FACULTE DE MEDECINE ET DE PHARMACIE RABAT
Année 2017 Numéro d’ordre : 22/16 CSVS
THÈSE DE DOCTORAT NATIONAL
Centre d’Etude Doctoral Science de la vie et de la Santé
Formation Doctorale : Biologie médicale, Pathologie Humaine et
Expérimentale et Environnementale
Intitulé de la thèse
Hajar LEMRISS
Présentée et soutenue le 25 juillet 2017
JURY :
Professeur Président
Faculté de Médecine et de Pharmacie, Université Mohammed V, Rabat
Professeur Azeddine IBRAHIMI Directeur de thèse
Faculté de Médecine et de Pharmacie, Université Mohammed V, Rabat
Professeur Chakib NEJJARI Rapporteur
Université Mohammed VI des Science de la Santé, Casablanca
Professeur Anas Kettani Rapporteur
Faculté des Sciences Ben M’Sik, Université Hassan II, Casablanca
Professeur Rachid EL JAOUDI Rapporteur
Faculté de Médecine et de Pharmacie, Université Mohammed V, Rabat
Analyse génomique de souches multi-résistantes
de Staphylococcus capitis isolées dans des services
Dédicace
Un simple merci ne suffira pas à remercier ma famille.
A mes parents pour leur amour, leur présence et leur soutien sans faille.
Vous avez été des piliers durant ces années d’étude parfois laborieuses, vos encouragements ont été bénéfiques et m’ont permis d’aller de l’avant.
Vous m’avez toujours poussé à faire ce que j’aimais. Trouvez ici le résultat de vos efforts, avec toute la reconnaissance et l’amour que je vous porte.
A mon frère pour ta présence pendant les moments difficile, pour ta générosité infinie et ton soutien. Je te dédie cette thèse et je te souhaite une vie pleine de joie, de bonheur et de succès. A mes sœurs, qui ont été présentes et compréhensives, leurs appels et leurs mots de soutien m’ont
beaucoup apportés. Que cette thèse soit le témoignage de ma profonde affection. A mon neveu, Youssouf, mon petit rayon de soleil.
A tout le reste de ma famille, ma grand-mère, ma tante, mon oncle,
mes cousins et mes cousines, pour vous encouragements je vous dédiez cette thèse avec ma profonde affection.
Cette thèse est le fruit de collaborations multiples entre le Laboratoire de Biotechnologie Médicale (MedBiotech) de la Faculté de Médecine et de Pharmacie de Rabat, Université Mohammed V, le Centre National de Référence des Staphylocoques (CNRS), Lyon, France et le Laboratoire de Recherche et d’Analyses Médicales de la Fraternelle de la Gendarmerie Royale (LRAM) Rabat. De plus, elle est le résultat d’un effort constant qui n’aurait pu aboutir sans la contribution d’un nombre de personnes. Ainsi se présente l’occasion de leur exprimer mes remerciements:
A Mon Directeur de thèse le Pr. Azeddine IBRAHIMI, Professeur d’Enseignement Supérieur et Chef du Laboratoire de Biotechnologie Médicale (Médbiotech) à la Faculté de Médecine et de Pharmacie de Rabat, Université Mohammed V, pour m’avoir accueilli dans votre équipe de recherche, pour votre confiance dès le départ, pour votre encadrement, pour votre disponibilité sans faille malgré un emploi du temps plus que chargé et pour votre patience. Je tiens également à vous remercier pour la liberté d’action que vous m’avez donnée à chaque étape, vos précieux conseils, vos connaissances scientifiques et vos encouragements tout au long des années que j’ai passé au sein de votre laboratoire afin d'aboutir à cette thèse d'analyse génomique. Veuillez trouver ici toute ma reconnaissance et mon profond respect.
A Monsieur le Dr Saâd EL KEBBAJ, Directeur du Laboratoire de Recherche et d’Analyses Médicales de la fraternelle de la Gendarmerie Royale (LRAM), pour m’avoir acceptée en tant que stagiaire à plusieurs reprises dans votre laboratoire afin d'apprendre les outils de séquençage haut débit et d'analyses bioinformatiques pour aboutir à ce travail. Je vous remercie pour l'intérêt que vous portez à la recherche scientifique et à l'avancée des sciences biomédicales au Maroc. Veuillez trouver ici le témoignage de ma reconnaissance, mes sincères remerciements et ma profonde gratitude.
A mon Co-encadrant de thèse le Pr. Frédéric Laurent, Professeur des Universités de Microbiologie à la Faculté de Pharmacie de Lyon au sein de l’Université Claude Bernard Lyon 1 et Co-directeur du Centre National de Référence des Staphylocoques, Lyon, France, pour m’avoir confié ce sujet qui me paraissait si vaste et complexe au départ et qui s’avère être si passionnant et enrichissant pour finir, et pour votre aide lors de la rédaction des articles et pour nous avoir fourni les souches utilisées dans le présent travail. Je vous remercie
discussions constructives que nous avons échangées tout au long des années de doctorat. J’espère sincèrement que ce travail sera l’occasion de pouvoir continuer à collaborer avec votre laboratoire. Veuillez trouver ici l’expression de mes sincères remerciements et ma profonde reconnaissance.
A ma Co-encadrante de thèse le Dr Sanaâ LEMRISS, Chef du Département de Biosécurité PCL3 au sein du LRAM. Pour m’avoir si chaleureusement accueillie dans votre laboratoire, pour m’avoir initié dans ce domaine et pour m’avoir guidée avec compétence. Je tiens également à vous remercier pour votre patience et soutien tout au long de mon stage de master et de ces années de thèse. Votre encadrement, vos encouragements et vos conseils précieux pour l’élaboration de ce travail resteront à jamais marqués dans mon cœur. Merci de m’avoir orientée vers ce sujet vaste et passionnant que représente le séquençage à haut débit et l’analyse bioinformatique. Veuillez trouver ici l’expression de ma profonde reconnaissance. A Mr. Le président de jury ainsi que les membres de jury, malgré vos multiples occupations vous me faites l’honneur d’évaluer mon travail en acceptant de participer à mon jury. Je vous exprime ma profonde gratitude.
A Monsieur le Pr. Jamal TAOUFIK, Professeur d’Enseignement Supérieure et Directeur du Centre d’Etudes Doctorales des Sciences de la Vie et de la Santé à la Faculté de Médecine et de Pharmacie de Rabat, Université Mohammed V, pour m’avoir inscrit au sein de cet établissement et pour votre aide administrative. Aussi de me faire l’honneur de présider le jury de cette soutenance de thèse malgré vos nombreuses occupations. Veuillez trouvez ici toute ma reconnaissance et mes sincères remerciements.
A Monsieur le Pr. Chakib NEJJARI, Professeur d’enseignement Supérieure et Président de l’Université Mohammed VI des Sciences de la Santé de Casablanca, pour m’avoir accordé du temps et d’avoir accepté de participer à ce jury en tant que rapporteur de cette thèse malgré votre emploi du temps plus que chargé. Veuillez trouver ici le témoignage de mes respectueuses considérations.
A Monsieur le Pr. Anas Kettani, Professeur d’Enseignement Supérieure à la Faculté des Sciences Ben M’Sik, Université Hassan II de Casablanca, pour m’avoir accordé du temps en participant à ce jury et d’avoir la gentillesse de bien vouloir juger ce travail en tant que rapporteur. Je tiens également à vous remercier pour vos encouragements tout au long de mes
A Monsieur le Pr. Rachid ELJAOUDI, Professeur Agrégé à la Faculté de Médecine et de Pharmacie de Rabat. Je vous suis reconnaissante d’avoir accepté de participer à ce jury et de bien vouloir juger ce travail en tant rapporteur. Veuillez trouver ici l’expression de ma profonde reconnaissance.
