HAL Id: hal-02806230
https://hal.inrae.fr/hal-02806230
Submitted on 6 Jun 2020HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Comment agit le gène BMP15 dans 2 espèces de
mammifères mono ou polyovulante : analyse statistique
de données RNA-seq de cellules de granulosa ?
Christèle Robert Granié, Liang Liang Ma
To cite this version:
Christèle Robert Granié, Liang Liang Ma. Comment agit le gène BMP15 dans 2 espèces de mammifères mono ou polyovulante : analyse statistique de données RNA-seq de cellules de granulosa ?. Sciences du Vivant [q-bio]. 2013. �hal-02806230�
RAPPORT DE STAGE
Stage de Quatrième année Génie Mathématique et Modélisation
Stage INRA :
Comment agit le gène BMP15 dans 2 espèces de mammifères
mono ou polyovulante : analyse statistique de données RNA-seq
de cellules de granulosa ?
Juillet-Septembre 2013
MA Liang
Responsables : Christèle Robert-Granié
Gwenola Tosser-Klopp
UR631-SAGA et UMR444-LGC INRA Toulouse
27, chemin de Borde Rouge 31326 Castanet
INSA Toulouse
135 Avenue de Rangueil 31400 Toulouse, France
RAPPORT DE STAGE
Stage de Quatrième année Génie Mathématique et Modélisation
Stage INRA :
Comment agit le gène BMP15 dans 2 espèces de mammifères
mono ou polyovulante : analyse statistique de données RNA-seq
de cellules de granulosa ?
Juillet-Septembre 2013
MA Liang
Responsables : Christèle Robert-Granié
Gwenola Tosser-Klopp
UR631-SAGA et UMR444-LGC INRA Toulouse
27, chemin de Borde Rouge 31326 Castanet
INSA Toulouse
135 Avenue de Rangueil 31400 Toulouse, France
Remerciements
Le stage de 3 mois que je viens de réaliser à l’INRA de Toulouse, restera pour moi une expérience professionnelle tout aussi enrichissante qu’agréable.
Je voudrais tout d’abord remercier mes deux maitres de stage : Christèle Robert-Granié et Gwenola Tosser-Klopp, pour m’avoir suivi et aidé, et pour leur disponibilité tout au long de cette période.
Je souhaite également remercier Sarah Maman, pour m’avoir présenté les aspects bio-informatiques.
Merci aussi à Félicien Shumbusho, mon partenaire de bureau qui m’a accueilli avec un grand sourire et beaucoup d’humour.
Pour finir, j’adresse un grand merci à toute l’équipe de la Station d’Amélioration Génétiques des Animaux (SAGA), pour leur accueil chaleureux.
J'ai pu, grâce à toutes ces personnes, vite intégrer l’équipe et effectuer un stage de qualité dans de très bonnes conditions de travail.
Table des matières
Introduction ... 1
I. Présentation de l’INRA ... 2
i. INRA ... 2
ii. Département de génétique animale ... 2
iii. Centre de Toulouse ... 3
iv. SAGA ... 3
v. LGC ... 4
II. Contexte et objectif du stage ... 5
III. Présentation des données ... 6
i. Séquençage Haut débit ... 6
ii. Traitements bio-informatiques ... 7
iii. Tableau de données ... 9
IV. Présentation des méthodologies et des techniques statistiques mises en œuvre ... 10
i. Analyses descriptives ... 10
ii. Analyses différentielles ... 11
a. Description de la méthode implémentée dans le package DESeq (Simon Anders & Huber, 2010) ... 11
b. Description de la méthode implémentée dans le package edgeR (M. D. Robinson, McCarthy, & Smyth, 2010) ... 13
c. Comparaisons DESeq et edgeR ... 17
V. Présentation des résultats obtenus... 18
i. Analyses des données de Porc ... 18
a. Analyses descriptives ... 18
b. Analyse différentielle des gènes pour les échantillons Porc ... 21
c. Comparaison des résultats obtenus avec les packages DESeq et edgeR ... 27
d. Lien entre les transcrits et les gènes de porc ... 29
ii. Analyses des données de Bovin... 31
a. Analyses descriptives ... 31
c. Comparaisons des résultats obtenus avec les packages DESeq et edgeR... 39
d. Lien entre les transcrits et les gènes de bovin ... 41
iii. Comparaisons entre espèces ... 43
a. Analyses des résultats obtenus avec DESeq ... 43
b. Analyse des résultats obtenus avec edgeR ... 47
c. Comparaison des 210 gènes obtenus avec DESeq et 126 gènes obtenus avec edgeR ……….51
Conclusion ... 52
Références ... 53
Table des illustrations ... 54
Annexes ... 57
i. Analyses des données de transcrits de Porc ... 57
a. Analyses descriptives ... 57
b. Analyse différentielle des transcrits pour les échantillons Porc ... 59
ii. Analyses des données de transcrits de Bovin ... 63
a. Analyses descriptives ... 63
Introduction
A la fin de la 4ème année à l’INSA de Toulouse, tous les étudiants doivent réaliser un stage en lien avec leur spécialité de l’INSA. Le but du stage est d’acquérir une première expérience en tant qu’un ingénieur en entreprise ou laboratoire.
J'ai réalisé cette expérience professionnelle durant 3 mois au sein de l'Institut National de la Recherche Agronomique (INRA) à Toulouse.
Dans une première partie de mon rapport, je présenterai l’INRA, son historique, ses activités et son organisation.
Dans un second temps, je décrirai davantage le contexte et l’objectif de mon stage, ainsi les données sur lesquelles j’ai travaillé.
J’aborderai ensuite des méthodologies et des techniques statistiques mises en œuvre pour les analyses descriptives et les analyses différentielles.
Je détaillerai enfin les résultats obtenus pour les analyses intra-espèces. Et Je finirai par une comparaison entre les résultats de porc et de bovin (étude inter-espèces).
I. Présentation de l’INRA
i. INRA
L'Institut National de la Recherche Agronomique (INRA) est un Établissement public à caractère scientifique et technologique (EPST) fondé en 1946, placé sous la double tutelle du Ministère de la Recherche et Ministère de l’Agriculture et de la Pêche. Premier institut de recherche agronomique en Europe concernant les domaines de l’agriculture, de l’alimentation et de l’environnement, deuxième dans le monde pour ces publications scientifiques en sciences agricoles, sciences de la plante et de l'animal, et deuxième institut de recherche publique français, l'INRA mène des recherches finalisées pour une alimentation saine et de qualité, pour une agriculture durable et compétitive, et pour un environnement préservé et valorisé.
Répartition : En 2013, l'INRA est présent sur 18 centres engagés dans 21 pôles
thématiques prioritaires et répartis en 148 sites de recherche et d’expérimentation en France métropolitaine et outre-mer.
Organisation : Les recherches de l'INRA s’articulent autour de différentes
thématiques réparties au sein de 13 départements scientifiques, 198 unités de recherche, dont 118 unités mixtes et 52 unités expérimentales.
Ressources humaines : Au 31 décembre 2010, l’INRA possède 8488 agents titulaires,
dont 1837 chercheurs, 2590 ingénieurs, 4061 techniciens et administratifs.
Missions:
Produire et diffuser des connaissances scientifiques
Concevoir des innovations et des savoir-faire pour la société
Éclairer, par son expertise, les décisions des acteurs publics et privés
Développer la culture scientifique et technique et participer au débat science-société
Former à la recherche et par la recherche
Budget : 881.61 millions euros en 2013
ii. Département de génétique animale
Le Département de Génétique Animale (GA) est l'un des 13 Départements de recherche de l'Institut National de la Recherche Agronomique (INRA). Il est construit en 9 unités de recherche dont la SAGA et le LGC et 12 unités expérimentales. En 2009, il possède environ 500 agents titulaires, 200 agents dans les unités expérimentales, 55 doctorants et 40 CDD.
Missions :
Comprendre le déterminisme génétique des caractères observés chez les animaux domestiques et plus généralement de contribuer au progrès des connaissances en biologie intégrative animale
Développer les méthodes et les outils d’amélioration et de conservation génétique des populations d’animaux d’élevage
Champs thématiques du département :
CT1 : Le génome : Quelle structure fine, quelle organisation fonctionnelle ? CT2 : L’animal : Quels leviers génétiques pour un élevage innovant et
durable ?
