une hépatopathie génétique dont la mutation la plus fréquente est celle du gène HFE1. Dans cette pathologie, l’absorption digestive de fer est excessive ce qui engendre des dépôts ferriques diffus intra tissulaires. Du point de vue rhumatologique, l’hémochromatose est responsable d’une ostéoporose et d’une arthropathie précoce, touchant 56% des patients dans l’étude de Pawlotsky44. Dans cette même série de 210 patients atteints d’hémochromatose, une chondrocalcinose était retrouvée chez 38% des patients hémochromatosiques homozygotes44. L’atteinte articulaire peut être révélatrice de l’hémochromatose (28% dans cette même étude)45. L’aspect radiologique typique se caractérise par une arthropathie des 2e et 3e métacarpo-phalangiennes. L’atteinte articulaire de l’hémochromatose est l’atteinte d’organe qui altère le plus la qualité de vie des patients46 La physiopathologie du rhumatisme hémochromatosique reste mal comprise. D’une part les dépôts synoviaux d’hémosidérine pourraient être à l’origine d’un infiltrat réactionnel de macrophages et de polynucléaires neutrophiles. D’autre part, un mécanisme de toxicité directe de la surcharge en fer est évoqué avec la production de radicaux libres exerçant une action catabolique sur le cartilage47. L’excès d’ion ferrique diminue l’activité de la TNAP ce qui augmente le taux de PPi extracellulaire et favorise la formation des cristaux de PPC. D’autres facteurs pouvant expliquer les dépôts de PPC sont avancés, comme l’élévation de la fraction 44-68 de la parathormone chez de nombreux patients atteints d’hémochromatose44.
Hyperparathyroïdie
L’hyperparathyroïdie primitive est l’étiologie la plus fréquente de maladie à dépôts de PPC secondaire. La prévalence de la chondrocalcinose au cours de l’hyperparathyroïdie primitive est en moyenne de 21 %, avec des valeurs extrêmes de 8 % à 57 % selon les études45,48–50. La prévalence de la chondrocalcinose chez les patients hyperparathyroïdiens augmente beaucoup avec l’âge : ainsi cette prévalence chez les cinquantenaires est évaluée à 4.4% contre 15.8% chez les soixantenaires et même 37.5% chez les plus de 70 ans51. Les patients ayant une hyperparathyroïdie et une chondrocalcinose sont le plus souvent des femmes et sont en général plus âgés que les patients sans chondrocalcinose. A l’inverse,
14 l’hyperparathyroïdie primitive est présente chez 3.9%52 à 5.6%43 des patients dans des petites cohortes de chondrocalcinose.
Les hyperparathyroïdies secondaires semblent beaucoup plus rarement associées aux dépôts de PPC. Cela s’explique pour certains auteurs par l’absence d’hypercalcémie chronique48 Sur le plan physiopathologique, l’hypercalcémie induite par l’hyperparathyroïdie diminue l’activité de la phosphatase alcaline et ainsi favorise l’accumulation du pyrophosphate extracellulaire qui en se liant au calcium aboutit à la formation des cristaux de PPC48.
Le traitement chirurgical de l’hyperparathyroïdie ne permet pas la régression des calcifications radiologiques, la parathyroïdectomie peut même s’accompagner d’accès d’arthrite aiguë à PPC51.
Hypophosphatasie
L’hypophosphatasie est une maladie génétique rare qui se caractérise par un défaut de minéralisation de l’os et des dents en lien avec la mutation du gène codant pour la phosphatase alcaline. L’expression clinique de la maladie est très variable et se caractérise par une perte précoce de la dentition primaire, une fragilité osseuse avec fractures récidivantes ou encore une chondrocalcinose. La formation des cristaux de PPC est favorisée par la baisse d’activité de la phosphatase alcaline et donc l’accumulation de pyrophosphate extracellulaire. Biologiquement, la phosphatase alcaline sérique est abaissée. Un traitement enzymatique substitutif est actuellement disponible pour les cas les plus sévères53,54.
Formes familiales
Plusieurs cas de formes familiales de maladie à dépôts de PPC ont été rapportés avec souvent un phénotype précoce et sévère. En 2000, Ho et al. ont souligné le rôle de la protéine transmembranaire ANKH dans le transport du PPi55. Des études génétiques menées chez des familles présentant une transmission autosomique dominante de la maladie à dépôts de PPC ont pu mettre en évidence des mutations sur le bras court du chromosome 5, à proximité du gène ANKH56. Une mutation activatrice du gène ANKH a été clairement identifiée dans plusieurs familles57,58. Récemment, une mutation activatrice du gène TNFRSF11B codant pour l’ostéoprotégérine (OPG) a été identifiée dans deux familles avec dépôts de PPC59,60. Le rôle exact de l’OPG dans la formation des cristaux de PPI n’est pas connu. La mutation identifiée ne module pas l’activité de la TNAP ni de la protéine ANKH60.
15
Autres
Il existe d’autres causes plus rares de maladie à dépôts de PPC secondaire. Parmi celles-ci on peut citer l’ochronose, l’hypercalcémie hypocalciurie familiale, la maladie de Wilson ou les causes post-traumatiques.