A Madame le Dr Patrícia Martins Simões, Ingénieur de recherche en bioinformatique au laboratoire de bactériologie au Centre International de Recherche en Infectiologie (CIRI), Hospices Civils de Lyon et fait partie de l’Equipe ”Pathogenèse des infections à staphylocoques” du Centre National de Référence des Staphylocoques, Lyon, France, pour avoir accepté de travailler en équipe l’analyse bioinformatique sur les données de séquençage, pour votre aide, vos suggestions et pour vos judicieux conseils au moment de la rédaction des articles. Je n’ai pu qu’apprécier votre réactivité au cours de nos discussions, souvent virtuelles, pour aboutir à ce travail collaboratif, preuve que la science est avant tout une histoire humaine. Bonne chance pour la suite et j’espère que nos chemins se recroiseront. Veuillez trouver ici l’expression de ma profonde reconnaissance.
A Monsieur le Dr Jean-Philippe Rasigade, Biologiste Médicale au Centre International de Recherche en Infectiologie (CIRI), Hospices Civils de Lyon, pour votre aide, pour votre participation et vos conseils avisés au cours de la correction des articles. Veuillez trouver ici l’expression de mes sincères remerciements.
A Madame le Dr Marine BUTIN, Pédiatre à Centre Hospitalier Universitaire de Lyon, France et thésard au laboratoire de bactériologie médicale, Hospices Civils de Lyon. J’ai beaucoup apprécié la qualité de nos échanges scientifiques et de travailler avec toi la partie génomique de ce projet, merci pour ta motivation, ton énergie et tes conseils. Veuillez trouver ici l’expression de mes sincères remerciements.
A Monsieur le Doctorant Yann Dumon, Thésard au laboratoire de bactériologie au Centre International de Recherche en Infectiologie (CIRI), Hospices Civils de Lyon, qui nous a rejoint en cours de route, merci pour ta contribution dans l’analyse bioinformatique et ton aide. Veuillez trouver mes sincères remerciements.
vos efforts et vous souhaitez la réussite que vous méritez.
A Mlle Amal SOURI, Chef adjoint du Département de Biosécurité PCL3 au sein du LRAM, pour votre aide et vos conseils durant toutes les années de ma thèse. Veuillez trouver ici l’expression de mes sincères remerciements et l’expression de ma profonde reconnaissance.
A Madame le Pr. Aicha RIFAI, Professeur Assistant à la Faculté des Sciences Université Chouaib Doukkali pour avoir pris le temps de relire mon manuscrit. Veuillez recevoir l’expression de ma profonde reconnaissance.
A l’équipe du Département de Biosécurité PCL3, LRAM, j’ai pu passer mes stages dans un cadre particulièrement agréable, grâce à l’ensemble des membres de l’équipe de Département de Biosécurité PCL3. Un immense merci pour leur aide, leur gentillesse, leur infinie patience…, et leur bonne humeur.
Un grand merci à mes collègues au sein du laboratoire de Biotechnologie Médicale (MedBiotech) pour leur soutien et leurs encouragements.
Ces remerciements ne seraient pas complets sans une pensée pour mes magnifiques collègues de la promotion doctorale mes amis de longue date. Merci de m’avoir aidé et encouragé, et pour m’avoir changé les idées quand j’en avais besoin.
Finalement, au terme de ce travail je tiens à remercier toutes les personnes que j'ai rencontrées lors de mes études doctorales et qui ont contribué de près ou de loin au bon déroulement de cette thèse.
ARTICLES
1. Lemriss, H., Martins Simões P., Lemriss, S., Butin, M., Ibrahimi, A., El Kabbaj, S.,
Rasigade JP., Laurent, F. (2014). Non-contiguous finished genome sequence of Staphylococcus capitis CR01 (pulsetype NRCS-A). Standards in Genomic Sciences, 9(3), 1118–1127.
2. Lemriss H., Dumont Y, Lemriss S., Martins-Simoes P., Butin M, Lahlou L., Rasigade JP,
El Kabbaj S., Laurent F., Ibrahimi A. (2015). Genome Sequences of Four Staphylococcus
capitis NRCS-A Isolates from Geographically Distant Neonatal Intensive Care
Units. Genome Announcements, 3(4), e00501–15.
3. Lemriss, H., Lemriss, S., Martins-Simoes, P., Butin, M., Lahlou, L., Rasigade, J.-P., El
Kabbaj, S. (2016). Genome Sequences of Multiresistant Staphylococcus capitis Pulsotype NRCS-A and Methicillin-Susceptible S. capitis Pulsotype NRCS-C. Genome
Announcements. 4(3):e00541-16.
4. Martins Simões, P., Rasigade, J.-P., Lemriss, H., Butin, M., Ginevra, C., Lemriss, S.,
Goering, R. V., Ibrahimi, A., Picaud, J. C., El Kabbaj, S., Vandenesch, F and Laurent, F. (2013). Characterization of a Novel Composite Staphylococcal Cassette Chromosome mec (SCCmec-SCCcad/ars/cop) in the Neonatal Sepsis-Associated
Staphylococcus capitis Pulsotype NRCS-A. Antimicrobial Agents and
Chemotherapy, 57(12), 6354–6357.
5. Martins-Simoes P., Lemriss, H., Dumont, Y., Lemriss, S., Rasigade, J.-P.,
Assant-Trouillet, S., Ibrahimi, A., El Kabbaj., Butin, M and Laurent, F. (2016). Single-Molecule Sequencing (PacBio) of the Staphylococcus capitis NRCS-A Clone Reveals the Basis of Multidrug Resistance and Adaptation to the Neonatal Intensive Care Unit Environment. Frontiers in Microbiology, 7, 1991.
F., Picaud JC and Laurent, F. (2015) Wide geographical dissemination of the multiresistant Staphylococcus capitis NRCS-A clone in neonatal intensive-care units.
Clinical Microbiology and Infection, vol. 22, no. 1, pp. 46-52.
COMMUNICATIONS ORALES
1- P. Martins-Simões, Y. Dumont, M. Butin, H. Lemriss, S. Lemriss, A. Ibrahimi, S.El
Kabbaj, F. Vandenesch, J.P. Rasigade, F. Laurent. Caractérisation moléculaire par séquençage PacBio du clone endémique mondial S. capitis NRCS-A responsable de sepsis en réanimation néonatale : résistances, virulence et avantage sélectif. RICAI, Paris, 14 Décembre 2015.
2- P. Martins Simoes, H. Lemriss, J.-P. Rasigade, S. Lemriss, F. Vandenesch, S. El Kabbaj,
F. Laurent. Staphylococcus capitis NRCS-A: SCC, SCCMEC and Their Evolutionary History. 5th Congrees of European Microbiologists, FEMS, Juillet 2013
3- M. Butin, P. Martins Simões, J.P. Rasigade, S. Lemriss, H. Lemriss, S. El Kabbaj, A.
Ibrahimi, S. Tigaud, F. Vandenesch, O. Claris, J.C. Picaud, F. Laurent. Diffusion mondiale du clone Staphylococcus capitis NRCS-A en réanimation néonatale : caractérisation moléculaire, analyse génomique et histoire évolutive. RICAI, Paris, 21 Novembre 2013.
4- Martins Simões P, Butin M, Lemriss H, Lemriss S, Deighton MA, Kearns A, Denis O,
Goering R, Ginevra C, Picaud JC, Ibrahimi A, EL Kabbaj S, Vandenesch F, Rasigade J-P, Laurent F. Worldwide distribution of an endemic multi-resistant NRCS-A Staphylococcus capitis clone in NICU: molecular typing, whole genome analysis and evolutionary history.10th International Meeting on Microbial Epidemiological Markers, Paris, 02-05 October 2013.
unexpected worldwide dissemination of an endemic multi-resistant clone. European Academy of Pediatric Societies. Barcelone, 17 - 21 October 2014.
La prévalence des infections staphylococciques, nosocomiales et communautaires, augmente régulièrement. Le traitement de ces infections est devenu de plus en plus difficile à cause de l’émergence de souches multirésistantes. Les Staphylocoques ont la capacité d'acquérir des déterminants de la résistance aux antimicrobiens rapidement, en particulier après l'introduction de nouveaux agents antimicrobiens dans la pratique clinique.