CT3 : Les populations : Comment mieux caractériser, améliorer et plus globalement gérer les populations ?
iii. Centre de Toulouse
Le centre INRA de Toulouse Midi-Pyrénées est l’un des 18 implantations de l’INRA en France. Avec plus de 850 chercheurs, ingénieurs et techniciens INRA donc 600 titulaires, le centre de Toulouse Midi-Pyrénées représente environ 10% des publications et près de 12% des brevets de l’INRA. Il privilège des activités de recherche et d’innovation en réponse à 3 grands enjeux :
Des systèmes de production agricoles végétaux, animaux et forestiers plus durables et adaptes au changement climatique
Une amélioration attentive aux questions de santé
De nouvelles filières de transformation des agro-ressources en faveur d’une valorisation du carbone renouvelable
iv. SAGA
La Station d'Amélioration Génétique des Animaux (SAGA, UR631) a été créée en 1970 suite à la décentralisation du département de Génétique Animale et à la création du Centre de Toulouse. En janvier 2013, l’unité se compose d’environ 45 agents permanents dont 39 agents INRA, 3 agents temporaires (CDD, apprenti) et 8 doctorants, répartis en 5 équipes de recherche.
Les champs d’activité se concentrent sur la méthodologie et outils de la génétique quantitative ; la caractérisation de la variabilité génétique des caractères d’intérêt chez les petits ruminants, lapins et canards ; la caractérisation et méthodes de gestion des populations animales.
v. LGC
Le Laboratoire de Génétique Cellulaire est une unité mixte de recherche qui dépend du Département de Génétique Animale de l’INRA et de l’Ecole Nationale Vétérinaire de Toulouse. Les recherches développées au sein du laboratoire ont pour objectif commun l’acquisition de connaissances sur la structure et le fonctionnement des génomes des espèces d’élevage (espèces avicole, caprine, ovine, porcine). Une de ces missions consiste à rechercher des mutations causales affectant des caractères d’intérêts agronomiques et ainsi participer à l’amélioration génétique des animaux domestiques. L’unité est composée de 54 agents INRA regroupés au sein de 5 équipes de recherche et 2 plateformes technologiques et méthodologiques.
II. Contexte et objectif du stage
Parmi les mammifères, il existe une forte variation du nombre d'ovulations dans les ovaires à chaque cycle sexuel et donc du nombre de nouveau-nés à chaque naissance. En effet, le nombre de petits peut aller de 1 à 2 chez la femme, la vache ou la brebis, à plus de 10 petits chez les rongeurs ou la truie. Des mutations naturelles chez la brebis indiquent que ce nombre d'ovulations peut être contrôlé par l'action d'un seul gène, BMP15 (Bone Morphogenetic Protein). Cette molécule est naturellement synthétisée par l'ovule et elle régule le fait que l'animal soit polyovulant ou mono-ovulant.
Nous souhaitons déterminer si la molécule BMP15 s'est spécialisée au cours de l'évolution des espèces pour contrôler strictement le nombre d'ovulations chez les mammifères. Le concept est basé sur le fait qu'un gène BMP15 pleinement actif conduirait à la mono-ovulation, tandis qu'un gène BMP15 inactif ou moins actif permettrait la poly-ovulation. Ce projet est mené dans un esprit de biologie comparée pour étudier les différences d'activité du gène BMP15 entre espèces mono-ovulante (vache) et poly-ovulante (truie).
L'objectif de ce projet est de comparer la vache (mono-ovulant) avec la truie (poly-ovulant). Pour mesurer l’impact de cette molécule, le gène cible (sur lequel BMP 15 agit) est recherché dans les cellules de la granulosa. Dans ce cadre, nous allons comparer le transcriptome de cellules de granulosa entre les deux espèces, après stimulation par une protéine BMP15 recombinante. Dix réplicats de culture cellulaires pour chacune des deux espèces et des deux conditions étudiés (avec ou sans traitement avec BMP15) ont été réalisés. Les ARN messagers sont séquencés par séquençage haut débit (RNASeq).
L’objectif de mon stage est donc de mettre en œuvre des méthodes statistiques adéquates pour analyser ces données et pour répondre à deux questions :
1. Quels sont les gènes ou les transcrits dont l’expression est modifiée par le traitement des cellules avec BMP15 dans chaque espèce ?
2. Comparer les gènes différentiellement exprimés dans les deux espèces pour voir si l’effet du traitement est différent selon l’espèce.
III. Présentation des données
i. Séquençage Haut débit
Pour chaque espèce (vache et truie), nous disposons de 10 réplicats (10 animaux) et 2 ovaires par animal. Des effets phénotypiques (d'étalement) ont été observés, mettant ainsi en évidence une variabilité entre animaux. Comme il y a un animal par culture, cette expérience pourra permettre d’étudier la variabilité individuelle. Des cellules folliculaires sont mises en culture avec de la BMP 15 (humaine), in vitro pendant 48H, pour cumuler des ARNm. Il y a ensuite extraction du matériel génétique (10 neg (non traités par BMP15) + 10 pos (traités par BMP15) = 20 ARNm par espèce). Les animaux utilisés sont des animaux d'abattoirs jeunes donc impubères, avec des cellules de granulosa à maturité identique.
Les 20 échantillons de porc et les 20 échantillons de bovin ont été séquencés automatiquement (avec un robot) mais avec des kits différents (un tampon spécifique qui contient des colonnes de fixation de l'ADN ou de l'ARN). Des différences entre bovins et porcs pourraient être observées mais aucune différence n’est attendue intra-bovins ou intra-porcs. Les 20 échantillons intra-espèce taggés ont étés mélangés pour un séquençage sur 2 lanes indépendantes afin de s’affranchir d’un effet lane éventuel. Chaque jeu contient environ la moitié des séquences d'un individu. Les données devront donc être normalisées avant toute analyse statistique. La méthode de RNAseq (séquençage à haut débit de l’ARN) est utilisée pour séquencer les ARN messagers. Elle consiste en plusieurs étapes. D’abord, l’ARN est converti en petits fragments d’ADN qui peuvent être séquencés. Puis, la librairie de cDNA d’un échantillon est placée sur 1 des 8 couloirs (8 lanes) de la flow-cell. Les fragments individuels de cDNA s’attachent à la surface du couloir et par suite subissent une étape d’amplification pour être convertis en clusters de double-brin d’ADN (Figure 1) (Bouchez & Marsaud, 2012).
La flow-cell est ensuite placée dans le séquenceur où chaque cluster est séquencé en parallèle. A chaque cycle (Figure 2), 4 nucléotides fluorescents sont ajoutés et le signal émis par chaque cluster est enregistré (Gall, 2011) .Un cluster est un ensemble de lecture. En théorie, la longueur de lecture est le nombre de cycles, mais il se peut que la synthèse ne soit pas 100% efficace.
Figure 2 : Cycle pour calculer le nombre de lecture
Le processus est répété pour un nombre donné de cycles. Les séquences de porc et de bovin sont déjà connues. Les lectures sont ensuite alignées sur ces séquences connues pour calculer le nombre de lectures par transcrits ou gènes. Le comptage pour un gène ou un transcrit est égal au nombre total de lectures.
ii. Traitements bio-informatiques
La plateforme PlaGe Toulouse a ensuite créé une banque à partir du séquençage du matériel génétique de chaque cellule : une banque pour les porcs, une banque pour les bovins. Les fichiers fastq issus du séquençage sont analysés sur la plateforme NG6. Le FASTQC Report donne des statistiques sur les lectures et les qualités des données. Ensuite, le programme TopHat est utilisé pour mapper les lectures de RNA-seq contre la banque de référence, puis nous analysons les résultats du mapping afin d’identifier des jonctions d’épissage parmi les exons (Figure 3).
Une étape de nettoyage est mise en œuvre en utilisant le programme SAMtools. Cette étape consiste à éliminer des répétitions et fusionner des alignements. Puis le programme Cufflinks est lancé pour assembler des alignements de lectures de RNA-seq et ainsi mettre en évidence les transcrits connus et les nouveaux transcrits. On fait de même pour les gènes. A la fin, les comptages bruts sont mesurés et sont sauvegardés dans des fichiers csv. Ces étapes de traitements bio-informatiques sont présentées dans la Figure 4.
Figure 4 : Etapes de traitements bioinformatiques
Suite à ces traitements, il y a des nouveaux transcrits ou gènes qui sont découverts par l’assemblage. Ce sont des gènes ou transcrits inconnus, qui sont modélisés par la bio-informatique et sont renommés par Cufflinks : XLOC pour les gènes et TCONF pour les transcrits. Les gènes connus sont comparables dans les deux espèces, ce n’est pas le cas pour les gènes inconnus. Un gène peut avoir plusieurs transcrits. La somme des comptages de transcrits associés à un même gène est égale au nombre de comptages de ce gène. Un gène est associé, au maximum, à une dizaine de transcrits (au vu de l’épissage alternatif). Lorsqu’un gène est associé à plus d’une dizaine de transcrits, ceci est dû à la répétition du génome.
Les gènes connus qui ont les mêmes noms dans les deux espèces sont comparables. Il s’agit des gènes connus, mais ils n’ont pas forcément le même nombre de transcrits dans chacune des espèces. Par contre, les transcrits ayant les mêmes noms dans les deux espèces ne sont pas comparables.