3) Homéostasie du magnésium dans l’organisme
L’ion magnésium Mg2+ est le second cation intracellulaire le plus abondant de l’organisme après le potassium. Il constitue le cofacteur de nombreuses enzymes impliquées dans des processus cellulaires clés tels que le métabolisme énergétique, la synthèse de l’acide désoxyribonucléique (ADN) et de l’acide ribonucléique (ARN) ou encore la synthèse protéique. Le magnésium joue également un rôle d’ostéoformation, de stabilité neuromusculaire et de contraction musculaire61,62
La magnésémie normale chez l’adulte est comprise entre 0.65 et 1.05 mmol/L. Cependant la fraction dosable du magnésium sanguin ne représente qu’un pourcentage très faible du stock de magnésium total de l’organisme. Le contenu de l’organisme en magnésium est d’environ 1000 mmol (25 g) dont plus de 60 % sont stockés dans l’os, 30 % à 40 % dans les muscles, érythrocytes et autres cellules, et seulement environ 1 % (10 mmol) dans le volume extracellulaire. Dans le plasma, seul 20% du magnésium est lié aux protéines plasmatiques. La magnésémie est donc moins affectée par les variations de protidémie que la calcémie (figure 2). La régulation du magnésium dépend de 2 déterminants majeurs : physiologiquement, le rein excrète l’équivalent de l’absorption nette intestinale de magnésium, et le bilan osseux sur 24h est nul (figure 3). En situation pathologique comme après une parathyroidectomie, une accrétion osseuse nette de magnésium peut induire une hypomagnésémie (hungry bone syndrome).
La ration journalière recommandée de magnésium chez l’adulte se situe entre 320 et 420 mg/j selon le sexe63.
16
Figure 2 : Représentation des différentes formes du magnésium sérique (ionisé, complexé ou lié aux protéines)(Jahnen-Dechent et Ketteler, CKJ 2012)
Figure 3 : Homéostasie du magnésium dans l’organisme figurant la balance entre l’absorption digestive et l’excrétion rénale journalière en Mg2+ (De Baaij et al, CKJ 2012)
17
Régulation digestive
L’absorption digestive du magnésium a lieu principalement dans l’intestin grêle et le colon. Il s’agit d’un processus adaptable et l’absorption digestive peut augmenter jusqu’à 80% lorsque les apports en magnésium sont bas. Deux mécanismes d’absorption coexistent : un transport passif paracellulaire du magnésium et un transport actif transcellulaire. L’absorption paracellulaire est responsable de 80 à 90% de l’apport intestinal en magnésium et est possible grâce à la haute concentration luminale en magnésium et au voltage transépithélial. L’absorption passive dépend également de la perméabilité des jonctions serrées mais leur fonctionnement est mal connu. L’iléon et le jéjunum distal sont les plus perméables du fait de leur expression faible en certains types de jonctions claudines. Les canaux TRMP6 et TRMP7 régulent de façon fine le reste de l’absorption intestinale en magnésium64. Ils sont exprimés à la membrane luminale des entérocytes. L’extrusion basolatérale du Mg2+ reste mal connue mais pourrait être couplée au gradient de sodium (figure 4). La 1-25OH vitamine D pourrait également stimuler l’absorption intestinale en magnésium63.
La malnutrition (notamment en cas d’alcoolisme chronique), les entéropathies (en particulier les maladies inflammatoires chroniques de l’intestin), les déficits génétiques d’absorption en magnésium ou les réductions du grêle peuvent mener à un défaut d’absorption du magnésium65–67. Les inhibiteurs de la pompe à protons (IPP) diminuent également l’absorption digestive du magnésium68.
18
Figure 4 : Régulation de l’absorption digestive du magnésium. TRPM6 : transient receptor potential cation channel subfamily M member 6 ; TRPM7 : transient receptor potential cation channel subfamily M member 7 (De Baaij et al, CKJ 2012)
Régulation rénale
Environ 2500 mg de magnésium sont filtrés quotidiennement par les glomérules rénaux dont 90 à 95% sont réabsorbés.
Dix à 25% du magnésium sont réabsorbés dans les tubes proximaux via un transport paracellulaire passif à la faveur de la réabsorption de l’eau et des solutés. Cela est possible grâce à un fort gradient de concentration en magnésium dû à la réabsorption importante de l’eau dans cette même région du rein.
La majorité du magnésium (50 à 60%) est réabsorbée dans la branche large ascendante de Henlé. A ce niveau, le magnésium est réabsorbé passivement par voie paracellulaire grâce au gradient de voltage transépithélial. Les claudines 16 et 19 sont impliquées dans ce processus et forment une jonction serrée sélective au calcium et magnésium facilitant le transport du
Mg2+63,69. L’énergie nécessaire à la réabsorption des cations divalents est fournie par la
différence de potentiel positive dans la lumière tubulaire, indirectement générée par la réabsorption sodée.
19 La régulation fine de la réabsorption du magnésium (environ 10%) est assurée par les cellules épithéliales des tubes contournés distaux. La réabsorption transcellulaire du magnésium suit des voies différentes de celle du sodium et du calcium. Le magnésium entre passivement dans la cellule à la faveur du potentiel transmembranaire. La concentration luminale du magnésium varie de 0.20 à 0.70 mmol/L en fonction des conditions d’étude alors que la concentration intracellulaire est de 0.50 mmol/L de telle sorte que dans certaines conditions l’entrée du magnésium se fait contre un gradient de concentration. Celle-ci se fait principalement par le canal TRPM6 (figure 5). L’activité de ce canal est dépendante d’un voltage transmembranaire apical assuré par les canaux potassiques Kv1.1 sur le versant apical, les canaux potassiques Kir4.1 au pôle basolatéral (recyclage du K+) et les canaux Na-K-ATPases basolatéraux. Si l’entrée apicale du Mg2+ commence à bien être décrite, sa voie de sortie basolatérale reste encore indéterminée.
Ainsi les diurétiques thiazidiques entrainent une hypomagnésémie en augmentant l’excrétion de Na+ par inhibition d’un cotransport sodium-chlore (canal NCC inactivé dans le syndrome de Gitelman (SG)) dans le tube contourné distal. La diminution intracellulaire du sodium inhibe alors à son tour l’expression des canaux TRPM6, à l’origine de l’hypomagnésémie63.
D’autres néphropathies génétiques ou acquises (néphrite interstitielle, syndrome de Fanconi) ou encore iatrogènes (cyclosporine, tacrolimus70, cisplatine….) peuvent induire une hypomagnésémie par défaut de réabsorption et excrétion accrue du Mg2+66. Le diabète augmente également l’excrétion urinaire du magnésium (hypomagnésémie d’origine multifactorielle)71.