Récemment, de nombreuses études ont décrit l’émergence dans les hôpitaux de plusieurs pays, et particulièrement dans les unités de soins intensifs néonatales (USIN), des isolats de
Staphylococcus capitis résistantes à la méthicilline et ayant une sensibilité réduites à la
vancomycine (VISA). Ces isolats de S. capitis ont causé des septicémies mortelles chez les nourrissons de faible poids (<1500 g), ce qui a entraîné une vague d'échecs thérapeutiques et des problèmes socio-économiques.
Une étude de séquençage de haut débit (NGS), d’assemblage, d’analyse bioinformatique des données NGS, de typage moléculaire et de comparaison des génomes complets de neuf souches de S. capitis isolées de différents USIN dans le monde à différentes périodes a été mis en place afin de contribuer à améliorer et à approfondir les connaissances sur le génome complet de cet espèce peu étudié dans le monde.
Les objectifs étaient d'analyser et de comparer les éléments génétiques communs de ces neuf souches aux six génomes de l’espèce S. capitis isolés chez des adultes et d'autres souches de staphylocoques d’une part et de caractériser le clone NRCS-A et de comprendre les origines de son succès d’autre part.
Les résultats de cette analyse et de comparaison de génomes ont permis d’avoir un premier génome de référence de la souche S. capitis CR01 et l’identification des transferts horizontaux des éléments génétiques mobiles (SCCmec-SCCcad / ars / cop et l’élément CRIPR, plasmide et phage), des gènes de résistance aux antibiotiques, des déterminants de virulences associés à la pathogénicité, de transposon, des séquences d’insertions et un gène nsr trouvé exclusivement dans les souches de S. capitis appartenant au clone NRCS-A fonctionnel et confère une résistance à la nisine.
Les caractéristiques génomiques trouvées mettent l'accent sur la contribution des éléments génétiques mobiles à l'émergence de la résistance multiple du clone de S. capitis NRCS-A et
NRCS-A n'a été détecté, ce qui ne soutient pas le rôle de la virulence en tant que force motrice de l'émergence du NRCS-A dans les USIN du monde entier.
En conclusion, notre étude a permis de caractériser, d’élucidé et d’enrichir les connaissances sur les génomes de S.capitis, ainsi que les mécanismes génétique impliqués à l’émergence, l’évolution, l’adaptation et la dissémination du clone endémique NRCS-A.
Mots-clés : Staphylococcus capitis, NGS, bioinformatique, élément mobile, cassette
SCCmec-SCCcad / ars / cop, résistance aux antibiotiques, virulence, phage, plasmide, transposon, évolution et dissémination clonale, NRCS-A.
The prevalence of staphylococcal nosocomial and community infections increases steadily. Treatment of these infections has become increasingly difficult due to the emergence of multidrug resistant strains. Staphylococci have the ability to acquire determinants of antimicrobial resistance quickly, especially after the introduction of new antimicrobial agents into clinical practice.
Recently, many studies have described the emergence of methicillin-resistant Staphylococcus
capitis isolates with reduced sensitivity to vancomycin (VISA) in hospitals in several
countries, particularly in the neonatal intensive care units (NICUs). These S. capitis isolates caused fatal septicemia in low-weight infants (<1500 g), which resulted in a wave of therapeutic failures and socio-economic problems.
A study using high-throughput sequencing (NGS), assembly, bioinformatics analysis of NGS data, molecular typing and complete genome comparison of nine S. capitis strains which are isolated from different NICUs worldwide at different period of time has been set up to contribute to the improvement and deepening of knowledge about the complete genome of this little studied species in the world.
The objectives were to analyze and compare the common genetic elements of these nine strains to six S. capitis genomes isolated from adults and other strains of staphylococci in one hand, and to characterize the NRCS-A clone and understand the origins of his success on the other hand.
The results of this analysis and comparison of genomes allowed to have a first reference genome of the S. capitis CR01 strain and the identification of the horizontal translocations of the mobile genetic elements (SCCmec-SCCcad / ars / CRIPR, plasmid and phage), antibiotic resistance genes, pathogenicity-associated virulence determinants, transposon, insert sequences and an nsr gene found exclusively in S. capitis strains belonging to clone NRCS- A which is functional and confers resistance to nisin.
The genomic characteristics found in this study emphasize the contribution of mobile genetic elements to the emergence of multiple resistance of the S. capitis NRCS-A clone and the uniqueness of the NRCS-A clone concerning antibiotic resistance, virulence factors and genetic elements.
around the world.
In conclusion, our study allowed us to characterize, elucidate and enrich the knowledge on the genomes of S.capitis, as well as the genetic mechanisms involved in the emergence, evolution, adaptation and spread of the clone Endemic NRCS-A.
Keywords: NSCS-A, Staphylococcus capitis, NGS, bioinformatics, mobile element, cassette
SCCmec-SCCcad / ars / cop, resistance to antibiotics, virulence, phage, plasmid, transposon, evolution and clonal dissemination.
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 1
INTRODUCTION
13
OBJECTIF DELA THESE
17
CHAPITRE I: Revue bibliographique
18
I.1 GENOMIQUE
18
I.1.1 Révolution biologique 18
I.1.2 Séquençage génomique 20
I.1.2.1 Historique 20
I.1.2.2 Constitution de banque d’ADN 22
I.1.2.3 Séquençage 1er Génération 24
I.1.2.3.1 Méthodes de séquençage 24
I.1.2.3.2 Automatisation de méthode de Sanger 26
I.1.2.3.3 Stratégies de séquençage de génome entier 27
I.1.2.4 Séquençage nouvelle génération (Next Generation Sequencing (NGS)) 30
I.1.2.4.1 Séquençage de 2ème génération 31
I.1.2.4.2 Séquençage de 3ème génération 39
I.1.2.4.3 Technologies émergentes 40
I.1.2.5 Application de séquençage à haut débit 41
I.1.2.5.1 Le séquençage de novo 41
I.1.2.5.2 Le reséquençage 41
I.1.2.5.3 La métgénomique 42
I.1.2.6 Génomique comparative et exploitation des génomes 42
I.1.2.6.1 Assemblages des génomes 42
I.1.2.6.2 Annotation des génomes 44
I.1.2.6.3 Apport de la génomique comparative 44
I.2 LES STAPHYLOCOQUES 47
I.2.1 Taxonomie 47
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 2
I.2.3 Physiopathologie 48
I.2.4 Infection 49
I.2.5 Caractéristiques intrinsèques 50
I.2.5.1 Génome 50
I.2.5.2 Enveloppe cellulaire 50
I.2.5.3 Capsule 50
I.2.5.4 Facteurs de virulence 51
I.2.5.5 Régulation des facteurs de virulence 52
I.2.6 Antibiothérapie 53
I.2.6.1 Mécanismes d’action des antibiotiques 53
I.2.6.2 Résistance aux antibiotiques 54
I.2.7 Cassettes SCCmec 56
I.2.7.1 Définition 56
I.2.7.2 Structure générale 57
I.2.7.3 Classification et typage 57
I.2.8 Staphylococcus capitis 60
I.2.8.1 Taxonomie 60
I.2.8.2 Habitat 60
I.2.8.3 Pouvoir pathogène 60
I.2.8.4 Résistance aux antibiotiques 60
I.2.8.5 Caractères bactériologiques 61
I.2.8.6 Caractéristiques génomiques 61
CHAPITRE II: Séquence génomique complète de Staphylococcus capitis
CR01 : Génome de référence
63
II.1 Abstract 65
II.2 Introduction 65
II.3 Classification and information 65
II.4 Genome sequencing information 70
II.5 Genome project history 70
