Development of a microfluidic platform for integrated DNA sequencing protocols
Texte intégral
Figure
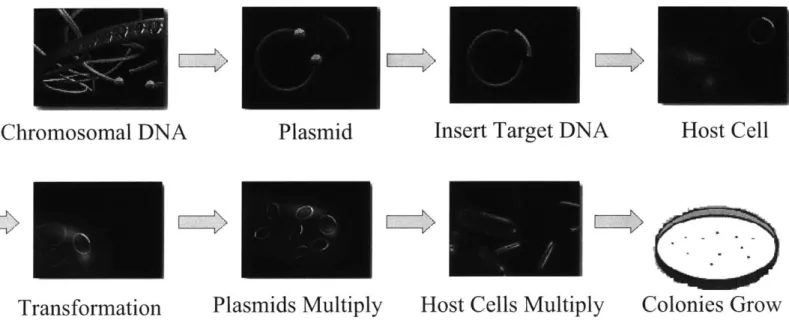
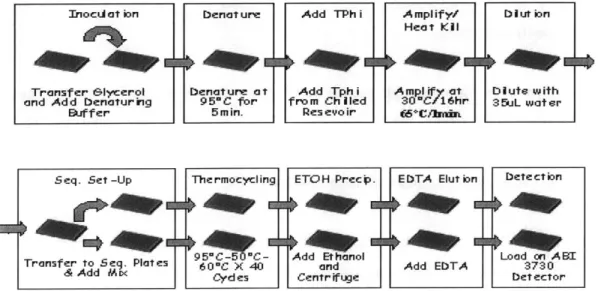
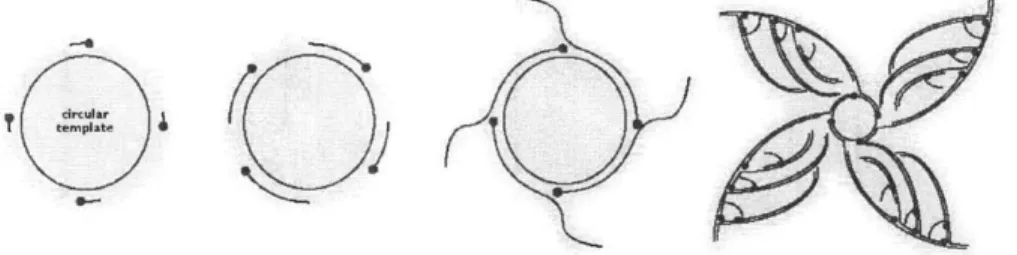
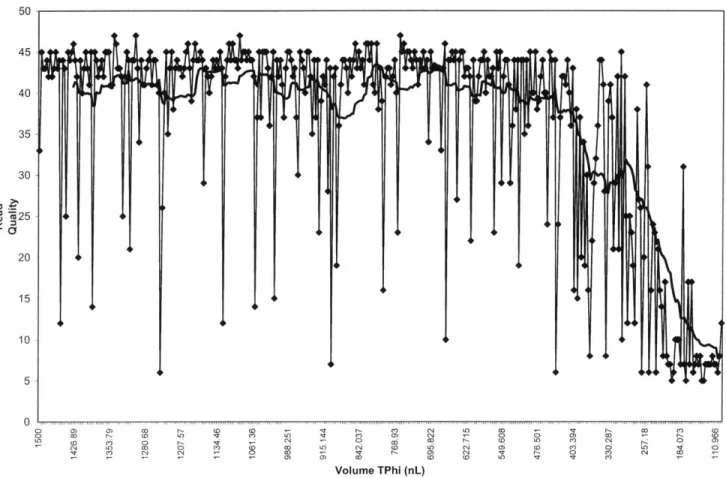





Documents relatifs
This platform editorializes semantic enrichments carried out by the TRIPLE project by proposing the discovery of scientific issues in the fields of SSH by taking advantage of
As such, genes such as KMT2D / MLL2 , although frequently mutated in mature B cell malignancies, were not selected due to the lack of clear evidence of their clinical significance,
In this thesis, we assume that the present value of a loan and the cost of its prepayment option are defined according to the intensity of default of the borrower, the short
In consequence subepithelial fibroblast network is necessary to integrate external signals and to provide the villi the adequate mechanical properties in response
Here, philosophy of education has no core in theories of pragmatism, there are no philosophical schools centering Dewey and research concentrates on national
These PDB structures are used as templates to guide the align- ment of the original sequences using structure-based sequence alignment methods like SAP or Fugue.. The final result is
Figure 4.9: Realistic packet ordering - Comparison of the distribution of packet sizes (a, b, c), the number of packets per bidirectional flow (d, e, f), the number of bytes
We observed that helping develop language re- sources (therefore, natural language processing applications) for one’s language is not enough of an incentive to produce the quantity