20
Figure 5 : Régulation de la réabsorption rénale du magnésium. ROMK : renal outer medullary potassium channel ; ClC-Kb : Chloride channel Kb ; NCC : Na/Cl cotransporteur ; TRPM6 : transient receptor potential cation channel subfamily M member 6 (adapté de Jeroen H. F. de Baaij et al, CKJ 2012)
Stockage osseux et musculaire
Les os et les tissus mous contiennent approximativement 66 et 33% du magnésium de l’organisme respectivement. Le magnésium extracellulaire ne représente qu’1% du magnésium total69. Dans les fibres musculaires, le magnésium régule la contraction en antagonisant l’action du calcium. Dans le tissu osseux, le magnésium en s’associant aux cristaux de carboxyapatite contribue à la densité et à la solidité du squelette72. Le magnésium osseux se situe principalement sur les surfaces osseuses ce qui suggère un échange continue du Mg2+ entre l’os et le sang73.
21
4) Complications rhumatologiques de l’hypomagnésémie
Maladie à dépôts de pyrophosphate de calcium
Une étude cas-témoins incluant 144 patients a relevé une association forte entre nutrition parentérale au long cours et survenue d’une chondrocalcinose au genou avec un odd ratio à 7. Or la nutrition parentérale est reconnue comme étant une cause d’hypomagnésémie d’origine digestive. Dans cette même étude, l’odd ratio pour la survenue d’une chondrocalcinose s’élevait même à 13 pour les patients qui présentaient un magnésium sérique bas74. Un autre cas d’arthrite aiguë à PPC a été rapporté dans un contexte de malabsorption digestive primitive du magnésium65
De nombreux case-reports de maladie à dépôts de PPC associée à une hypomagnésémie d’origine rénale ont aussi été publiés. Une trentaine de cas ont été rapportés dans les années 90 chez des patients étiquetés avec un syndrome de Bartter associé à une hypomagnésémie75,76. En 2018 une nouvelle revue de la littérature a identifié 42 cas rapportés de maladie à dépôts de PPC ou chondrocalcinose secondaires à une hypomagnésémie d’origine rénale. Vingt-trois de ces patients étaient atteints du syndrome de Bartter, cependant les auteurs soulignent que ces patients étaient probablement étiquetés à tort du syndrome de Bartter sans certitude génétique alors qu’ils avaient un tableau clinico-biologique évocateur de syndrome de Gitelman77. Parmi ces 42 cas, 5 avaient un syndrome de Gitelman prouvé génétiquement. Le cas rapporté par Ea et al. était celui d’un patient atteint du syndrome de Gitelman prouvé génétiquement qui présentait des arthrites récidivantes des poignets et genoux dans un contexte de myalgies, asthénie et crampes. Ses radiographies retrouvaient des dépôts de chondrocalcinose articulaire au niveau des ménisques du genou, de la symphyse pubienne et des ligaments triangulaires des carpes78. Les 4 autres cas de syndrome de Gitelman ont été rapportés par Punzi et al : 3 des patients présentaient des atteintes des genoux et 1 une atteinte des épaules. Des cristaux de PPC ont été mis en évidence dans le liquide articulaire de 3 patients79. Deux autres cas d’arthrite aiguë à PPC ont été rapportés chez des transplantés hépatiques avec hypomagnésémie sous tacrolimus70
Physiopathologie de la maladie à dépôts de pyrophosphate de calcium dans l’hypomagnésémie
Les mécanismes à l’origine de la formation des cristaux de PPC dans l’hypomagnésémie sont mal connus. Cependant, il a été décrit in vitro que l’ion magnésium augmenterait directement la solubilité des cristaux de PPC80. D’autre part, le magnésium est un cofacteur de la
22 phosphatase alcaline qui hydrolyse le PPi en Pi81,82. Les mécanismes de formation des cristaux de PPC dans l’hypomagnésémie seraient donc liés d’une part à une solubilité diminuée des cristaux et d’autre part à une accumulation de PPi extracellulaire par défaut enzymatique de la phosphatase alcaline.
Complications neuromusculaires de l’hypomagnésémie
La déplétion en magnésium s’accompagne d’autres signes notamment neuromusculaires. Au-delà de l’asthénie qui est aspécifique mais fréquente, on observe des crampes, des crises de tétanie, une fatigabilité musculaire, des fasciculations voire des paralysies flasques. Les patients peuvent également présenter des spasmes carpopédaux ou un signe de Trousseau. Le signe de Chvostek encore appelé signe du facial a également été décrit chez les patients présentant une hypomagnésémie ou une hypocalcémie traduisant une hyperexcitabilité neuro-musculaire 61,66,83.
Impact sur la densité minérale osseuse
Des études épidémiologiques ont démontré une corrélation entre apports en magnésium élevés et densité minérale osseuse élevée et inversement. Cependant ces études sont hétérogènes, ont intéressé des populations différentes avec des méthodes d’investigation diverses et ont peu intéressé les hommes. D’autre part, le régime pauvre en magnésium est souvent également déficient en autres nutriments, notamment le calcium, ce qui peut aussi affecter la densité minérale osseuse et donc constituer un biais d’interprétation84.
Chez le rat, le régime pauvre en magnésium a révélé des défauts de croissance osseuse et une ostéopénie par diminution du nombre d’ostéoblastes et augmentation de la résorption ostéoclastique85.
L’hypomagnésémie semble également corrélée chez l’homme et le rat à un défaut de sécrétion de la PTH et a un taux de 1-25 OH vitamine D bas ce qui pourrait conduire à un turnover osseux diminué et un os adynamique66,85.