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 3
II.9 Genome properties 71
II.10 Conclusion 73
II.11 Acknowledgments 74
II.12 References 74
CHAPITRE III: Séquençage de huit isolats de Staphylococus capitis isolées
de différentes régions géographiques dans le monde
77III.1 Genome Sequences of Four Staphylococcus capitis NRCS-A Isolates from
Geographically Distant Neonatal Intensive Care Units 79
III.2 Genome Sequences of multiresistant Staphylococcus capitis pulsotype NRCS-A and
methicillin- 2 susceptible Staphylococcus capitis pulsotype NRCS-C 83
CHAPITRE IV: Génomique comparative de souches de Stapylococcus
capitis
88
IV.1 Characterization of a Novel Composite Staphylococcal Cassette Chromosome mec (SCCmec-SCCcad/ars/cop) in the Neonatal Sepsis-Associated Staphylococcus capitis
Pulsotype NRCS-A 91
IV.2 Single-Molecule Sequencing (PacBio) of the Staphylococcus capitis NRCS-A Clone Reveals the Basis of Multidrug Resistance and Adaptation to the Neonatal Intensive
Care Unit Environment 107
IV.2.1 Abstract 107
IV.2.2 Introduction 108
IV.2.3 Methods 109
IV.2.4 Results 113
IV.2.5 Discussion 122
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 4
IV.2.9 References 124
IV.2.10 Supplementary data 129
CHAPITRE V: Diffusion mondiale du clone Staphylococcus capitis NRCS-A
en réanimation néonatale : caractérisation moléculaire, analyse génomique
et histoire évolutive
136
V.1 Abstract 138
V.2 Introduction 138
V.3 Materials and Methods 139
V.4 Results 142 V.5 Discussion 146 V.6 Conclusion 147 V.7 Transparency declaration 148 V.8 Acknowledgements 148 V.9 References 148 V.10 Supplementary data 150
CHAPITRE VI : DISCUSSION GENERALE ET PERSPECTIVES
154
REFERENCES BIBLIOGRAPHIQUES
166
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 5
ADN : Acide désoxyribonucléique ADNc : ADN complémentaire Agr : accessory gene regulator ARN : Acide ribonucléique ARNm : ARN messager ARNt : ARN de transfert
AST : Antimicrobial Susceptibility Testing
BAC : Chromosomes Artificiels Bactériens
BBH : Bidirectional best hits
BLAST : Basic Local Alignment Search Tool
BMR : Bactéries Multirésistantes
bp : Base pair (paire de base)
CARD : Comprehensive Antibiotic Resistance Database
ccr : Cassette Chromosome Recombinase
CDS : Coding Sequence (sequence codante)
CHUV : Centre Hospitalier Universitaire Vaudois
CMI : Concentration Minimale Inhibitrice
COG : Cluster of Orthologous Groups (Groupe de Cluster Orthologues)
CoNS : Coagulase-negative staphylococci
CRISPR : Clustered Regularly Interspaced Short Palindromic Repeat CRT : Cyclic Reversible Termination
dru-typing : Direct repeat units typing
ddNTP : Didésoxyribonucéotides triphosphate
dNTP : Désoxyribonucéotides triphosphate
emPCR : PCR en émulsion
FC : Flow Cell
FRET : Fluorescence Resonance Emission Transfer
GIs : Genomic Islands
GISA : Glycopeptide intermidiate Staphylococcus aureus
GTR : General Time Reversable
IHGSC : International Human Genome Sequencing Consortium
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 6
LOS : late-onset sepsis
NICU : neonatal intensive care units
MALDI-TOF : Matrix-Assisted Laser Desorption Ionization Time-Of-Flight
MID : Multiplex Identifier
MGE : Mobile Genetic Elements
ML : Maximum Likelihood
MLST : Multi Locus Sequence Typing
MSCRAMM : Microbial Surface Components Recognizing Adhesive Matrix Molecules
NCBI : National center for biotechnology information
NGS : Next-generation sequencing
NTS : Nouvelles Techniques de Séquençages
ORF : Open Reading Frames
ORF : Open Reading Frame
pb : paire de bases
PBP : Penicillin Binding Proteins
PCR : Polymerase Chain Reaction
PFGE : Pulsed-field gel electrophoresis
PGM : Personal Genome Machine
PMS : Phenol Soluble Modulin
PTP : PicoTiterPlate
PVL : Leucocidine de Panton-Valentine
SARM : S. aureus résistants à la méticilline
SARM-C : S. aureus résistants à la méticilline d’origine communautaire SARM-H : Staphylococcus aureus Résistant à la Méticilline Hospitalier SASM : S. aureus sensible à la méticilline
SCC : Staphylococcal Chromosomal Cassette
SCCmec : Staphylococcal Chromosomal Cassette mec SCN : Staphylocoques à Coagulase Négative SCP : Staphylocoques à Coagulase Positive SCNRM : SCN résistants à la méticilline SMS : Single Molecule Sequencing
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 7
SMRT : Single Molecule Real Time
SNN : Septicémies nosocomiales néonatales
SNP : Single Nucleotide Polymorphism
SOLiD : Sequencing by Oligonucleotide Ligation and Detection
RAST : Rapid annotation using subsystem technology
RDP : Ribosomal Database Project
VFDB : Virulence Factors database
VISA : Vancomycin Intermediate Staphylococcus aureus
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 8
Figure I.1 : Dogme central de la biologie moléculaire.
Figure I.2 : Evolution du nombre de génomes complets disponible
(http://www.genomesonline.org).
Figure I.3 : Quelques étapes importantes démontrant l’évolution des progrès du séquençage (Blow, 2008).
Figure I.4 : Schéma simplifié de préparation de banques d’ADN génomique et d’ADNc (http://www.britannica.com/).
Figure I.5 : Schéma récapitulatif de la méthode de séquençage de Maxim et Gilbert (http://departments.oxy.edu/biology/Stillman/bi221/092200/lecture_notes.htm). Figure I.6 : Séquençage d’ADN par synthèse enzymatique avec chimie dye primer et dye
terminator (http://www.appliedbiosystems.com/).
Figure I.7 : Schéma de la stratégie du séquençage aléatoire global
(www.bio.davidson.edu/COures/genomics/method/shotgun.html). Figure I.8 : Schéma simplifiée de la stratégie de séquençage "clone par clone". Figure I.9 : Chronologie de la commercialisation des séquenceurs à haut-débit Figure I.10 : Etapes du pyroséquençage (Ahmadianet al., 2006).
Figure I.11 : Préparation de la libraire d’ADN (http://www.454.com/).
Figure I.12 : Préparation des billes où sont fixées les sstDNA pour le séquenceur 454 (http://www.454.com/).
Figure I.13 : Schéma simplifié de Pyroséquençage sur PTP 454 (http://www.454.com/). Figure I.14 : Amplification en pont de la technologie Illumina ((http://www.illumina.com/). Figure I.15 : Les principales infections staphylococciques.
Figure I.16 : Facteurs de virulence exprimés à la surface ou excrétés par S. aureus en fonction de la densité bactérienne (Ferry T et al., 2005).
Figure I.17 : L’histoire d’apparition des antibiotiques et des SARMs (Kos et al.,2012). Figure I.18 : Représentation des 4 types de cassettes SCCmec (Hiramatsuet al., 2002). Figure I.19 : Représentation schématique des 8 cassettes SCCmec (Van den Eede et al.,
2009).
Figure II.1 : Reference mass spectrum from Staphylococcus capitis strain (CR01).
Figure II.2 : Transmission electron microscopy of Staphylococcus capitis strain (CR01) using a JEOL 1400. The scale bar represents 200 nm.
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 9
Figure II.3 : 16S rRNA Phylogenetic tree highlighting the position of Staphylococcus
capitis strain CR01 (indicated by the yellow circle) relative to other type strains
within the genus Staphylococcus.
Figure II.4 : Graphical circular map of the chromosome
Figure IV.1.1 : Comparative structure analysis of the composite staphylococcal chromosome cassette SCCmec-SCCcad/asr/cop element of Staphylococcus capitis strain CR01.
Figure IV.1.2 : Phylogenetic analysis of whole genome-sequenced S. capitis strains and proposed evolutionary history of SCC elements acquisition in S. capitis.