Beaucoup de gènes ou transcrits ont des comptages à zéros pour un ou plusieurs échantillons (Figure 5), indiquant qu’aucune lecture n’a été identifiée pour ces transcrits. S’agissant de comptages, la distribution de ces données est particulière, elle se rapproche souvent d’une distribution de Poisson ou d’une Binomiale Négative.
Figure 5 : Histogramme de comptages bruts pour 3 échantillons de porc
iii. Tableau de données
Suite au traitement bio-informatique, nous disposons de 4 tableaux de données : Un tableau des comptages pour les 48 220 gènes de porcs,
Un tableau des comptages pour les 78 670 transcrits de porcs, Un tableau des comptages pour les 47 052 gènes de bovins, Un tableau des comptages pour les 74 251 transcrits de bovins.
Pour chaque tableau de données, nous avons en ligne les noms de transcrits ou gènes, et en colonne les 40 échantillons. Les 40 échantillons sont issus de 10 animaux pour les deux conditions (non traitée (NT) et traitée (T) par BMP15) et les 2 réplicats par condition pour chaque animal. Pour les tableaux de transcrits, nous avons une colonne supplémentaire contenant les noms de gènes associés pour chaque transcrit. Les échantillons notés ‘neg’ correspondent à la condition non traitée (NT), et Les échantillons notés ‘pos’ correspondent à la condition traitée (T). On dispose également des longueurs de chaque transcrit pour chacune des espèces. Ces 2 fichiers contiennent 2 colonnes (nom du transcrits et sa longueur). Les transcrits sont de longueurs variables. Pour un gène, les transcrits associés à ce gène peuvent avoir des longueurs différentes. Comme nous ne pouvons pas calculer la longueur d'un gène, nous considérons que le plus long des transcrits nous indique la longueur du gène.
IV. Présentation des méthodologies et des
techniques statistiques mises en œuvre
i. Analyses descriptives
L’objectif des outils de statistique descriptive élémentaire est de fournir, si possible graphiquement, des résumés synthétiques des observations réalisées (Baccini et al., 2008).
Une étape de transformation logarithme de données est mise en œuvre pour les analyses descriptives en utilisant la fonction log10(x+1), où x représente le nombre de comptages brutes. Cette étape est nécessaire puisque le nombre de comptage d’un gène ou un transcrit peut varier de 0 à 90000 (cf. Figure 5).
Au cours de cette analyse, plusieurs outils graphiques sont utilisés pour décrire, c'est-à-dire résumer ou représenter les données disponibles.
Diagramme en boîtes à moustaches : Un graphique très simple qui résume les données à partir de quelques caractéristiques de position du caractère étudié (médiane, quartiles, valeurs extrêmes)
Histogramme : Un graphique permettant de représenter la répartition d'une variable
Nuage de points : Une représentation des données dépendant de deux variables quantitatives qui permet de mettre en évidence le degré de corrélation entre ces deux variables
Heatmap : Une représentation graphique pour étudier les corrélations entre les échantillons de chaque espèce
Diagramme de Venn : Un schéma représentant les relations entre les ensembles étudiés.
Les corrélations entre les échantillons sont calculées pour tracer les heatmaps. Les moyennes, les variances et les médianes de tous les échantillons de données sont aussi calculées. Le test de student est appliqué pour tester l’égalité entre les moyennes de deux échantillons de la même condition d’un animal. Le test de Fisher est appliqué pour tester l’égalité entre les variances de deux échantillons de la même condition d’un animal.
ii. Analyses différentielles
Il existe beaucoup de packages disponibles dans R qui peuvent être appliqués pour analyser ce type de données (données de séquençage haut – débit de ARN), comme DESeq, edgeR, GPseq, BaySeq, NBPseq, etc. Les méthodes implémentées dans le package DESeq et edgeR sont les méthodes les plus performantes au niveau de la normalisation des données et le contrôle du FDR (false discovery rate) pour détecter les gènes différentiels (Dillies et al., 2012). En conséquent, nous avons étudié et utilisé ces deux packages pour analyser les données pendant mon stage.
a. Description de la méthode implémentée dans le package DESeq (Simon Anders & Huber, 2010)
Le nombre de comptage d’un gène ou d’un transcrit dans une condition donnée est modélisé via une distribution binomiale négative (NB), dont les paramètres (moyenne et variance) sont à estimer. Une des difficultés est d’estimer ces paramètres lorsque le nombre de réplicats est faible. D’autre part afin que les échantillons soient comparables entre eux, il sera nécessaire de s’intéresser à la normalisation les données.
L’hypothèse faite lors du développement de cette procédure est de supposer que la plupart des gènes ou transcrits ne sont pas différentiellement exprimés.
Le modèle :
Soit 𝑌𝑖,𝑗 le nombre de comptage correspondant au gène i (i=1,…,n) et à l’échantillon
j (j=1,…,m). Il est possible que pour un même échantillon, plusieurs réplicats soient disponibles (c’est le cas dans notre analyse). Nous disposons de 2 réplicats par échantillon biologique.
On suppose:
𝑌𝑖,𝑗 ~ 𝑁𝐵(𝜇𝑖,𝑗, 𝜎𝑖,𝑗2 )
Trois hypothèses :
1) La valeur attendue du nombre de comptage 𝜇𝑖,𝑗 du gène i, échantillon j peut
s’écrire comme suit :
𝐸[𝑌𝑖,𝑗] = 𝜇𝑖,𝑗= 𝑠𝑗𝑞𝑖,𝑝(𝑗)
avec
𝑠𝑗 : Le « size factor » de l’échantillon j qui représente la couverture ou
profondeur de l’échantillon j.
𝑞𝑖,𝑝(𝑗) : Un paramètre proportionnel à la valeur attendue de la concentration
du gène i sous la condition ρ dont dépend l’échantillon j.
(« shot noise ») et la variance brute : 𝜎𝑖,𝑗2 = 𝜇
𝑖,𝑗+ 𝑠𝑗2𝑣𝑖,𝑝(𝑗)
3) Le paramètre 𝑣𝑖,𝑝(𝑗) associé à la variance brute du gène est une fonction
lissée de la forme :
𝑣𝑖,𝑝(𝑗)= 𝑣𝑝(𝑞𝑖,𝑝(𝑗))
C’est une fonction linéaire locale de l’abondance du gène. Cette hypothèse est nécessaire car le nombre de réplicats par condition est souvent trop faible pour bien estimer la variance.
Remarques :
(1) Chaque échantillon j est ainsi standardisé grâce au paramètre 𝑠𝑗 (« size factor »).
L’objectif de cette transformation est de rendre comparable les échantillons entre eux. Si on divise chaque colonne de la table de comptages par le « size factor » associé, les valeurs de comptages sont mises sur la même échelle, elles sont alors comparables (S Anders & Huber, 2013).
(2) Le ratio 𝐸[𝑌𝑖,𝑗]
𝐸[𝑌𝑖,𝑗′] devrait être égal au ratio
𝑠𝑗
𝑠𝑗′ , si le gène i n’est pas
différentiellement exprimé entre les 2 conditions ou si j et j’ sont des réplicats. En effet dans ces deux cas, nous aurons 𝑞𝑖,𝑝(𝑗)= 𝑞𝑖,𝑝(𝑗′), donc :
𝐸[𝑌𝑖,𝑗] 𝐸[𝑌𝑖,𝑗′] = 𝑠𝑗𝑞𝑖,𝑝(𝑗) 𝑠𝑗𝑞𝑖,𝑝(𝑗′) = 𝑠𝑗 𝑠𝑗′
Estimation des estimateurs :
Le « size factor » (𝑠𝑗) de chaque échantillon est estimé par la médiane du ratio
suivant : 𝑠̂𝑗 = median 𝑖 𝑦𝑖,𝑗 (∏𝑚 𝑦𝑖,𝑣 𝑣=1 ) 1/𝑚
avec m : le nombre d’échantillons.
Le dénominateur peut être interprété comme un pseudo échantillon de référence obtenu en prenant la moyenne géométrique sur tous les échantillons. Si les gènes ne sont pas exprimés différemment, ce ratio doit être proche de 1.
On estime 𝑞𝑖,𝑝(𝑗) par 𝑞̂𝑖𝑝= 𝑚1
𝑝∑
𝑦𝑖,𝑗
𝑠̂𝑗
𝑗:𝑝(𝑗)=𝑝 , avec 𝑚𝑝 le nombre d’échantillon dans
la condition p.
𝑞𝑖,𝑝′
𝑞𝑖,𝑝. L’analyse différentielle est ainsi réalisée sur les paramètres 𝑞𝑖,𝑝(𝑗).