De façon intéressante, des travaux chez la souris mutée Ncc-/- et chez les patients atteints de Gitelman ont retrouvés à l’inverse une densité minérale osseuse augmentée. Les mécanismes évoqués sont ceux d’une hypoparathyroïdie relative chez l’homme et d’une résistance osseuse à la PTH chez la souris ; causées par la déplétion en magnésium, et ce avec une calcémie normale ou basse malgré une réabsorption accrue du calcium86.
23 Partie II
1) Définition du syndrome de Gitelman et épidémiologie
Le syndrome de Gitelman est une tubulopathie génétique autosomique récessive. Il s’agit d’une maladie rare dont la prévalence est estimée à 1/40 000 ; cependant il s’agit de la tubulopathie génétique la plus fréquente. Le syndrome est dû à une mutation inactivatrice du gène SLC12A3 codant pour un transporteur sodium-chlore sensible aux thiazidiques (NCC) exprimé au niveau apical des cellules épithéliales bordant le tube contourné distal du rein. La mutation inactivatrice en cause (inactivation bi allélique) touche le gène SLC12A3 et peut être homozygote ou hétérozygote. Plus de 350 mutations ont été décrites à ce jour. La prévalence des personnes hétérozygotes pour ce gène dans la population caucasienne est estimée à 1%87.
La majorité des patients atteints du syndrome de Gitelman sont hétérozygotes composites mais certains portent un seul type de mutation88.
2) Historique de la description du syndrome de Gitelman
L’hypokaliémie hypomagnésémie familiale a été décrite pour la première fois par le Docteur Gitelman en 1966 chez 3 patients dont 2 étaient soeurs89,90. Le syndrome clinico-biologique a ensuite été baptisé du nom de son premier descripteur. La caractérisation génétique et moléculaire de la maladie a été réalisée en 199688.
3) Physiopathologie du syndrome de Gitelman
Le syndrome de Gitelman est une tubulopathie avec perte de magnésium et de sodium. Celle-ci entraine une activation du système rénine angiotensine aldostérone avec une augmentation des taux de rénine et d’aldostérone. L’élévation de l’aldostérone permet une réabsorption accrue du sodium dans le tube collecteur via les canaux ENaC aux dépens d’une fuite urinaire du potassium et des ions H+ responsables de l’hypokaliémie et de l’alcalose métabolique87.
L’hypocalciurie est variable et peut s’expliquer par une réabsorption passive accrue du Ca2+ dans le tubule proximal91. L’augmentation de la réabsorption rénale de calcium pourrait expliquer en partie la formation des cristaux PPC de même que l’augmentation de la densité minérale osseuse observée chez les patients avec syndrome de Gitelman ou sous thiazidiques au long cours (par baisse du remodelage osseux).
L’hypomagnésémie est fréquente et peut s’expliquer par deux principaux mécanismes. Premièrement, la mutation du canal NCC et la fuite sodique s’accompagnent d’une diminution
24 du nombre de canaux TRPM6 responsables de l’absorption rénale du magnésium au pôle apical des cellules du tube contourné distal91,92. Cette down-régulation des canaux TRPM6 pourrait être liée à l’augmentation des taux d’aldostérone, réactionnelle à la fuite sodique93. Il est aussi probable que le type de mutation du gène SLC12A3 influence la régulation des canaux TRPM6 comme suggéré dans l’étude de Fujimura et al.94. Deuxièmement, la dépolarisation membranaire induite par la fuite potassique ne permettrait plus d’assurer un voltage transmembranaire efficace pour la réabsorption rénale du magnésium95,96.
4) Diagnostic et présentation clinique
Le plus souvent la découverte du syndrome de Gitelman est fortuite, devant la découverte d’une hypokaliémie sur un bilan de routine comme l’a précisé l’étude japonaise de Fujimura et al. (figure 6). La maladie peut aussi être à l’origine de symptômes divers et aspécifiques tels que la faiblesse musculaire, l’asthénie, l’appétence pour le sel, la soif, la nycturie, les crampes, les paresthésies, les douleurs abdominales… Parmi ces symptômes, la tétanie était à l’origine de 32% des diagnostics de syndrome de Gitelman dans l’étude de Fujimura et al. Le diagnostic est porté le plus souvent chez l’adolescent ou l’adulte jeune. Les patients sont normo ou hypotendus.
Les arguments en défaveur du syndrome de Gitelman sont une histoire familiale de malformation rénale, une notion de poly-hydramnios ou de rein fœtal hyperéchogène, un début des symptômes avant 3 ans, un traitement chronique par diurétiques ou laxatifs, une hypokaliémie inconstante ou absente, une hypertension artérielle (HTA) ancienne ou encore des manifestations d’hyperhydratation extracellulaire88.
25
Figure 6 : Circonstances menant au diagnostic du Syndrome de Gitelman (Fujimura et al., Kidney Int Rep. 2019)
5) Pénétrance, formes sévères et complications
La pénétrance du syndrome de Gitelman est très variable et les phénotypes cliniques sont divers même chez des patients présentant les mêmes mutations97. Cette variabilité pourrait s’expliquer par des différences génotypiques, le sexe, des facteurs environnementaux, des phénomènes compensateurs comme suggéré dans l’étude de Riveira-Munoz où un sous-groupe de patients de sexe masculin présentant une mutation spécifique du gène SLC12A3 présentait un phénotype sévère98.
Outre les symptômes aspécifiques déjà décrits, le syndrome peut parfois mener à des manifestations cliniques sévères avec un début des symptômes avant l’âge de 6 ans, un retard de croissance ou une puberté retardée (par dysrégulation de l’axe GH/IGF1).