Figure IV.1.3 : Schematic diagram and comparative analysis of the clustered regularly inter-spaced short palindromic region (CRISPR) region in Staphylococcus capitis strain CR01 SCCmec element with ST398 LA-MRSA strain 08BA02176. Figure IV.2.1 : Comparison of the genetic content of the strain CR01 plasmid with the draft
genomes of the other three NRCS-A strains
Figure IV.2.2 : Comparison of the genetic content of the strain CR01 intact prophage present in the other three draft genomes of NRCS-A strains.
Figure V.1 : Dendrogram and schematic representation of pulsotypes and dru types of 100 Staphylococcus capitis isolates from neonatal intensive-care units (NICUs) (n = 86) or other settings (n = 14).
Figure V.2 : Single-nucleotide polymorphism (SNP)-based maximum-likelihood tree with whole genome sequences of neonatal Staphylococcus capitis isolates from four distinct countries and publicly available S. capitis genomes.
Figure V.S1 : Molecular characterization of S. capitis bloodstream isolates from neonates (n = 12) and adult patients (n = 2).
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 10
Tableau I.1 : Mécanismes d’action des principales familles d’antibiotiques utilisées chez l’Humain.
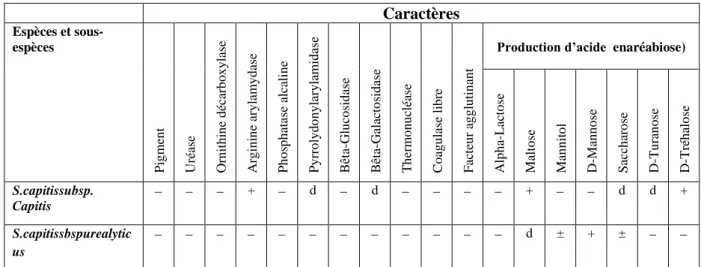
Tableau I.2 : Identification biochimique de S. capitis
Tableau I.3 : Lieu, origine et date de séquençage des 3 souches S. capitis.
Tableau I.4 : Caractéristiques générales du génome des trois souches de S. capitis (http://www.ncbi.nlm.nih.gov/genome/genomes/2054?details=on&
http://www.ncbi.nlm.nih.gov/genome/genomes/155?details=on&project_id=53 751).
Table II.1 : Classification and general features of Staphylococcus capitis strain CR01, pulsetype-NRCS-A according the MIGS recommendation.
Table II.2 : Project information.
Table II.3 : Nucleotide content and gene count levels of the genome.
Table II.4 : Number of genes associated with general COG functional categories. Table III.1 : Summary of genome sequencing in the present study.
Table IV.1.S1 : Open reading frames of the novel composite staphylococcal chromosome cassette (SCC) found in methicillin-resistant Staphylococcus capitis NCRS-A prototype strain CR01. The first 42 ORFs (orf1 to orf42) were found in the SCCmec element, while orf43 to orf70 were found in the SCCcad/ars/cop element.
Table IV.1.S2 : Highest probable matches for the 15 interspersed DNA sequences (spacers) identified in the clustered regularly inter-spaced short palindromic repeats (CRISPR) region within the SCCmec element found in S. capitis strain CR01. Table IV.2.1 : Baterial strains used in nisin susceptibility test.
Table IV.2.2 : General genomic features of S. capitis strain CR01 compared with another three WGS NRCS-A genomes.
Table IV.2.3 : Comparison of virulence factors in S. capitis NRCS-A, non-NRCS-A, and S.
epidermidis after exclusion of virulence factors present only in S. aureus.
Table IV.2.4 : Genomic profiles of antibiotic resistance for clone NRCS-A strains from four different countries.
Table IV.2.S1 : Genes found exclusively in the S. capitis NRCS-A clone and not in other S.
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 11
Table IV.2.S2 : Methylated motifs detected for S. capitis NRCS-A strain CR01.
Table V.1 : Antimicrobial susceptibility profile of Staphylococcus capitis bloodstream isolates from neonates (n = 12) and adults (n = 2).
Table V.S1 : Characteristics of 7 house-keeping genes and related primers 389 used for the determination of the genetic relationships between 14 S. capitis isolates.
Table V.S2 : Profiles of the MLST-like approach of S. capitis bloodstream isolates from neonates (n=12) and 409 adults (n=2).
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V-Rabat⎹ 12
Annexe 1 : Chronologie non exhaustive des événements marquants survenus en
biologie (Rose), et en bioinformatique (Incolore).
Annexe 2 : Espèces et sous-espèces du genre Staphylococcus et hôtes associés. Annexe 3 : Principales caractéristiques des différentes techniques de séquençage.
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 13
Le Staphylocoque est observé pour la première fois par Pasteur en 1879 dans un pus de furoncle, et ensuite identifié par Ogston en 1881 à travers la description d’observations cliniques et bactériologiques relatives à son rôle dans le sepsis et la formation d’abcès (Vandenesch et al., 2007). Actuellement, 50 espèces et sous espèces sont répertoriées dans le genre Staphylococcus.
La prévalence des infections staphylococciques, nosocomiales et communautaires, augmente régulièrement. Le traitement de ces infections est devenu de plus en plus difficile à cause de l’émergence de souches multirésistantes. Les Staphylocoques ont la capacité d'acquérir des déterminants de la résistance aux antimicrobiens rapidement, en particulier après l'introduction de nouveaux agents antimicrobiens dans la pratique clinique.
A partir de 1950, dix ans après la découverte de la pénicilline, les Staphylococcus aureus deviennent un problème thérapeutique majeur par l’acquisition de la résistance plasmidique à la pénicilline. Peu après l’introduction d’un nouvel antibiotique, la méticilline (découverte en 1959), apparaissent des souches de S. aureus résistantes appelées S. aureus résistants à la méthicilline (SARM). En fait, les SARM sont résistants non seulement à la méticilline mais également aux autres antibiotiques de la même classe. Ces souches de SARM sont avant tout responsables d’infections nosocomiales pouvant évoluer sur un mode épidémique (Mishaan et
al., 2005). En outre, des infections à SARM d’origine communautaire (SARM-C) ont été
également décrites (Dufour et al., 2002, Okuma et al., 2002).
Récemment, de nombreuses études ont décrit l’émergence dans les hôpitaux de plusieurs pays, et particulièrement dans les unités néonatales de soins intensifs (NICU), des isolats de
Staphylococcus capitis résistantes à la méticilline et ayant une sensibilité réduites à la
vancomycine (Van Der Zwet, 2002 ; Gras-Le Guen et al., 2007). Ces isolats de S. capitis ont causé des septicémies mortelles chez les nourrissons de faible poids (<1500 g), ce qui a entraîné une vague d'échecs thérapeutiques et des problèmes socio-économiques (Rasigade et
al, 2012).
Une étude menée dans des NICU françaises a démontré la propagation d'une seule population clonale de S. capitis (pulsotype NRCS-A) résistante à la méthicilline, associée à une sensibilité réduite à la vancomycine, antibiotique de première ligne pour le traitement de ces sepsis néonataux (Rasigadeet al, 2012). De plus, ce même clone a été récemment identifié
dans les NICU en Belgique, au Royaume-Uni et en Australie, ce qui suggère une distribution mondiale (Van Der Zwet et al., 2002 ; Venkatesh et al., 2006 ; D'mello et al.,2008 ;Rasigade
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 14
et al., 2012 ; Boghossian et al., 2013 ; Cui et al., 2013). En revanche, dans la bactériémie chez
l'adulte, S. capitis est rarement trouvée et lorsqu'elle est détectée, elle présente une plus grande diversité en terme de génotypes et de profils de sensibilité aux antimicrobiens que chez les nouveau-nés (Qin N et al., 2012).
Ce clone présente un profil atypique. En plus d'une résistance à l’ensemble des bêta-lactamines, il possède une résistance à l’ensemble des aminosides, ainsi qu'une sensibilité diminuée (hétérorésistance) aux glycopeptides qui constituent le traitement probabiliste de première intention en cas de sepsis chez le prématuré. Cette combinaison de résistance est très rarement retrouvée parmi les souches présentes chez l'adulte et reflète la pression de sélection des antibiotiques utilisés dans les services de néonatologie.