Pour la variance, on définit 𝑤𝑖𝑝=𝑚1
𝑝−1∑ ( 𝑦𝑖,𝑗 𝑠̂𝑗 − 𝑞̂𝑖,𝑝) 2 𝑗:𝑝(𝑗)=𝑝 et 𝑧𝑖𝑝 =𝑞̂𝑚𝑖𝑝 𝑝∑ 1 𝑠̂𝑗 𝑗:𝑝(𝑗)=𝑝
𝑤𝑖𝑝− 𝑧𝑖𝑝 est alors un estimateur sans biais de la variance brute 𝑣𝑖,𝑝. En utilisant
une régression locale, on obtient 𝑣̂𝑝(𝑞̂𝑖𝑝) = 𝑤𝑝(𝑞̂𝑖𝑝) − 𝑧𝑖𝑝 qui est donc l’estimation
de la variance brute.
Pour les gènes ayant des valeurs nulles pour tous les échantillons, ils sont mis en NA pour tous les calculs.
Les gènes ayant des valeurs nulles mais pas pour tous les échantillons ne sont pas pris en compte pour les calculs de size factor, mais ils sont pris en compte pour la suite de l’analyse.
Test d’hypothèses:
Supposons que nous ayons deux conditions A et B. Pour chaque gène i, on teste les hypothèses suivantes, 𝐻0: 𝑞𝑖,𝐴 = 𝑞𝑖,𝐵 contre 𝐻1: 𝑞𝑖,𝐴 ≠ 𝑞𝑖,𝐵.
Pour chaque gène i et dans chaque condition on définit : 𝑌𝑖𝐴= ∑𝑗:𝑝(𝑗)=𝐴𝑌𝑖𝑗 et
𝑌𝑖𝐵 = ∑𝑗:𝑝(𝑗)=𝐵𝑌𝑖𝑗, et la somme 𝑌𝑖𝑆= 𝑌𝑖𝐴+ 𝑌𝑖𝐵.
Sous l’hypothèse 𝐻0, les observations sont supposées indépendantes, on peut
définir la statistique de test suivante : 𝑝(𝑎, 𝑏) = Pr(𝑌𝑖𝐴= 𝑎) Pr(𝑌𝑖𝐵 = 𝑏) . En
utilisant la fonction nbinomTest et le test exact de Fisher, on peut calculer la p-valeur associée à chaque gène i :
𝑝𝑖 =
∑ 𝑎+𝑏=𝑦𝑖𝑆 𝑝(𝑎, 𝑏)
𝑝(𝑎,𝑏)≤𝑝(𝑦𝑖𝐴,𝑦𝑖𝐵)
∑𝑎+𝑏=𝑦𝑖𝑆𝑝(𝑎, 𝑏)
Cette fonction calcule également les p-valeurs ajustées pour les tests multiples selon la procédure de Benjamini-Hochberg (Benjamini & Hochberg, 1995) pour contrôler le False Discovery Rate (FDR).
b. Description de la méthode implémentée dans le package edgeR (M. D. Robinson, McCarthy, & Smyth, 2010)
Comme pour l’approche précédente, l’hypothèse faite lors du développement de cette procédure est de supposer que la plupart des gènes ou transcrits ne sont pas différentiellement exprimés. Les approches DESeq et edgeR diffèrent par la méthode de normalisation des données et les méthodes utilisées pour l’estimation des paramètres du modèle.
Le modèle
Soit 𝑌𝑖,𝑗 le nombre de comptage correspondant au gène i (i=1,…,n) et l’échantillon j
(j=1,…,m).
On suppose: 𝑌𝑖,𝑗 ~ 𝑁𝐵(𝜇𝑖,𝑗, 𝜙𝑖)
La moyenne 𝜇𝑖,𝑗 s’exprime comme suit : 𝐸[𝑌𝑖,𝑗] = 𝜇𝑖,𝑗 = 𝑁𝑗𝑝𝑖,𝑘 où
𝑁𝑗 : Le nombre total de comptage de l’échantillon j
𝑝𝑖,𝑘 : L’abondance relative de gène i dans la condition k dont l’échantillon j fait partie
La variance des observations : 𝑣𝑎𝑟(𝑌𝑖,𝑗) = 𝜎𝑖,𝑗2 = 𝜇𝑖,𝑗(1 + 𝜇𝑖,𝑗𝜙𝑖) où 𝜙𝑖 est la
dispersion (il représente en d’autres termes le coefficient de variation biologique). Si 𝜙𝑖 = 0, la distribution est une distribution de Poisson.
Le package edgeR est basé sur une méthode bayésienne empirique pour estimer les paramètres du modèle. Il utilise la méthode TMM (Trimmed Mean of M values), celle-ci sélectionne un échantillon 𝑗′ comme référence et calcule un « normalisation factor » pour les autres échantillons. On peut choisir l’échantillon de référence, il peut être spécifié par l’argument refColumn dans la fonction calcNormFactors. Si cela n’est pas spécifié, l’échantillon dont le 3ème quantile est plus proche du 3ème quartile de moyenne est sélectionné comme l’échantillon de référence.
Pour le gène i, on définit 𝑀𝑖,𝑗𝑗′ pour l’échantillon j relative à l’échantillon 𝑗′ : 𝑀𝑖,𝑗𝑗′ = 𝑙𝑜𝑔2
𝑌𝑖,𝑗 /𝑁𝑗 𝑌𝑖,𝑗′ /𝑁𝑗′
Le niveau d’expression absolue : 𝐴𝑖,𝑗𝑗′ = 12𝑙𝑜𝑔2(𝑌𝑖,𝑗 /𝑁𝑗∗ 𝑌𝑖,𝑗′ /𝑁𝑗′) ∀ 𝑌𝑖,∙ ≠ 0
Les gènes appartenant aux 30% extrêmes des 𝑀𝑖 et aux 5% extrêmes pour 𝐴𝑖 sont
exclus du calcul. Les cas où 𝑌𝑖,𝑗= 0 ou 𝑌𝑖,𝑗′ = 0 sont enlevés dans les calculs
puisque le logFC ne peut pas être calculé.
Le « normalisation factor » est alors obtenu par la formule suivante (M. Robinson & Oshlack, 2010): 𝑙𝑜𝑔2(𝑇𝑀𝑀𝑗𝑗′) =∑ 𝑤𝑖,𝑗 𝑗′𝑀 𝑖,𝑗𝑗 ′ 𝑖∈𝐺∗ ∑𝑖∈𝐺∗ 𝑤𝑖,𝑗𝑗′
où la pondération par les 𝑤𝑖,𝑗𝑗′ dépend de l’inverse de la variance asymptotique qui est obtenue par : 𝑤𝑖,𝑗𝑗′ =𝑁𝑁𝑗−𝑌𝑖,𝑗
𝑗𝑌𝑖,𝑗 +
𝑁𝑗′−𝑌𝑖,𝑗′
𝑁𝑗′𝑌𝑖,𝑗′ et 𝐺∗ représente l’ensemble des gènes
Remarque : Les gènes ayant des valeurs nulles pour tous les échantillons ne sont pas
pris en compte pour le calcul de « normalisation factor » par TMM.
Coefficient de variation biologique (BCV) (Davis, Yunshun, & Gordon, 2012)
La variance : 𝑣𝑎𝑟(𝑌𝑖,𝑗) = 𝜎𝑖,𝑗2 = 𝜇𝑖,𝑗(1 + 𝜇𝑖,𝑗𝜙𝑖)
Si on divise par 𝜇𝑔𝑖2 l’équation précédente, on obtient 𝑣𝑎𝑟(𝑌𝑖,𝑗 )
𝜇𝑖,𝑗2 = 𝐶𝑉2(𝑌𝑖,𝑗) =
1 𝜇⁄ 𝑖,𝑗+ 𝜙𝑖. Le premier terme 1 𝜇⁄ 𝑖,𝑗 représente le carré de coefficient de variation
(CV) de la distribution de Poisson (que l’on peut appeler le 𝐶𝑉2 technique). Le deuxième terme représente le carré du coefficient de variation des valeurs des expressions non observées (que l’on peut appeler le 𝐶𝑉2 biologique). √𝜙𝑖 est
donc le 𝐶𝑉 biologique (BCV). Il représente le CV qui pourrait rester entre les réplicats biologiques si la profondeur de séquençage devenait infinie. Quand le nombre de lecture augmente, le CV technique diminue, mais le BCV reste stable. Il est donc la source dominante des incertitudes pour les gènes ayant de grands comptages.
Estimation des estimateurs (M. D. Robinson & Smyth, 2007) :
Supposons qu’on dispose de n échantillons et 2 conditions. Pour chaque condition k, on a 𝑛𝑘 échantillons, et on note 𝑒𝑘 l’ensemble des échantillons de la condition k.