La plupart des complications de la maladie est liée aux perturbations électrolytiques. Sur le plan articulaire on peut observer une maladie à dépôts de PPC liée à l’hypomagnésémie ou encore des calcifications scléro-choroïdales. Sur le plan cardiaque, le QT est souvent allongé ce qui peut favoriser la survenue des troubles du rythme. Sur le plan néphrologique on peut observer des protéinuries glomérulaires ou des néphropathies chroniques. Les douleurs abdominales sont également possibles du fait d’une parésie intestinale. Enfin l’insulino-résistance et/ou l’intolérance au glucose ont été décrits chez certains patients atteints du
26 syndrome de Gitelman99. Cependant il faut préciser que ces phénotypes sévères ont été décrits parfois chez des patients sans confirmation génétique de la maladie.
L’altération de la qualité de vie peut être similaire à celle rencontrée dans l’insuffisance cardiaque ou le diabète100.
L’évolution de la maladie est difficilement appréciable notamment en ce qui concerne les risques de chondrocalcinose, de néphropathie chronique, d’HTA secondaire ou encore d’arythmie cardiaque.
6) Tableau biologique
Le tableau biologique typique du syndrome de Gitelman associe le plus souvent :
• une hypokaliémie chronique <3.5 mmol/L avec kaliurèse inadaptée (K+/créatinine urinaire > 2) en l’absence de traitement hypokaliémiant (sinon ce dernier doit être arrêté 48h avant l’analyse)
• une alcalose métabolique
• une hypomagnésémie <0.7 mmol/L d’origine rénale avec fraction d’excrétion urinaire du Mg > 4% (en l’absence de traitement hypomagnésémiant)
• une hypocalciurie (calcium/créatinine urinaire < 0.2) • une rénine élevée (activité ou fraction plasmatique) • une fraction d’excrétion du chlore > 0.5%
Cependant, les caractéristiques clinico-biologiques de la maladie ne sont pas toujours suffisantes pour différencier le syndrome de Gitelman des autres néphropathies avec fuite rénale de sodium. Un phénotype SG-like a déjà été décrit avec des mutations du gène CLCNKB codant pour un canal chlore ClC-Kb, une cause classique du syndrome de Bartter de type III. La localisation du canal ClC-Kb dans le tube contourné distal explique le chevauchement phénotypique avec le syndrome de Gitelman.
7) Diagnostic génétique
Le diagnostic génétique (séquençage du gène) est à proposer systématiquement devant une suspicion clinico-biologique de syndrome de Gitelman ou bien dans le cadre du dépistage d’un cas apparenté. Les patients adultes atteints ont un faible risque de transmettre la maladie à leurs enfants (environ 1 cas sur 400) en l’absence de consanguinité du couple87. Le test
27 diagnostique à l’hydrochlorothiazide n’est plus recommandé et le plus souvent la biopsie rénale n’est pas nécessaire88.
8) Diagnostics différentiels
Le syndrome de Bartter est le principal diagnostic différentiel. L’âge de début est souvent plus précoce (<3 ans) avec un retard de croissance associé, une polyurie et une magnésémie normale.
Les abus de laxatifs ou de diurétiques et les vomissements chroniques sont aussi à l’origine d’hypokaliémie. Dans ces cas, un screening urinaire des diurétiques et laxatifs et un dosage du chlore urinaire (<25 mEq/l si vomissements) permettent de faire le diagnostic.
La mutation KCNJ10 codant pour canal potassique KCNJ10/Kir4.1 peut également être à l’origine d’une hypokaliémie mais le tableau clinique associe volontiers épilepsie, ataxie et surdité101.
La mutation HNF1B peut mimer l’hypomagnésémie du syndrome de Gitelman mais le tableau se caractérise par un diabète et une insuffisance rénale chronique précoces avec une histoire familiale de maladie génétique autosomique dominante, une cytolyse hépatique, une hyperuricémie, des malformations rénales ou urogénitales102.
Les autres diagnostics différentiels à envisager sont la fibrose kystique, le traitement par cisplatine ou les maladies auto-immunes comme le syndrome de Goujerot Sjögren.
9) Prise en charge thérapeutique
La prise en charge thérapeutique est tirée de l’expérience clinique, d’études observationnelles et de cas rapportés. Il n’existe pas de recommandations clairement établies et la prise en charge doit être individualisée. Premièrement, un régime riche en sel, en potassium et en magnésium est recommandé. Une supplémentation en K+ et Mg2+ est indiquée en cas d’hypokaliémie et d’hypomagnésémie persistantes en fractionnant si possible les prises orales pour une meilleure tolérance digestive (diarrhée sous supplémentation du magnésium). Une supplémentation intra-veineuse peut être proposée lorsque les symptômes liés aux perturbations électrolytiques sont sévères. Les objectifs de la supplémentation sont de corriger au moins en partie les carences ioniques afin d’améliorer les symptômes de la maladie. Un équilibre doit être trouvé entre la correction des pertes et la tolérance de la
28 supplémentation. En cas d’hypokaliémie ou d’hypomagnésémie symptomatiques réfractaires, les diurétiques épargnant le potassium, les inhibiteurs de l’enzyme de conversion (IEC) et les AINS sont parfois utilisés. L’éducation thérapeutique et un suivi néphrologique réguliers sont indispensables à la prise en charge. Enfin les médicaments responsables d’hypokaliémie, d’hypomagnésémie (comme les IPP) ou allongeant le QT sont à proscrire88.
29
Objectifs
L’objectif principal de l’étude est de dépister la prévalence de la maladie à dépôts de PPC ou de la chondrocalcinose dans une cohorte de patients atteints du syndrome de Gitelman. En effet, aucune étude de prévalence n’a été publiée à ce jour dans cette population et seuls des case-reports isolés ont été rapportés.
Les objectifs secondaires sont de décrire les caractéristiques cliniques et biologiques associées à maladie à dépôts de PPC ou la chondrocalcinose dans cette population afin de mieux comprendre la physiopathologie de la maladie microcristalline et de tenter d’identifier des facteurs de risques de survenue de maladie à dépôts de PPC dans cette population.