Aujourd’hui, la connaissance scientifique de cette population clonale multirésistantes de S.
capitis (pulsotype NRCS-A), très peu étudiée et disséminée dans les NICU à travers le
monde, devient une urgence primordiale afin de réduire son endémie et limiter sa propagation. Le séquençage complet du génome (WGS) est devenu un outil essentiel pour comprendre le pouvoir des bactéries pathogènes. Des avancées majeures ont eu lieu dans le domaine du séquençage au cours de cette dernière décennie avec le développement de plateformes de séquençage nouvelle génération, caractérisées par un haut débit et la possibilité d’analyser simultanément de nombreux échantillons. La génomique comparative des S. aureus est désormais possible, car plusieurs génomes de S. aureus ont été séquencés et disponibles dans les bases de données (Holden et al., 2004; Azarian et al., 2015 ; Sabat et al., 2017) . Des travaux d’analyse génomique ont été également réalisés pour S. epidermidis RP62a et S.
epidermidis ATCC12228 en comparant leurs génomes à ceux des espèces staphylocoques les
plus pathogènes. Ces études ont permis de découvrir de manière approfondie les gènes qui contribuent probablement à la virulence de S. epidermidis, en particulier la formation de biofilm et la défense immunitaire (Zhang et al., 2003; Gill et al., 2005). Par la suite, des projets de génomique comparative similaires ont été effectués pour d'autres espèces de Staphylocoques à Coagulase Négative (SCN) telles que S. saprophyticus (Kuroda et al., 2005), S. haemolyticus (Takeuchi et al., 2005 ; Cavanagh et al., 2014) et S. lugdunensis (Heilbronner et al., 2011).
En revanche, très peu de travaux de recherche ont été entrepris concernant le séquençage du génome complet de S. capitis. La disponibilité de ce dernier va permettre d’explorer les caractéristiques génomiques (résistance aux antibiotiques, facteurs de virulence, éléments
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 15
génétiques mobiles (MGE) et gènes spécifiques) ayant un rôle dans l'émergence du phénotype multirésistant du clone NRCS-A dans les différents NICU, et de comprendre l’histoire de son évolution, son adaptation et sa dissémination dans le monde.
Dans ce contexte et en collaboration avec le Centre Nationale de Référence des staphylocoques, Lyon, France, notre travail de thèse est un projet de séquençage, d’analyse bioinformatique et de comparaison génomique de souches multirésistantes de S. capitis (pulsotype NRCS-A) d'origines géographiques différentes et isolées à différentes périodes. Le manuscrit est organisé de la façon suivante :
Un premier chapitre présente une revue bibliographique sur la génomique et les staphylocoques.
Un second chapitre concerne le séquençage, l’assemblage et l’annotation de S. capitis CR01 (pulsotype NRCS-A) qui représente le premier génome complet de référence de souches isolées en neonatologie.
Un troisième chapitre traite le séquençage, l’assemblage et l’annotation de huit souches de S. capitis d'origines géographiques différentes dans le monde et isolées à différentes périodes.
Le chapitre 4 décrit le reséquençage, par un séquenceur de 3ème génération PacificBio (SMRT), et l’assemblage du génome de la souche de S. capitis CR01 (pulsotype NRCS-A), afin d’avoir un génome de référence fermé. De plus, ce chapitre présente également le travail d’analyse et de comparaison de ce génome de référence par rapport aux huit autres génomes séquencés de S. capitis, aux six génomes de S. capitis disponibles sur les bases de données (souches adultes), et aux souches non S. capitis, en utilisant différents outils et logiciels bioinformatiques, afin de caractériser et de localiser les éléments génétiques communs (gènes de résistances, facteurs de virulences, îlots de pathogénicité, îlots génomiques et MGE).
Le chapitre 5 illustre les diverses méthodes moléculaires utilisées (PFGE Sma et SacII, typage SCCmec, MLST, Dru typing et analyse phylogénétique des génomes complet) pour prouver la diffusion mondiale et l’unicité du clone épidémique NRCS-A de la souche S. capitis.
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 16
Le chapitre 6 est consacré à une discussion générale des résultats et aux perspectives ouvertes de ce travail.
OBJECTIF DE LA THESE
H. Lemriss⎹ Thèse de doctorat⎹ Université Mohammed V⎹ 17
L’objectif principal de mon projet de thèse est de contribuer à améliorer et à approfondir les connaissances du génome complet de l’espèce S. capitis peu étudié dans le monde, et de caractériser le clone endémique NRCS-A de cette espèce afin de comprendre les origines de son succès de dissémination et d’adaptation dans les NICU de quatre pays (France, Belgique, Angleterre et Australie).
La méthodologie du travail s’articule autour de quatre grands axes :
Séquençage, assemblage et annotation d’un premier génome de référence de l’espèce
S. capitis CR01 isolée de l’NICU en France.
Séquençage, assemblage et annotation de huit autres génomes de S. capitis isolées des NICU de quatre pays du monde (France, Belgique, Angleterre et Australie).
Identification, analyse et comparaison des éléments génétiques communs des neuf souches séquencées pendant mon travail de thèse aux six génomes de l’espèce S.
capitis isolés chez des adultes et d'autres souches de staphylocoques disponibles sur
NCBI.
Caractérisation du clone endémique NRCS-A de cette espèce, afin de comprendre les mécanismes génétique de résistances aux antibiotiques impliqués à son émergence, son évolution, son adaptation et sa dissémination dans le monde.
CHAPITRE I
Revue bibliographique
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 18 I.1 GENOMIQUE
En l’espace de 50 ans seulement, la biologie est passée de l’étude d’un seul gène à l’étude de génomes, de transcriptomes ou de protéomes d’organismes complets, avec une granularité d’analyse allant de la simple molécule isolée à l’organisme dans sa globalité. Ceci a été rendu possible par les avancées technologiques réalisées conjointement en biologie et en bioinformatique qui ont été motivées par la volonté des scientifiques de soutirer toujours davantage d’informations de notre génome. L’annexe 1 regroupe les événements majeurs qui sont intervenus en biologie, et en bioinformatique depuis 1944.
I.1.1 Révolution biologique
La révolution biologique a été initiée par quatre événements majeurs : la découverte de l’ADN en tant que support de l’information génétique (Avery et al., 1944) et de sa structure en double hélice (Watson et Crick, 1953), ainsi que la mise en place du dogme central de la biologie moléculaire et le déchiffrage du code génétique (Matthaei et al., 1962). Ces événements constituent les fondements de la génomique.
Le dogme central est la modélisation simplifiée du flux de l’information génétique à travers différentes molécules de la cellule et se résume en trois processus (Figure I.1).
ADN ARNm Protéine
Figure I.1:Dogme central de la biologie moléculaire
Grâce à ces principes, de nouvelles techniques de biologie moléculaire ont pu être développées, et en synergie avec la montée en puissance et la disponibilité des ordinateurs, une nouvelle discipline a émergé : la bioinformatique.
L’apparition de deux nouvelles approches de séquençage de l’ADN (Maxam et al., 1977; Sanger et al., 1977) a permis à la bioinformatique prend réellement son envol. La production de séquences par ces méthodes est l’occasion de créer les nouvelles banques de données EMBL (Cochrane et al., 2009) et GenBank (Benson et al., 2009), afin de répertorier ces séquences nucléiques, et de développer de nouveaux algorithmes
Réplication
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 19
permettant de traiter les données biologiques. Ces derniers ont abouti aux outils majeurs de la bioinformatique que sont FASTA (Pearson et Lipman, 1988), CLUSTALW (Thompson
et al., 1994), et BLAST (Altschul et al., 1997).
Avec l’apparition des séquenceurs automatiques et de nouveaux outils de biologie moléculaire, la production des séquences s’accélère et l’on voit apparaître des projets de séquençage de génomes complets qui aboutissent vers la fin du siècle.