L’estimation du paramètre 𝜙 se fait en décomposant : - La Log-vraisemblance de 𝜙 pour le gène i :
𝑙𝑖(𝜙) = ∑ [ ∑ 𝑙𝑜𝑔Γ(𝑦𝑖𝑗 + 𝜙−1) 𝑗∈𝑒𝑘 + 𝑙𝑜𝑔Γ(𝑛𝑘𝜙−1) − 𝑛 𝑘𝑙𝑜𝑔Γ(𝜙−1) 2 𝑘=1 − 𝑙𝑜𝑔Γ(𝑧𝑖𝑘+ 𝑛𝑘𝜙−1)] avec 𝑧𝑖𝑘 = ∑𝑗∈𝑒𝑘𝑦𝑖𝑗
- La Log-vraisemblance commune de 𝜙 : 𝑙𝐶(𝜙) = ∑𝐺𝑖=1𝑙𝑖(𝜙), où G est le
nombre total de gène.
On définit la log-vraisemblance conditionnelle pondérée (WL) pour 𝜙 pour le gène i : 𝑊𝐿𝑖(𝜙) = 𝑙𝑖(𝜙) + 𝛼𝑙𝐶(𝜙)
où 𝛼 est la pondération associée à la log-vraisemblance commune. Si 𝛼 = 0, on obtient la qCML (quantile adjusted conditional maximum likelihood) méthode pour la «tag-wise» dispersion 𝑙𝑖(𝜙) qui est conditionné par le comptage total de chaque
Test d’hypothèses :
On utilise un test exact (fonction exactTest) similaire au test de Fisher (M. D. Robinson, Davis, Yunshun, & Gordon, 2013), par contre ce test est adapté à des données surdispersées.
Supposons que nous avons deux conditions A et B. Pour chaque gène i, on teste les hypothèses suivantes, 𝐻0: 𝑞𝑖,𝐴 = 𝑞𝑖,𝐵 contre 𝐻1: 𝑞𝑖,𝐴 ≠ 𝑞𝑖,𝐵. Etant donné que la
somme de variables aléatoires iid de loi binomiale négative suit aussi une loi binomiale négative, on peut calculer la p-valeur comme la probabilité de comptage supérieur à ce qu’on a observé(M. D. Robinson & Smyth, 2007).
La fonction topTags implémente également les tests multiples selon la procédure de Benjamini-Hochberg (Benjamini & Hochberg, 1995) pour contrôler le False Discovery Rate (FDR).
c. Comparaisons DESeq et edgeR Les points communs aux deux méthodes sont :
qu’elles supposent que la plupart des gènes ou transcrits ne sont pas différentiellement exprimés
qu’elles sont basées sur une distribution binomiale négative
qu’elles utilisent des facteurs de normalisation dans les modèles statistiques pour les analyses différentielles
qu’elles implémentent des procédures de Benjamini-Hochberg (Benjamini & Hochberg, 1995) pour contrôler le False Discovery Rate (FDR)
qu’elles contrôlent le FDR (false discovery rate) pour détecter les gènes différentiels (Dillies, et al., 2012)
Les principales différences entre les deux méthodes sont :
les gènes ou transcrits ayant des comptages nuls pour tous les échantillons sont mis en NA pour DESeq. Ils ne sont pas pris en compte pour l’analyse. Pour edgeR, ils sont pris en compte pour l’analyse, mais ils ont de p-valeurs à 1 pour le test différentiel, et des log2FC égaux à 1.
la fonction pour estimer les « size factors » est plus robuste dans l’approche DESeq. (Simon Anders & Huber, 2010)
pour un petit nombre de réplicats, edgeR propose d’estimer une dispersion commune. DESeq utilise une régression locale dépendant de la moyenne qui est plus flexible. (Simon Anders & Huber, 2010)
Il semble que le package DESeq est plus performant que la package edgeR, mais ces deux méthodes donnent des résultats très similaires (Dillies, et al., 2012) .
V. Présentation des résultats obtenus
Nous avons analysé séparément les données de transcrits et de gènes de chaque espèce. Pour chaque tableau de données, nous avons utilisé et comparé les résultats obtenus avec les packages DESeq et edgeR. Nous présentons dans une première partie l’analyse des données Porc ; puis celle des données Bovin et nous terminons par une comparaison des résultats entre espèces.
i. Analyses des données de Porc
Etant donné que les analyses des données de gènes et de transcrits de porc ont des résultats semblables, nous ne présentons ici que les analyses des données de gènes porc. Les analyses de données de transcrits sont présentées en annexe du rapport.
a. Analyses descriptives
Après avoir normalisé les données en utilisant la fonction log10(x+1), le Figure 6 présente les distributions de 8 échantillons de gènes de porc. Ce sont les échantillons des animaux 10 et 3 dans les deux conditions. Les distributions ne sont pas normales puisque les valeurs de comptages sont des entiers positifs et beaucoup de gènes ont des comptages à zéro. Tous les échantillons de gènes de porc ont des distributions semblables à celles présentées ici.
Figure 6 : Histogramme de quelques échantillons pour les gènes de porc
Animal 10
Les boîtes à moustaches des 40 échantillons de gènes de porc sont présentées sur la Figure 7. Pour chaque animal, nous disposons de 4 boîtes (2 dans la condition non-traitée et 2 dans la condition traitée). Les échantillons d’un même animal dans la même condition ont des médianes identiques et des boîtes à moustaches similaires, sauf pour les échantillons 4_pos_2_t et 4_pos_3_t qui ont une différence de médiane (ellipse en rouge, figure 7).
Figure 7: Boîtes à moustaches des 40 échantillons pour les gènes de porc
Les moyennes des 40 échantillons varient entre 0.912 à 1.045 et les variances varient entre 1.278 à 1.455. Les échantillons de la même condition d’un même animal ont des moyennes et variances très proches avec une différence de quelques millièmes. Nous effectuons les tests de comparaison de moyennes et de variances entre les échantillons de la même condition d’un même animal. Les p-valeurs de tous les tests de moyennes et de variances sont supérieures au seuil de 5%, on ne rejette pas l’hypothèse que les moyennes (variances) de deux échantillons d’un même animal dans la même condition sont égales. Il faut noter que les tests de student et de Fisher sont basés sur l’hypothèse de normalité des données. Nous avons vu que cette hypothèse n’est pas vérifiée sur nos données (cf Figure 6). Cependant nous pouvons admettre que les 2 réplicats d’un même animal dans la même condition sont identiques et peuvent donc être considérés comme de ‘‘vrai réplicats’’.
log1 0(c o mp tag es + 1) Echantillons Animal 10 11 12 1 2 3 4 5 7 8
La Figure 8 représente les corrélations entre les 40 échantillons pour les gènes de porc. Nous constatons que toutes les corrélations sont supérieures à 0.96. Les corrélations entre les échantillons de même condition d’un même animal sont supérieures à 0.98. Les échantillons de la même condition d’un même animal sont classés ensemble. On remarque également que les 20 échantillons relatifs à la condition traitée d’une part et les 20 échantillons non traitées se regroupent ensemble.
Figure 8 : Heatmap de corrélations entre 40 échantillons pour les gènes de porc
Comme les échantillons de même condition d’un même animal ont de fortes corrélations (>0.98) et des moyennes et variances identiques, nous supposons donc qu’il n’y a pas d’effet lanes. Nous décidons donc de sommer les échantillons des comptages bruts des gènes de même condition d’un même animal. Nous disposons alors d’un tableau de données de gènes pour 20 échantillons (10 animaux * 2 conditions). Nous allons rechercher les gènes différentiels entre les deux conditions en utilisant les deux packages DESeq et edgeR.
Les conclusions faites sur les gènes sont identiques pour les analyses descriptives des transcrits de porc. Elles sont donc transposables aux données des transcrits.
Echantillons traités Echantillons non traités
Ech an tillon s n o n t rait és Ech an tillon s t rait és
b. Analyse différentielle des gènes pour les échantillons Porc
Après avoir sommé les échantillons, nous avons donc 10 réplicats par condition. L’analyse différentielle se fait sur ces 20 échantillons de comptages bruts pour trouver les gènes qui sont différentiellement exprimés entre les deux conditions (T vs NT).
Analyse à partir du package DESeq :
Les « size factors » pour chaque échantillon sont estimés (Figure 9). Les échantillons non traités sont en rouges et les échantillons traités sont en bleus. Pour tous les animaux, les échantillons non traité ont des « size factors » plus grand que ceux des échantillons non traité, sauf pour l’animal 5. Etant donné que les gènes non différentillement exprimés ont des expressions similaires pour tous les échantillons, et qu’on suppose que la majorité des gènes sont non différentiellement exprimés, les « size factors » doivent être proche de 1. Ici les valeurs varient de 0.722 à 1.341.