30
Matériels et méthodes
Population
Les patients ont été inclus à partir du centre de référence des tubulopathies (dont le syndrome de Gitelman) basé à l’hôpital européen Georges Pompidou (filière de soin ORKID). Le critère d’inclusion principal était le diagnostic phénotypiquement établi de syndrome de Gitelman. Tous les patients éligibles ont été invités à bénéficier d’une consultation de dépistage de la maladie à dépôts de PPC auprès d’un rhumatologue senior de l’hôpital Lariboisière. Lors de la consultation chaque patient a bénéficié d’un interrogatoire médical et d’un examen physique standardisé.
Données recueillies
Les données anamnestiques ont été récupérées de façon rétrospective à partir des comptes-rendus de consultations néphrologiques et de façon prospective lors de la consultation de rhumatologie. Les antécédents du patient, les antécédents familiaux, le typage génétique
SLC12A3, le traitement en cours, les arthralgies ou rachialgies actuelles ou anciennes, l’horaire
des douleurs ostéoarticulaires, les traitements rhumatologiques reçus et les antécédents de ponction articulaires étaient recherchés.
Sur le plan physique, le poids, la taille, la présence d’un trouble de la statique rachidienne, la présence de synovites ou d’enthésites étaient notifiés. Les patients présentant un trouble de la statique rachidienne ont bénéficié d’un EOS.
Sur le plan biologique, le bilan néphrologique initial au moment du diagnostic de Gitelman a été recueilli ainsi que le dernier bilan effectué auprès du néphrologue référent. Dans le service de rhumatologie ou en laboratoire de ville, tous les patients ont bénéficié d’un nouveau prélèvement sanguin pour contrôle du bilan phosphocalcique, de la magnésémie et afin d’éliminer les autres causes de maladies à dépôts de PPC (ferritinémie, CST, PTH, PAL). Un ionogramme urinaire était également prescrit dans le même temps.
Dépistage de la maladie à dépôts de PPC
Chaque patient a bénéficié d’un dépistage de la chondrocalcinose par des radiographies systématiques de toutes les articulations périphériques (épaules, coudes, mains, poignets, bassin, hanches, genoux, chevilles et pieds) et du rachis cervical. Les radiographies étaient relues par le rhumatologue senior et un radiologue senior à la recherche de dépôts
31 caractéristiques de la chondrocalcinose. Chaque patient a également bénéficié d’une échographie de dépistage à la recherche de dépôts microcristallins hyperéchogènes évocateurs, en ciblant les poignets, les genoux et les chevilles ainsi que les autres articulations si elles étaient symptomatiques. Les échographies ont été effectuées dans le service hospitalier par un échographiste senior, en aveugle des données cliniques. Les patients souffrant de cervicalgies ont bénéficié d’un scanner cervical à la recherche de dépôts microcristallins rachidiens. Quelques patients ont également bénéficié au cas par cas d’un scanner des pieds et chevilles devant des arthralgies invalidantes des membres inférieurs.
Relecture radiologique
L’ensemble des examens radiologiques (radiographies et scanners) effectués dans le centre hospitalier ont été relus en double aveugle par un radiologue et un rhumatologue seniors. La présence ou non de dépôts et leur localisation précise étaient notifiées.
Analyse statistique
Une corrélation entre les symptômes articulaires ou la présence d’arthropathies et le nombre de sites articulaires touchés par les dépôts de chondrocalcinose a été étudiée par test de Spearman ou T de Student.
Une corrélation entre âge, kaliémie, calcémie ou magnésémie et chondrocalcinose disséminée a été étudiée par régression linéaire.
La présence d’une arthropathie en fonction de la présence de dépôts de chondrocalcinose, de l’âge et de l’indice de masse corporelle a été étudiée après régression logistique.
32
Résultats
Caractéristiques de la population d’inclusion
Cinquante-sept patients ont été inclus, il s’agissait en majorité de femmes (63.2%, n=36) d’âge moyen 46.5 ± 12.4 ans. L’âge au diagnostic du syndrome de Gitelman était de 29.1± 14.1 ans en moyenne. Comme attendu, la kaliémie au diagnostic du syndrome de Gitelman était basse, en moyenne de 2.6 ± 1.1 mM, de même que la magnésémie qui était en moyenne de 0.6 ± 0.3 mM. La majorité des patients présentait une mutation hétérozygote composite du gène
SLC12A3 (77.2%, n=44). Neuf patients étaient apparentés au premier degré : 6 étaient issus
de la même fratrie et 5 avaient un lien mère-enfant. Chez 18 patients de la cohorte, un antécédent de syndrome de Gitelman chez un apparenté du 1er degré était retrouvé (tableau 1).
Hommes, n (%) 21 (36.8)
Age moyen, moyenne ± SD (années) 46.5 ± 12.4
Taille, moyenne ± SD (m) 1.64 ± 0.1
Poids, moyenne ± SD (kg) 64.8 ± 13.3
IMC, moyenne ± SD (kg/m2) 24.0 ± 4.1
Age au diagnostic du SG, moyenne ± SD (années) 29.1± 14.1
Kaliémie au diagnostic, moyenne ± SD (mM) 2.6 ± 1.1
Magnésémie au diagnostic, moyenne ± SD (mM) 0.6 ± 0.3
Type de mutation
-hétérozygote composite SCL12A3 -homozygote SLC12A3 -autres 44 (77.2) 6 (10.5) 6 (10.5) Supplémentation en K+, n (%) 54 (94.7) Supplémentation en Mg2+, n (%) 51(89.5)
Tableau 1 : caractéristiques de la population (SG : syndrome de Gitelman, IMC ; indice de masse corporelle)
La majorité des patients présentait une symptomatologie articulaire (93.0%) puisque seulement 4 patients n’avaient aucune plainte douloureuse ostéoarticulaire. Les arthralgies inflammatoires étaient retrouvées chez 42.1% des patients (n=24) avec une prédominance des atteintes mono et oligo-articulaires. Les cervicalgies étaient rapportées chez 47.4% des patients (n=27). Le nombre moyen d’articulation douloureuse était de 3.7 ± 3.0 (tableau 2).