Après dix années d’efforts, l’arrivée des premières séquences préliminaires du génome Humain (Lander e tal., 2001; Venter et al., 2001) marque la fin de l’ère génomique et l’entrée dans l’ère post-génomique. Cependant, il a fallu attendre jusqu’en 2004 pour obtenir de la part de l’IHGSC Consortium une version que l’on peut considérer comme finalisée (IHGSC, 2004).
Actuellement, grâce aux techniques de séquençage haut-débit, les projets de séquençage se sont multipliés (environ 60000) de sorte que la communauté scientifique a accès à 1530 génomes complets et publiés (Figure I.2).
Figure I.2 : Evolution du nombre de génomes complets disponible
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 20 I.1.2 Séquençage génomique
Le séquençage de l’ADN constitue une méthode dont le but est de déterminer la succession linéaire des bases A, C, G et T prenant part à la structure de l’ADN. La lecture de cette séquence permet d’étudier l’information biologique contenue par celle-ci. Étant donné l’unicité et la spécificité de la structure de l’ADN chez chaque individu, la séquence de l’ADN permet de nombreuses applications dans le domaine de la médecine, comme, par exemple, le diagnostic, les études génétiques, l’étude de paternité, la criminologie, la compréhension de mécanismes physiopathologiques, la synthèse de médicaments, les enquêtes épidémiologiques. L’objectif de la partie ci-dessous est de décrire l’évolution du séquençage manuel jusqu’aux séquenceurs haut débit qui sont les plus utilisées à l'heure actuelle.
I.1.2.1 Historique
En 1965, Holley et ses collaborateurs ont séquencé les deux premiers acides nucléiques de l’histoire, l’ARNt de l’alanine de la bactérie Escherichia coli, puis celui de la levure
Saccharomyces cerevisiae. C’est grâce à la capacité de purifier des ARNt particuliers et à la
connaissance de RNAses, dont la spécificité était connue, que ces premiers séquençages ont pu avoir lieu. De plus, il a été possible de déterminer la structure secondaire de l’ARNt, puisque l’hybridation entre les bases était connue à l’époque. C’est en 1971 que la première molécule d’ADN a été séquencée. Cette molécule consistait en une séquence de 12 nucléotides, soit la séquence des extrémités cohésives du phage lambda (Wu, 1970). Ces premières séquences ont été obtenues à l’aide de réactions chimiques spécifiques, comme la dépurination. Ces méthodes permettaient d’obtenir des séquences longues de 10 à 20 nucléotides.
En 1975, Sanger et Coulson ont introduit la méthode de terminaison des chaînes pour le séquençage de l’ADN (Figure I.3). En 1977, Maxam et Gilbert ont conçu une méthode similaire à celle de Sanger, mais ils utilisaient plutôt des nucléotides qui ne permettaient pas l’élongation des chaînes. La même année, Sanger a introduit la méthode des didéoxynucléotides, méthode qui permettait de séquencer jusqu’à 100 nucléotides. Cette technique a permis le séquençage du génome du phage PhiX (Sanger et al., 1977).
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 21
La grande innovation suivante dans l’histoire du séquençage a été l’automatisation des protocoles et de l’analyse (Première génération) (Hutchison, 2007). Cette avancée importante a permis de démocratiser le séquençage, jusqu’à permettre le séquençage de génomes complets (95%), dont le génome humain en février 2001 (Lander et al., 2001, Venter et al., 2001) et le génome des TriTryp en 2005.
La seconde génération d’outils de séquençage est apparue en 2005 en réponse au prix élevé et au faible débit du séquençage de première génération. Ici, des dizaines de milliers de séquences sont traitées ensemble et en parallèle. C’est l’apparition du séquençage haut-débit (« high throughput sequencing » ou encore le « next-generation sequencing »).
Alors que le projet de séquençage du génome humain a coûté trois milliards de dollars et a duré 13 ans (achevé en 2006), celui de James Watson (âgé de 79 ans, co-découvreur de la structure de l’ADN) a coûté un million de dollars et a été réalisé en deux mois. Il a été effectué sur un séquenceur FLX de Roche (société 454 Life Sciences, Baylor College of Medicine, Houston, Texas, États-Unis,). Quatre mois après, l’institut Craig Venter publiait le génome complet de Craig Venter (Levy et al., 2007). Contrairement à celui de James Watson, celui-ci a été séquencé selon la technique classique de Sanger (Figure 3).
En 2009, un des co-fondateurs de la société Helicos Bionsciences, Stephen R. Quake, séquence son génome (Pushkarev et al., 2009) avec une profondeur de 28x et une couverture de génome de 90% pour un coût de 48000 dollars. La même année (2009), quatre autres génomes humains ont été décrits : ceux d’un homme yoruba du Nigeria (Bentley et al., 2008) séquencé à une profondeur de 30x, de 2 coréens (Ahn et al., 2009 ; Kim et al ., 2009) à une profondeur de 28 et 29x et d’un chinois Han (Wang et al., 2008) à une profondeur de 36x. Ces séquençages individuels constituent une étape majeure vers la médecine personnelle
Les plateformes NGS actuellement disponible dans le marché utilisent des technologies de séquençage à haut débit de la seconde génération proposées par Roche 454 Life Sciences, Illumina, Solid et Ion Torrent et la troisième génération (« next-next-generation sequencing ») proposé par Pacific Biosciences (PacBio RS) (Metzker, 2010 ; McAdam et al., 2014).
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 22 Figure I.3 : Quelques étapes importantes démontrant l’évolution des progrès du séquençage
(Blow, 2008).
I.1.2.2 Constitution de banque d’ADN
Tout projet de séquençage commence par la constitution d’une ou plusieurs banques d’ADN. Cette banque est une collection de fragments d’ADN à séquencer qui ont été intégrés au génome de cellules hôtes (généralement des microorganismes) à des fins de stockage et de réplication. L’intégration est réalisée par l’intermédiaire d’une molécule d’ADN, appelée vecteur de clonage, à l’intérieur de laquelle a été placé un fragment de l’ADN que l’on veut séquencer (appelé dans le cas présent ‘insert’).
Il existe deux types de banques d’ADN : les banques d’ADN génomique dont les inserts sont issus de la fragmentation du matériel génétique initial à séquencer, et les banques d’ADNc dont les inserts sont des ARNm qui ont été « recopiés » en ADN sous l’effet d’une enzyme de rétrovirus, la transcriptase inverse. Les banques génomiques sont utilisées dans le cadre de séquençage de génomes, alors que les banques d’ADNc sont utilisées dans des études d’expression de gènes.
Mis à part une première étape différente entre la construction d’une banque génomique et celle d’une banque d’ADNc, les autres étapes sont communes aux deux (Figure I.4).
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 23
Première étape :
Banque génomique : consiste à fractionner l’ADN génomique par digestion partielle
avec une endonucléase, ou une enzyme de restriction, mais les méthodes physiques sont préférées car elles sont plus reproductibles et les fragmentations sont plus aléatoires. Ces méthodes physiques peuvent mettre en jeu la sonication, la nébulisation sous haute pression, ou la force de cisaillement (Levinthal et Davison, 1961).
Banque d’ADNc : aucune fragmentation n’est nécessaire. Au contraire, une attention toute particulière est apportée pour préserver les molécules d’ARN qui sont plus fragiles que l’ADN, puisqu’elles ne sont constituées que d’un seul brin. Lors de cette première étape, l’ARNm est rétro-transcrit en ADN sous l’action de la transcriptase inverse, une enzyme de rétrovirus.
Les autres étapes :
Elles consistent en une séparation par électrophorèse sur gel d’agarose des fragments d’ADN ou d’ADNc afin de sélectionner et d’extraire du gel les fragments de taille désirée, puis de les intégrer au vecteur de clonage choisis. Les cellules hôtes sont ensuite transformées par l’insertion d’un vecteur à leur matériel génétique. Finalement, les cellules ayant été transformées sont cultivées et isolées en colonies bien distinctes. Les cellules ayant intégré un vecteur sont sélectionnées à l’aide d’un des marqueurs du vecteur, généralement un gène de résistance à un antibiotique présent dans le milieu de culture, et qui empêche la multiplication des cellules non transformées. Chaque colonie est ensuite repiquée, conservée et étiquetée par un identifiant unique en vue de son séquençage.