Figure 9 : « Size factors » des 20 échantillons de gènes chez le porc
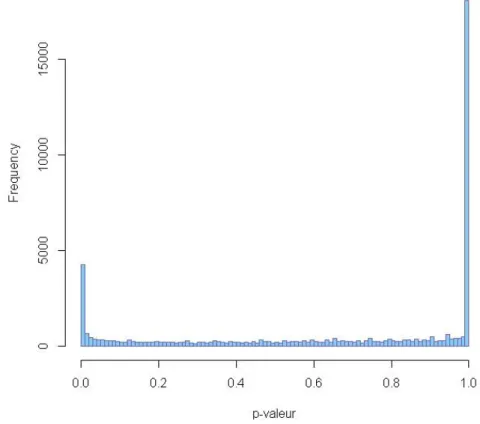
Après avoir testé si les gènes sont différentiellement exprimés, on trace l’histogramme des p-values ajustées pour les tests multiples (Figure 10). Comme nous avons supposé que la plupart de gènes ne sont pas différentiellement exprimés, nous les retrouvons pour les gènes qui ont des p-valeurs ajustées à 1. Les
Echantillons « Siz e fact o rs »
gènes ayant de petites p-valeurs ajustées (autour de 0) sont des gènes différentiellement exprimés. Ce sont des gènes qu’on cherche à étudier. Les gènes ayant des comptages nuls pour tous les échantillons (NA) sont exclus du test différentiel.
Figure 10 : Histogramme de p-valeurs ajustées de gènes chez le porc
En fixant le seuil à 0.01 (1%), on cherche les gènes qui ont des p-valeurs ajustées inférieures à ce seuil. On trouve 5899 gènes différentiellement exprimés dont 2898 avec des valeurs de log2FC positives et 3001 avec des valeurs de log2FC négatives. Si la valeur de log2FC est positive, le ratio entre la moyenne dans la condition traitée et celle dans la condition non traitée est supérieur à 1, donc les expressions de ces gènes sont plus fortes dans la condition traitée que dans la condition non traitée. C’est l’inverse pour les gènes qui ont des valeurs de log2FC négatives.
Ensuite, on trace le MA plot (Figure 11). Le MA plot trace pour chaque gène, la valeur de log2FC en fonction de la moyenne des comptages normalisés. Par contre, les gènes ayant des comptages nuls pour tous les échantillons (NA) ne sont pas présentés sur la figure. Les points en rouge sont des gènes qui sont différentiellement exprimés. Les gènes ayant des log2FC hors l’intervalle [-3, 3] sont représentés au bord du graphe (ellipses bleus). Les gènes de log2FC à ‘+Inf’ ou ‘-Inf’ sont aussi inclus dans les ellipses bleus. Ce sont des gènes qui ne sont exprimés que dans une des deux conditions.
Figure 11 : MA plot des gènes chez le porc avec DESeq
On représente le boxplot des comptages bruts dans les 2 conditions pour les 6 gènes ayant les plus petites p-valeurs avec DESeq (Figure 12). Ces boxplots ne sont pas sur la même échelle. Les variations de ces gènes ne sont pas les mêmes. Il y a des gènes pour lesquels les expressions sont plus grandes dans la condition non traitée que dans la condition traitée, et il y a des gènes qui varient en sens inverse.
Analyse à partir du package edgeR :
Nous avons réalisé la même analyse qu’avec DESeq mais en utilisant le package edgeR. Nous étudierons les différences observées entre ces deux procédures. On calcule les « normalisation factors » pour chaque échantillon donné (Figure 13). Comme les « size factors », les « normalisations factors » sont aussi estimées pour rendre comparables différents échantillons entre eux. Si on divise chaque échantillon par le « normalisation factors » associé, les comptages sont alors sur la même échelle, et sont comparables.
Les « normalisations factors » doivent être proche de 1 en raison du nombre important de gènes non différentillement exprimés et donc des expressions similaires attendues pour tous les échantillons. Ici les valeurs varient de 0.972 à 1.035.
Figure 13 : « Normalisation factors » des gènes chez le porc avec edgeR
Après avoir testé si les gènes sont différentiellement exprimés entre les deux conditions, on trace l’histogramme des p-valeurs ajustées des tests (Figure 14). Comme avec DESeq, nous trouvons des gènes qui ont de petites p-valeurs (autour de 0). Ce sont des gènes différentiellement exprimés. Nous trouvons plus de gènes ayant des p-valeurs ajustées à 1 avec edgeR qu’avec DESeq, puisque les gènes qui ont des comptages nuls pour tous les échantillons ne sont pas pris en compte avec DESeq. Pour edgeR, ces gènes ont des p-valeurs ajustées à 1 et des log2FC à 0.
Echantillons « N o rma lisa tion f ac to rs »
Figure 14 : Histogramme de p-valeurs ajustées de gènes chez le porc avec edgeR
Si on fixe le seuil à 0.01, on a 4250 gènes différentiellement exprimés, dont 1970 avec des valeurs de log2FC positives et 2280 avec des valeurs négatives. Comme avec DESeq, si la valeur de log2FC est positive, le ratio entre la moyenne dans la condition traitée et celle dans la condition non traitée est supérieur à 1, donc les expressions de ces gènes sont plus fortes dans la condition traitée que dans la condition non traitée. C’est l’inverse pour les gènes qui ont des valeurs de log2FC négatives.
On utilise la fonction plotSmear pour tracer le Figure 15. Elle trace pour chaque gène, la valeur de log2FC en fonction de la moyenne des log2 de comptages par million (CPM). Le CPM d’un gène dans un échantillon est calculé par le comptage de ce gène dans cet échantillon divisé par le comptage total de cet échantillon fois un million.
Les points en rouge sont des gènes qui sont différentiellement exprimés dans les deux conditions. Les gènes ayant des comptages nuls pour tous les échantillons sont représentés dans le cercle bleu. Ils ont des valeurs de log2FC à 0 et des moyennes des log2CPM négatives. Les gènes qui ne sont exprimés que dans une des deux conditions sont représentés dans les ellipses vertes. Ils ont des valeurs de log2FC grandes en valeur absolue, et des moyennes des log2CPM négatives.
Figure 15 : plot MA de gènes chez le porc avec edgeR
On représente le boxplot des comptages bruts des 2 conditions pour les 6 gènes ayant les plus petites p-valeurs avec edgeR (Figure 16). Ils ont des comptages différents entre les 2 conditions. Ces boxplots ne sont pas sur la même échelle. Les variations de ces gènes ne sont pas les mêmes. Il y a des gènes pour lesquels leurs expressions sont plus grandes dans la condition non traitée que dans la condition traitée, et il y en a pour lesquels les variations sont inverses. Pour ces gènes, les valeurs de log2FC sont toutes supérieures à 2 en valeur absolues.
c. Comparaison des résultats obtenus avec les packages DESeq et edgeR
En comparant les gènes différentiels obtenus avec DESeq et edgeR au seuil 0.01 (Tableau 1), on trouve 4021 gènes différentiellement exprimés en commun. Avec la méthode DESeq, les NAs sont des gènes ayant des comptages à zéro pour tous les échantillons. Ils sont éliminés de l’analyse. Alors dans edgeR, ces gènes sont comptabilisés dans les gènes non différentiellement exprimés.
Tableau 1 : Comparaison de gènes différentiels de porc entre deux méthodes
DEseq edgeR
Gènes Non différentiels
Gènes
Différentiels NA Nombre total Gènes Non différentiels 38140 1878 3952 43970
Gènes Différentiels 229 4021 0 4250 Nombre total 38369 5899 3952 48220 On compare les p-valeurs de ces 4021 gènes en commun avec les deux méthodes (Tableau 2 et Tableau 3). Pour chaque méthode, la 1ère colonne contient les intervalles de p-valeurs ajustées, la 2ème colonne contient le nombre de gènes différentiels (DE) obtenus avec la méthode considérée, la 3ème contient le nombre de gènes différentiels en commun avec l’autre méthode, et la 4ème colonne contient l’écart entre la 2ème et la 3ème colonne. Ce sont des gènes trouvés différentiels avec une méthode mais pas l’autre au seuil de 0.01.
Tableau 2: P-valeurs ajustées des gènes chez le porc avec DESeq
P-valeur ajustée Nb gènes DE avec DESeq Nb gènes DE en commun entre DESeq et edgeR Nb de gènes DE mais non en commun [10^(-3), 10^(-2) [ 1748 614 1134 [10^(-4), 10^(-3) [ 1144 726 418 [10^(-6), 10^(-4) [ 1298 1055 143 [10^(-10), 10^(-6) [ 957 886 71 ] -Inf, 10^(-10) [ 752 740 12 Total 5899 4021 1778
Tableau 3: P-valeurs ajustées de gènes de porc pour edgeR
P-valeur ajustée Nb gènes DE avec edgeR Nb gènes DE en commun entre DESeq et edgeR Nb de gènes DE mais non en commun [10^(-3), 10^(-2) [ 1421 1231 190 [10^(-4), 10^(-3) [ 746 727 19 [10^(-6), 10^(-4) [ 905 889 16 [10^(-10), 10^(-6) [ 665 661 4 ] -Inf, 10^(-10) [ 513 513 0 Total 4250 4021 229
On constate que pour les deux méthodes, les gènes qui ne sont pas en communs ont des p-valeurs proche de 0.01. Presque tous les gènes qui ont de p-valeurs ajustées petites (inférieur à 10^ (-6)) sont trouvés différentiellement exprimés avec les deux méthodes au seuil de 0.01.