33
Nombre d’articulation(s) douloureuse(s), moyenne ± SD 3.7 ± 3.0
Arthralgies inflammatoires, n (%) Mono-articulaire Oligo-articulaire Polyarticulaire Toutes 22 (38.6) 14 (24.6) 4 (7.0) 24 (42.1) Arthralgies mécaniques, n (%) 34 (59.6) Rachialgies, n (%) Cervicalgies Lombalgies Toutes 27 (47.4) 34 (59.6) 41 (71.9) Douleurs tendineuses, n (%) 16 (28.1) Crampes, n (%) 31 (54.4) Paresthésies, n (%) 23 (40.4)
Tableau 2 : Symptômes ostéoarticulaires et neuromusculaires des patients inclus
Sept patients avaient déjà bénéficié d’une ponction de liquide articulaire au cours de leur suivi médical antérieur. Le plus souvent la recherche de cristaux par anatomopathologie n’avait pas été réalisée ou les résultats n’étaient pas retrouvés a posteriori. Cependant la présence de cristaux de pyrophosphate de calcium a pu être documentée chez 3 patients.
Mise en évidence des dépôts de chondrocalcinose
Des dépôts de chondrocalcinose ont été mis en évidence chez 45 patients soit 79% des patients de la cohorte. Ces dépôts ont été retrouvés sur les radiographies standard (n=33), par l’échographie articulaire (n=26) et grâce au scanner cervical (n=27). Quatre patients ont bénéficié d’un scanner des chevilles et pieds : des dépôts de chondrocalcinose qui n’étaient pas visibles sur les radiographies standard ont été mis en évidence chez 3 d’entre eux.
Le scanner cervical a permis de révéler des dépôts chez 13 patients avec radiographies cervicales étiquetées normales. Chez 7 patients, l’échographie des mains et poignets a permis d’identifier une chondrocalcinose alors que les radiographies articulaires étaient normales, ces dépôts visualisés uniquement à l’échographie siégeaient principalement en intra-carpien ou dans le ligament triangulaire du carpe. Aux genoux, seuls 2 patients ont présenté une échographie pathologique malgré des radiographies normales avec des dépôts constatés aux ligaments collatéraux et aux ménisques. Aux pieds et aux chevilles, l’échographie était pathologique chez 3 patients avec radiographies normales : les dépôts visualisés étaient tendineux (achilléen, tibial postérieur).
34
Localisation des dépôts de chondrocalcinose
Figure 7 : Répartition par site articulaire des dépôts de chondrocalcinose radiologiques (dépôts radiographiques, échographiques et scanographiques confondus) dans la cohorte de patients avec syndrome de Gitelman
Les dépôts siégeaient uniquement au niveau des articulations périphériques chez 13 patients (22.8%), uniquement au niveau du rachis cervical chez 9 patients (15.8%) et à la fois en axial et en périphérie chez 23 patients (40.4%).
Les atteintes anatomiques préférentielles des dépôts de chondrocalcinose étaient par ordre de fréquence décroissante le rachis cervical (n=32), les genoux (n=30), les mains et poignets (n=29), les chevilles et pieds (n=22). De façon moins fréquente, des dépôts ont été retrouvés aux épaules (n=19), aux hanches (n=13), au bassin (n=11) et enfin aux coudes (n=8). Le plus souvent, les atteintes articulaires périphériques étaient bilatérales et symétriques (figure 7). Au rachis cervical, les dépôts siégeaient de façon préférentielle dans les disques cervicaux (n=25). La seconde localisation la plus fréquente était la région C1-C2 : cette dernière était pathologique chez 24 patients issus de la cohorte avec des dépôts localisés le plus souvent au
35 niveau du ligament rétro odontoïdien dans sa portion horizontale ou verticale et au niveau des ligaments alaires. Onze patients avaient des dépôts dans les articulations temporo-mandibulaires et 11 patients avaient des dépôts ligamentaires autres (ligament vertébral antérieur ou postérieur, ligament jaune). Enfin, 5 patients avaient des dépôts calciques en regard d’une épineuse cervicale, le plus souvent C7 et un patient présentait des dépôts d’une articulaire postérieure cervicale (figure 8).
Figure 8 : Scanner cervical en coupe sagittale (A) retrouvant des dépôts calciques diffus intra discaux, péri-odontoïdiens et ligamentaires chez un patient atteint de syndrome de Gitelman ; en (B) coupe sagittale centrée sur l’articulation temporo-mandibulaire retrouvant également des dépôts calciques intra-articulaires
Aux genoux, les dépôts siégeaient dans les ménisques chez 25 patients, dans les cartilages hyalins des condyles, de la trochlée ou des plateaux tibiaux chez 19 patients et dans les tendons ou ligaments périarticulaires chez 18 patients. Des dépôts capsulaires postérieurs diffus ont été observés chez 9 patients (figure 9).
36
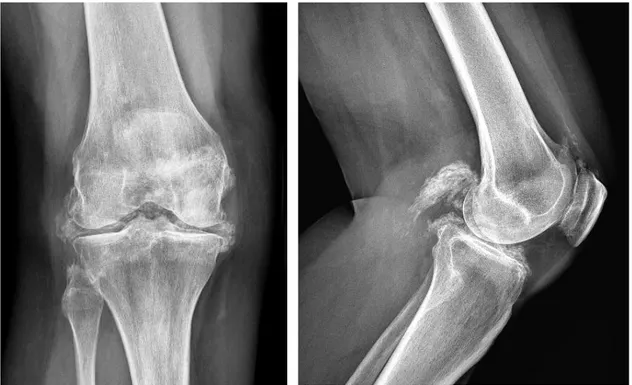
Figure 9 : Radiographies de face et de profil d’un genou présentant des dépôts calciques diffus méniscaux, ligamentaires, hyalins et capsulaires chez un patient avec syndrome de Gitelman
Au niveau des mains et poignets, les atteintes préférentielles étaient le ligament triangulaire du carpe (n=20) et en intra carpien (n=18). Les dépôts des métacarpo-phalangiennes, interphalangiennes proximales ou distales ont été retrouvés chez 10 patients (figure 10).