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 24 Figure I.4 : Schéma simplifié de préparation de banques d’ADN génomique et d’ADNc
(http://www.britannica.com/)
I.1.2.3 Séquençage 1er génération I.1.2.3.1 Méthodes de séquençage
Il existe deux méthodes de séquençage dites « classiques » : la méthode de Maxam et Gilbert, par dégradation chimique sélective et la méthode De Sanger par synthèse enzymatique. Alors que l’utilisation de la première est restée confidentielle, la deuxième a été largement développée et constitue aujourd’hui la technique de référence.
Méthode chimique (Maxam et al., 1977)
Cette méthode est basée sur une dégradation chimique de l'ADN et utilise les réactivités différentes des quatre bases A, T, G et C, pour réaliser des coupures sélectives. En reconstituant l'ordre des coupures, on peut remonter à la séquence des nucléotidesde l'ADN correspondant. On peut décomposer ce séquençage chimique en six étapes successives :
Marquage : Les extrémités des deux brins d'ADN à séquencer sont marquées par un
traceur radioactif (32P). Cette réaction se fait en général au moyen d'ATP radioactif et de polynucléotide kinase.
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 25
Isolement du fragment d'ADN à séquencer : Celui-ci est séparé au moyen d'une
électrophorèse sur un gel de polyacrylamide. Le fragment d'ADN est découpé du gel et récupéré par diffusion.
Séparation de brins : Les deux brins de chaque fragment d'ADN sont séparés par
dénaturation thermique, puis purifiés par une nouvelle électrophorèse.
Modifications chimiques spécifiques : Les ADN simple-brin sont soumis à des
réactions chimiques spécifiques des différents types de base. Walter Gilbert a mis au point plusieurs types de réactions spécifiques, effectuées en parallèle sur une fraction de chaque brin d'ADN marqué : par exemple, une réaction pour les G (alkylation par le sulfate de diméthyle), une réaction pour les G et les A (dépurination), une réaction pour les C, ainsi qu'une réaction pour les C et les T (hydrolyse alcaline).
Coupure : Après ces réactions, l'ADN est clivé au niveau de la modification par réaction
avec une base, la pipéridine.
Analyse : Pour chaque fragment, les produits des différentes réactions sont séparés par
électrophorèse en conditions dénaturantes et analysés pour reconstituer la séquence de l'ADN. Cette analyse est analogue à celle que l'on effectue pour la méthode de Sanger (Figure I.5).
Les produits chimiques utilisés dans les milieux réactionnels lors des coupures spécifiques étant excessivement dangereux pour la santé, cette méthode a été abandonnée au profit de la méthode par synthèse enzymatique.
Figure I.5 : Schéma récapitulatif de la méthode de séquençage de Maxim et Gilbert
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 26
Méthode enzymatique (Sanger et al., 1977).
Cette méthode, encore appelée méthode De Sanger en raison de son inventeur, est basée sur l’activité de l’ADN polymérase qui permet de polymériser un brin d’ADN complémentaire à un brin matrice, à partir d’un oligonucléotide, appelé amorce. Cette capacité est utilisée pour synthétiser un brin complémentaire, mais de façon incomplète, en arrêtant aléatoirement la réaction de manière à obtenir statistiquement des produits issus de réaction interrompue à chacune des bases du fragment à séquencer.
Le mix réactionnel est constitué du vecteur de clonage contenant le fragment à cloner, de la polymérase, des amorces et des dNTP. Pour arrêter aléatoirement la réaction, une faible concentration de ddNTP est ajoutée. Ces ddNTP ne comportant pas de groupement 3’-OH, ils agissent comme des terminateurs de la réaction de polymérisation en empêchant l’accomplissement d’une liaison 5'-3' phosphodiester ultérieure.
A l’instar de la méthode chimique, un marquage des produits de réaction est nécessaire pour pouvoir détecter ces derniers après leur séparation par électrophorèse sur gel. Pour ceci, il existe deux chimies : dyeprimer et dyeterminator(Figure I.6).
Figure I.6 : Séquençage d’ADN par synthèse enzymatique avec chimie dye primer et
dyeterminator (http://www.appliedbiosystems.com/)
I.1.2.3.2 Automatisation de méthode de Sanger
La lecture manuelle de gels de séquençage est fastidieuse et sujette à de multiples erreurs d’interprétation, surtout après avoir lu les premiers milliers de bases. C’est pourquoi cette tâche a été automatisée à l’aide des séquenceurs.
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 27
L’étape de séquençage par synthèse enzymatique est toujours nécessaire. Elle est déléguée à des robots qui la réalisent dans des microplaques de 96 ou 384 puits, correspondant à autant de clones séquencés. Ces plaques sont ensuite transférées dans un séquenceur, qui va réaliser l’électrophorèse tout en enregistrant sur ordinateur les profils d’intensités lumineuses des fluorochromes. Ces profils sont appelés chromatogrammes.
Il existe deux types de séquenceurs automatiques de technologie Sanger :
Les séquenceurs à gel plat disposent d’un gel enserré entre deux plaques de verre en haut duquel sont disposés des puits.
Des séquenceurs à capillaires tel que ABI 3730 DNA Analyser de Life technologies. Cette technologie a permis des progrès en génomique depuis sa commercialisation en 2002. En 2004, en utilisant le séquenceur à capillaire automatique ABI3700, dans le cadre d'un projet collaboratif, le génome de P. atrosepticum SCRI1043 a été séquencé, ce qui a permis la caractérisation des facteurs de virulence de cette espèce par comparaison aux pathogènes de plantes ou d'animaux de la famille des
Enterobacteriaceae (Bell et al., 2004).
I.1.2.3.3 Stratégies de séquençage de génome entier
On distingue deux méthodes de séquençage des génomes entiers : Séquençage aléatoire globale (ou whole-génome shotgun)
Séquençage par ordonnancement hiérarchique
Dans les deux cas, l'ADN génomique est tout d'abord fragmenté par des méthodes soit enzymatiques (enzymes de restriction), soit physiques (ultrasons).
Séquençage aléatoire globale « whole-génome shotgun »
Le séquençage du premier génome bactérien, Haemophilus influenzae, a introduit la méthode du séquençage aléatoire global (Fleischman et al., 1995). Cette dernière avait été proposée en 1982 par Sanger et ses collaborateurs et avait été utilisée, à plus petite échelle, pour le séquençage du génome du phage lambda. C’est la stratégie retenue aujourd’hui pour tous les génomes bactériens. On l’associe plus volontiers aux sociétés privées, telles que Celera Genomics ou Syngenta, parce qu’elle est rapide et économique. Celera a ainsi accompli le séquençage de plusieurs organismes par une stratégie aléatoire globale. Toutefois, il faut remarquer que, pour de grands génomes comme celui de l'Homme, ces sociétés se sont
H. Lemriss Thèse de doctorat Université Mohammed V-Rabat 28
souvent appuyées sur des données de cartographie établies par les chercheurs académiques. En outre, pour ces grands génomes, on tend aujourd’hui à utiliser des stratégies mixtes, mêlant séquençage aléatoire global et "clone par clone".
Le séquençage du génome humain a donné lieu à une lutte médiatique entre Celera Genomics et le consortium international responsable du « projet Genome humain », qui utilisait quant à lui une stratégie « clone par clone »
Principe
La technique "shotgun" repose sur un principe simple : découper un génome en un grand nombre de fragments de petite taille (Figure I.7). Les extrémités d'une partie de ces fragments sont ensuite séquencées, puis ces séquences sont assemblées sur la base de leurs chevauchements grâce à des programmes informatiques pour essayer de produire une séquence complète (Ameziane et al., 2005). Les difficultés d'une telle technique sont à deux niveaux :
1- avoir assez de fragments pour couvrir le génome dans son entier 2- réussir l'assemblage
Figure I.7: Schéma de la stratégie du séquençage aléatoire global (www.bio.davidson.edu/COures/genomics/method/shotgun.html)