On compare les valeurs des log2FoldChanges obtenues avec les deux méthodes (Figure 17). On trace un nuage de points (en noir) pour les log2FC de DESeq en fonction de logFC d’edgeR pour tous les gènes (48220 gènes). Vu que les points sont alignés sur la 1ère bissectrice, ils ont les mêmes logFC avec les deux méthodes. On colore les gènes différentiels. Les points en jaune représentent les gènes ayant des p-valeurs ajustées inférieures à 0.01 avec la méthode DESeq (5899 gènes). Les points en vert représentent les gènes ayant des p-valeurs ajustées inférieures à 0.01 avec la méthode edgeR (4250 gènes). Les points en rouge représentent les gènes ayant des p-valeurs ajustées supérieures à 0.01 avec DESeq et inférieures à 0.01 avec edgeR (229 gènes). Les points en bleu représentent les gènes ayant des p-valeurs ajustées inférieures à 0.01 avec DESeq et supérieures à 0.01 avec edgeR (1778 gènes). Avec edgeR, les gènes différentiellement exprimés au seuil 1% n’ont pas de log2FC proches à 0 (cercle rouge). Les gènes différentiellement exprimés au seuil 1% avec DESeq et non détectés avec edgeR ont des log2FC plus concentrés autour de 0.Une étude plus approfondie doit être réalisée pour détecter les différences entre les deux méthodes.
d. Lien entre les transcrits et les gènes de porc
Nous avons effectué les mêmes analyses pour les transcrits de porc (l’étude et les résultats sont présentés en annexe). Nous avons trouvé 12957 transcrits différentiels avec DESeq et 9684 transcrits différentiels avec edgeR. Les 12957 transcrits avec DESeq sont associés à 6626 gènes. Les 9684 transcrits avec edgeR sont associés à 4955 gènes.
On voudrait savoir (1) si tous les gènes associés aux transcrits différentiels sont aussi différentiellement exprimés, et (2) si tous les transcrits associés aux gènes différentiellement exprimés sont aussi différentiellement exprimés. Nous analysons ces 2 questions pour chacune des méthodes étudiés.
Analyse avec DESeq
En traçant le diagramme de Venn (Figure 18), on trouve que sur les 5899 gènes différentiellement exprimés au seuil 1%, 41 gènes différentiels n’ont aucun transcrit différentiel. On remarque également que 768 gènes sont trouvés non différentiels alors qu’au moins un de leur transcrit est détecté comme différentiellement exprimé. 5858 gènes différentiels ont au moins un transcrit différentiellement exprimé. Parmi les 5858 gènes différentiellement exprimés, il y a 4792 gènes pour lesquels tous les transcrits associés sont différentiellement exprimés et 1066 gènes qui ont des transcrits différentiels mais pas tous.
Figure 18 : Diagramme de Venn pour les gènes différentiels et les gènes associés aux transcrits différentiels chez le porc avec DESeq
Pour les 41 gènes (Figure 18), ils sont associés par 119 transcrits. Parmi ces 119 transcrits, 75 transcrits ont des p-valeurs ajustés comprises entre 0.01 et 0.05, 14 transcrits ont des p-valeurs ajustés comprises entre 0.05 et 0.1.
transcrits, 966 transcrits ont des p-valeurs ajustés inférieurs à 0.01, 564 transcrits ont des p-valeurs ajustées comprises entre 0.01 et 0.05, 188 transcrits ont des p-valeurs ajustées comprises entre 0.05 et 0.1.
Comme les variations de transcrits pour ces gènes ne sont pas tous dans le même sens, c'est-à-dire pour un gène il y a des transcrits qui sont exprimés plus fortement dans la condition non traitée et aussi des transcrits qui sont exprimés plus fortement dans la condition traitée, quand on fait la somme des comptages, ils se compromettent. Dece fait, ces gènes ne sont pas différentiels.
Analyse avec edgeR
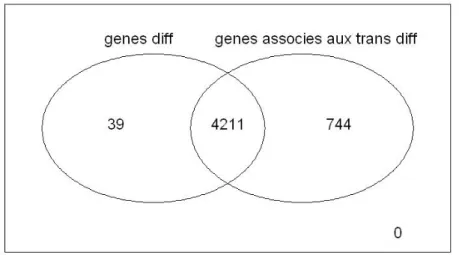
A l’aide du diagramme de Venn (Figure 19), on trouve que 39 gènes différentiels n’ont aucun transcrit différentiel. On remarque également que 744 gènes sont trouvés non différentiels alors qu’au moins un de leur transcrit est détecté comme différentiellement exprimé. 4211 gènes différentiels ont au moins un transcrit différentiellement exprimé. Parmi les 4211 gènes différentiellement exprimés, il y a 3560 gènes pour lesquels tous les transcrits associés sont différentiellement exprimés et 751 gènes qui ont des transcrits différentiels mais pas tous.
Figure 19 : Diagramme de Venn pour les gènes différentiels et les gènes associés aux transcritps différentiels de porc par edgeR
Les 39 gènes (Figure 19) sont associés à 125 transcrits. Parmi ces 125 transcrits, 85 transcrits ont des p-valeurs ajustées comprises entre 0.01 et 0.05, 7 transcrits ont des p-valeurs ajustées comprises entre 0.05 et 0.1.
Les 744 gènes (Figure 19) sont associés aux 3255 transcrits. Parmi ces 3255 transcrits, 962 transcrits ont des p-valeurs ajustées inférieures à 0.01, 397 transcrits ont des p-valeurs ajustées comprises entre 0.01 et 0.05, 119 transcrits ont des p-valeurs ajustées comprises entre 0.05 et 0.1.
ii. Analyses des données de Bovin
En faisant la même étude que sur les données porc, nous analysons les données de bovins. Nous ne présentons ici que les analyses des données de gènes de bovin. Les analyses de données de transcrits sont présentées en annexe.
a. Analyses descriptives
Après avoir normalisé les données en utilisant la fonction log10(x+1), la Figure 20 présente les distributions de 8 échantillons de gènes de bovin. Ce sont les échantillons obtenus des animaux 11 et 2 dans deux conditions. Comme les distributions des échantillons de porc (Figure 6), les distributions ne sont pas normales puisque les valeurs de comptages sont des entiers positifs et beaucoup de gènes ont des comptages à zéro. Tous les échantillons de gènes de bovin ont des distributions semblables à celles présentées ci-dessous.
Figure 20 : Histogramme de quelques échantillons pour les gènes de bovin
Les boîtes à moustaches des 40 échantillons de gènes de bovin sont présentées sur la Figure 21. Comme pour le porc, nous disposons de 4 boîtes pour chaque animal. Les échantillons d’un même animal dans la même condition ont des médianes identiques sauf pour les échantillons 1-2_neg_7_b et 1-2_neg_8_b, 2_pos_7_b et 2_pos_8_b (ellipses en rouge, figure 21).
Echantillons non traités Echantillons traités Animal 11
Figure 21 : Boîtes à moustaches des 40 échantillons pour les gènes de bovin
Les moyennes des 40 échantillons varient entre 0.834 à 1.002, les variances varient entre 1.278 à 1.484. Les échantillons de la même condition d’un même animal ont des moyennes et variances proches (différence entre 0.02 et 0.04).
Nous effectuons les tests de comparaison de moyennes et de variances entre les échantillons de la même condition d’un même animal. Les p-valeurs de tous les tests sont inférieures au seuil de 5%, donc on rejette l’hypothèse que les moyennes et les variances de deux échantillons d’un même animal dans la même condition sont égales. Cependant les tests de student et de Fisher sont basés sur l’hypothèse de normalité de données. Nous avons vu que cette hypothèse n’est pas vérifié sur nos données (Figure 20).
La Figure 22 représente les corrélations entre les 40 échantillons pour les gènes de bovin. Pour les bovins, les échantillons de même condition d’un animal sont classés ensemble comme chez le porc. Les corrélations entre ces échantillons sont supérieures à 0.98. Par contre, les échantillons bovins ne se regroupent pas par condition. Ils sont regroupés par animal, c’est-à-dire les échantillons d’un animal sont classés ensemble, sauf pour l’animal 1-1 (ellipses en rouge).
log1 0(c o mp tag es + 1) Echantillons Animal 11 12 14 16 1 2 3 5 6 1 8
Nous pouvons supposer qu’il y a plus de variabilité individuelle chez le bovin que chez le porc. En plus, les données de bovin semblent moins homogènes que les données de porc. Les données de porc ont été séquencées sur la même machine le même jour, alors que pour les données bovines, quelques problèmes techniques sont survenus lors de l’acquisition des données.