Figure 10 : Radiographies des mains et poignets de face présentant des dépôts calciques intra-carpiens et des ligaments triangulaires des carpes
37 Aux chevilles et aux pieds, les tendons achilléens (n=14) et les autres tendons et ligaments périarticulaires (n=14) étaient les localisations préférentielles des dépôts calciques. Le tarse était touché chez 11 patients et les métatarso-phalangiennes chez 10 patients. L’articulation tibio-talienne était siège de dépôts chez 5 patients et l’aponévrose plantaire chez 6 patients (figures 11 et 12).
Figure 11 : Radiographies d’un pied et d’une cheville droites présentant des dépôts calciques du tendon achilléen et du fascia plantaire
38
Figure 12 : Scanner des pieds et chevilles chez un patient atteint de syndrome de Gitelman : en coupe coronale (A) présence de dépôts calciques du cartilage hyalin talo calcanéen et probablement en intratendineux (tibio-taliens) ; en coupe sagittale (B) dépôts calciques retrouvés dans le sinus du tarse
Aux épaules, les dépôts siégeaient le plus souvent au niveau du cartilage gléno-huméral (n=13), dans l’acromio-claviculaire (n=8) ou dans les tendons de la coiffe (n=6) de même que le cartilage coxofémoral était le plus souvent touché aux hanches. Au bassin (hors coxo-fémorale), la symphyse pubienne était le siège de dépôts calciques chez 11 patients.
A
39
Chondrocalcinose diffuse et facteurs de risque
Les dépôts de chondrocalcinose étaient diffus touchant au moins 3 articulations différentes chez 28 patients soit 49.1% des patients de la cohorte. Onze patients avaient au moins 10 atteintes articulaires, 19 patients entre 3 et 9 articulations atteintes, 15 patients entre 1 et 2 articulation(s) atteinte(s) et 12 n’avaient pas de dépôts de chondrocalcinose.
Les patients présentant des dépôts diffus étaient plus souvent symptomatiques que les autres puisque le nombre d’articulations douloureuses était significativement corrélé à la présence de dépôts (p<0.0001). La présence d’arthralgies inflammatoires était également significativement corrélée à la présence de dépôts de chondrocalcinose (p<0.0001).
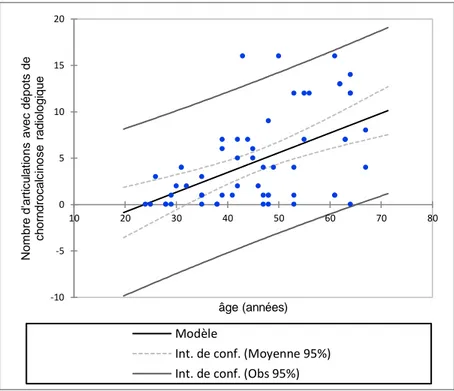
L’âge était un facteur de risque de présenter une chondrocalcinose disséminée. Ainsi les moins de 35 ans avaient en moyenne 1.2 ± 1.5 articulations touchées, ceux entre 35 et 49 ans 3.5 ± 3.7 articulations touchées et ceux de plus de 49 ans avaient en moyenne 8.2 ± 5.5 articulations avec dépôts de PPC (p<0.0001) (figure 13).
Figure 13 : Nombre d’articulations atteintes par la chondrocalcinose en fonction de l’âge des patients atteints du syndrome de Gitelman (régression linéaire ; R2=0.277 ; p<0.0001)
-10 -5 0 5 10 15 20 10 20 30 40 50 60 70 80 N o m b re d 'a rti cu la ti o n s a v e c d é p o ts d e ch o rn d ro ca lc in o se r a d io lo g iq u e âge (années) Modèle
Int. de conf. (Moyenne 95%) Int. de conf. (Obs 95%)
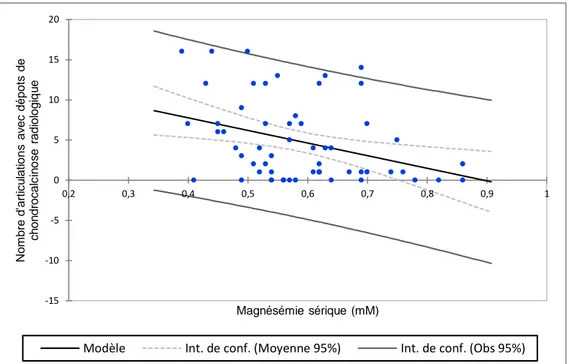
40 Le taux de magnésium sérique était inversement corrélé au nombre de sites articulaires présentant des dépôts de chondrocalcinose (p=0.007) (figure 14) et au type de structure anatomique atteinte dans chaque articulation (que ce soit le cartilage, les tendons ou les ligaments) (p=0.005). L’hypomagnésémie restait un facteur de risque indépendant d’atteinte diffuse de chondrocalcinose après ajustement sur l’âge (p<0.0001).
Figure 14 : Nombre d’articulations atteintes par la chondrocalcinose en fonction la magnésémie sérique chez les patients atteints du syndrome de Gitelman (régression linéaire ; R2=0.125 ; p<0.0001)
La présence de dépôts n’était pas corrélée au niveau de kaliémie (p =0.593) ni au taux de calcémie (p=0.083) sériques (tableau 3).
-15 -10 -5 0 5 10 15 20 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1 N o m b re d 'a rti cu la ti o n s a v e c d é p o ts d e ch o n d ro ca lc in o s e r a d io lo g iq u e Magnésémie sérique (mM)