Figure 22 : Heatmap de corrélation entre 40 échantillons pour les gènes chez le bovin
Etant donné que les échantillons des gènes de la même condition d’un même animal ont de fortes corrélations (>0.98), des moyennes et variances proches, nous avons considéré qu’il n’y a pas d’effet de lanes. Nous avons décidé de sommer les échantillons des comptages bruts de gènes de la même condition d’un même animal. Nous disposons alors d’un tableau de données de gènes pour 20 échantillons (10 animaux * 2 conditions). En utilisant les deux packages DESeq et edgeR, nous allons rechercher les gènes différentiels entre les deux conditions.
Les conclusions faites sur les gènes sont identiques pour les analyses descriptives des transcrits de bovin (analyses présentées en annexe).
b. Analyse différentielle des gènes pour les échantillons Bovin
Après avoir sommé les échantillons, nous avons donc 10 réplicats par condition. L’analyse différentielle se fait sur ces 20 échantillons de comptages bruts pour trouver les gènes qui sont différentiellement exprimés entre les deux conditions (T vs NT).
Analyse à partir du package DESeq :
Les « size factors » pour chaque échantillon de gènes de bovin sont estimés (Figure 23). Les échantillons non traités sont en rouges et les échantillons traités sont en bleus. Les animaux 1-4, 2, 3 et 8 ont des « size factors » dans la condition traitée plus grands que ceux dans la condition non traitée. Ici, les «size factors » sont proches de 1. Les valeurs varient de 0.785 à 1.402.
Figure 23 : « Size factors » des 20 échantillons de gènes chez le bovin
Après avoir testé si les gènes sont différentiellement exprimés entre les deux conditions, on trace l’histogramme des p-valeurs ajustées pour les tests multiples (Figure 24). Nous n’avons pas autant de gènes différentiels (p-valeurs ajustées autour de 0) que chez le porc, mais beaucoup plus de gènes non différentiellement exprimés (p-valeurs ajustées proche de 1).
Cela peut s’expliquer par une variabilité individuelle plus importante chez les bovins que chez le porc.
Echantillons « siz e fact o r »
Figure 24 : Histogramme de p-valeurs ajustées de gènes chez le bovin avec DESeq
En fixant le seuil à 0.01 (1%), on trouve 956 gènes différentiellement exprimés dont 575 avec des valeurs de log2FC positives et 381 des valeurs de log2FC négatives. Ensuite, on trace le MA plot (Figure 25). Les points en rouge sont des gènes qui sont différentiellement exprimés. Les gènes ayant des log2FC hors de l’intervalle [-3, 3] sont représentés au bord du graphe (ellipses bleus).
On représente le boxplot des comptages bruts dans les 2 conditions pour les 6 gènes ayant les plus petites p-valeurs avec DESeq dans (Figure 26). Les boxplots ne sont pas sur la même échelle. Ces gènes varient dans le même sens. Les moyennes de ces gènes sont plus grandes dans la condition traitée que dans la condition non traitée.
Figure 26 : Boxplot des 6 gènes bovins ayant les plus petites p-valeurs ajustées avec DESeq
Analyse à partir du package edgeR :
Nous avons réalisé la même analyse qu’avec DESeq mais en utilisant le package edgeR. Nous étudierons les différences observées entre ces deux procédures. On calcule les « normalisation factors » pour chaque échantillon donné (Figure 27). Ici, les « normalisation factors » sont tous proches de 1. Les valeurs varient de 0.963 à 1.064.
Figure 27 : « Normalisation factors » des gènes chez le bovin avec edgeR
Après avoir testé si les gènes sont différentiellement exprimés entre les deux conditions, on trace l’histogramme des p-valeurs ajustées des tests. Les gènes qui ont de petites p-valeurs (autour de 0) sont des gènes différentiellement exprimés. Comme avec la procédure DESeq, on trouve moins de gènes différentiellement exprimés chez le bovin que chez le porc.
Figure 28 : Histogramme de p-valeurs ajustées de gènes chez le bovin avec edgeR
Echantillons « N o rma lisa tion f ac to r »
Si on fixe le seuil à 0.01, on a 665 gènes différentiellement exprimés, dont 399 avec des valeurs de log2FC positives et 266 avec des valeurs négatives. On utilise la fonction plotSmear pour tracer la Figure 29. Les points en rouge sont des gènes qui sont différentiellement exprimés dans les deux conditions. Les gènes ayant des comptages nuls pour tous les échantillons sont représentés dans le cercle bleu. Les gènes qui ne sont exprimés que dans une des deux conditions sont représentés dans les ellipses vertes.
Figure 29 : plot MA de gènes chez le bovin avec edgeR
On représente le boxplot des comptages bruts des 2 conditions pour les 6 gènes ayant les plus petites p-valeurs ajustées avec edgeR (Figure 30). Les boxplots ne sont pas sur la même échelle. Ces gènes varient dans le même sens. Les moyennes de ces gènes sont plus grandes dans la condition traitée que dans la condition non traitée.
Figure 30 : Boxplot des 6 gènes de bovin ayant les plus petites p-valeurs ajustées avec edgeR
c. Comparaisons des résultats obtenus avec les packages DESeq et edgeR
En comparant les listes de gènes différentiels obtenus avec DESeq et edgeR au seuil 0.01, on trouve 506 gènes différentiellement exprimés en commun.
Tableau 4 : Comparaison de gènes différentiels chez le bovin entre les deux méthodes
DESeq
edgeR Non différentiels Différentiels NA Nombre total Non différentiels 41600 450 4337 46387
Différentiels 159 506 0 665 Nombre total 41759 956 4337 47052
NA : les gènes qui ont des comptages à zéro pour tous les échantillons en NA pour DESeq
On compare les p-valeurs de ces 506 gènes en commun avec les deux méthodes (Tableau 5 et Tableau 6). Pour chaque méthode, la 1ère colonne contient les intervalles de p-valeurs ajustées, la 2ème colonne contient le nombre de gènes différentiels obtenus avec la méthode considérée, la 3ème contient le nombre de
gènes différentiels en commun avec l’autre méthode, et la 4ème colonne contient l’écart entre la 2ème et la 3ème colonne. Ce sont des gènes trouvés différentiels avec une méthode mais pas l’autre au seuil de 0.01.
Tableau 5 : p-valeurs ajustées des gènes différentiels chez le bovin avec DESeq
P-valeur ajusté Nb gènes DE avec DESeq Nb gènes DE en commun entre DESeq et edgeR Nb de gènes DE mais non en commun [10^(-3), 10^(-2) [ 452 158 294 [10^(-4), 10^(-3) [ 208 117 91 [10^(-6), 10^(-4) [ 147 96 51 [10^(-10), 10^(-6) [ 95 82 13 ] -Inf, 10^(-10) [ 54 53 1 Total 956 506 450
Tableau 6 : p-valeurs ajustées des gènes différentiels chez le bovin avec edgeR
P-valeur ajusté Nb gènes DE avec edgeR Nb gènes DE en commun entre DESeq et edgeR Nb de gènes DE mais non en commun [10^(-3), 10^(-2) [ 304 179 125 [10^(-4), 10^(-3) [ 108 90 18 [10^(-6), 10^(-4) [ 137 124 13 [10^(-10), 10^(-6) [ 70 68 2 ] -Inf, 10^(-10) [ 46 45 1 Total 665 506 159
On constate que pour les deux méthodes, les gènes qui ne sont pas en communs ont des p-valeurs proche de 0.01. Presque tous les gènes qui ont de p-valeurs ajustées petites (inférieur à 10^ (-6)) sont en communs pour les deux méthodes.
On compare les valeurs de log2FoldChange obtenus avec les deux méthodes. On trace un nuage de points (en noire) pour les log2FC de DESeq en fonction de logFC d’edgeR pour tous les gènes (47052 gènes). Vu que les points sont alignés sur la première bissectrice, ils ont les mêmes valeurs de logFC avec les deux méthodes. On colore les gènes différentiels. Les points en jaune représentent les gènes ayant des p-valeurs ajustées inférieures à 0.01 pour DESeq (956 gènes). Les points en vert représentent les gènes ayant des p-valeurs ajustées inférieures à 0.01 pour edgeR (665 gènes). Les points en rouge représentent les gènes ayant des p-valeurs ajustées supérieures à 0.01 pour DESeq et inférieures à 0.01 pour edgeR (159 gènes). Les points en bleu représentent les gènes ayant des p-valeurs ajustées inférieures à 0.01 pour DESeq et supérieures à 0.01 pour edgeR (450 gènes).