Autonomously Responsive Membranes
for Chemical Warfare Protection
The MIT Faculty has made this article openly available.
Please share
how this access benefits you. Your story matters.
Citation
Li, Yifan et al. "Autonomously Responsive Membranes for Chemical
Warfare Protection." Advanced Functional Materials 30, 25 (April
2020): 2000258 © 2020 Wiley
As Published
http://dx.doi.org/10.1002/adfm.202000258
Publisher
Wiley
Version
Author's final manuscript
Citable link
https://hdl.handle.net/1721.1/128130
Terms of Use
Creative Commons Attribution-Noncommercial-Share Alike
DOI: 10.1002/ ((please add manuscript number))
Article type: Full Paper
Title: Autonomously Responsive Membranes for Chemical Warfare Protection
Yifan Li†, Chiatai Chen†, Eric R. Meshot, Steven F. Buchsbaum, Myles Herbert, Rong Zhu,
Oleg Kulikov, Ben McDonald, Ngoc Bui, Melinda L. Jue, Sei Jin Park, Carlos A. Valdez, Saphon Hok, Qilin He, Christopher J. Doona, Kuang Jen Wu, Timothy M. Swager*, Francesco Fornasiero*
Dr. Y. Li, Dr. M. Herbert, Dr. R. Zhu, Dr. O. Kulikov, Dr. B. McDonald, Q. He, Prof. T. M. Swager
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
E-mail: [email protected]; [email protected]
C. Chen, Dr. E. R. Meshot, Dr. S. F. Buchsbaum, Dr. N. Bui, Dr. M. L. Jue, Dr. S. J. Park, Dr. C. A. Valdez, Dr. S. Hok, Dr. K. J. Wu, Dr. F. Fornasiero
Physical and Life Sciences,Lawrence Livermore National Laboratory, Livermore, CA 94550, USA
Dr. C. J. Doona
US Army Combat Capabilities Development Command - Soldier Center, Natick, MA 01760-5020, USA
†Equally contributing
Keywords: breathable and protective membranes, carbon nanotube pores, chemical-warfare-agent responsive polymers, color and volume changes, live chemical-warfare-agents.
[Abstract]
Stimuli-responsive materials offer new opportunities to resolve long-standing materials challenges and are rapidly gaining pivotal roles in diverse applications. For example, smart protective garments that rapidly transport water vapor and autonomously block chemical threats are expected to enable an effective new paradigm of adaptive personal protection. However, the incorporation of these seemingly incompatible properties into a single responsive system remains elusive. Herein, we demonstrate a bistable membrane that can rapidly, selectively, and reversibly transition from a highly breathable state in a safe environment to a chemically protective state when exposed to organophosphate threats such
2
as sarin. Dynamic response to chemical stimuli is achieved through the physical collapse of an ultrathin copolymer layer on the membrane surface, which efficiently gates transport through membrane pores composed of single-walled carbon nanotubes (SWNT). The adoption of nanometer-wide SWNTs for ultrafast moisture conduction enables a simultaneous boost in size-sieving selectivity and water-vapor permeability by decreasing nanotube diameter, thereby overcoming the breathability/protection trade-off that limits conventional membrane materials. Adaptive multifunctional membranes based on this platform greatly extend the active use of a protective garment and present exciting opportunities in many other areas including separation processes, sensing, and smart delivery.
1. Introduction
The creation of integrated materials systems that autonomously and dynamically respond to external stimuli represents a major challenge in materials science with enormous
technological potential in many areas[1] including: controlled release of active ingredients[2] and gating of molecular transport,[3] modulation of surface adhesion and wetting,[4] dynamic switching of optical properties,[5] energy harvesting,[6] droplet manipulation and assembly,[7] biomimetic materials,[8] sensor actuators,[9] and signal transducers.[10]
One area wherein stimuli-responsive materials promise to solve a significant materials challenge is the development of advanced fabrics that simultaneously protect wearers against biochemical agents and maintain high breathability (water vapor transport rate) to achieve efficient thermoregulation of the human body by evaporative cooling.[11] Conventional protective materials are passive systems based on impermeable barriers or heavy-weight adsorptive laminates and sacrifice breathability to prevent exposure to harmful agents. This leads to rapid overheating of the wearer in hot climates and/or during strenuous activities.[12] Smart dynamic materials that rapidly transition from a breathable state to a locally-actuated
protective state in response to environmental threats are expected to be particularly effective in mitigating this physiological burden.[12a] The localized material response allows for the unaffected part of a protective garment to remain highly breathable. In addition to enhancing thermal comfort, autonomously responsive systems provide protection even when the threat has not been otherwise detected.[13]
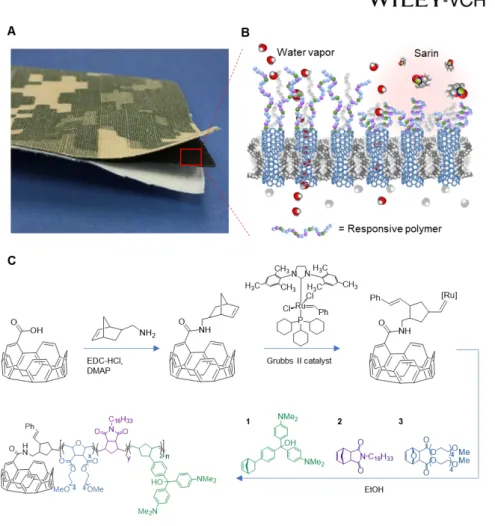
Herein we report the realization of this new paradigm of protective yet breathable adaptive garments that mimic the moisture regulation and barrier functions of the human skin as well as its ability to respond to the environment. Our concept is based on a bistable, chemical-threat-responsive membrane that integrates a highly breathable, bioprotective, nanoporous membrane with an ultrathin, responsive polymer layer that reversibly blocks the membrane pores upon contact with a chemical warfare agent (CWA) (Figure 1.B).
Combining high water-vapor permeability and high rejection of toxic chemicals in advanced fabrics must address conflicting material requirements. Optimal protection via efficient, threat-triggered pore closure requires small pore sizes and a high density of actuating polymer chains. On the other hand, large pores/high porosity and short, loosely packed polymer chains would boost breathability. To overcome the diametric selectivity-permeability trade-off, we select single-walled carbon nanotubes (SWNT) as vertically-oriented nanopores spanning the membrane thickness. These nanochannels can sustain water-vapor transport rates that exceed those of other equally-sized nanopores by more than an order of magnitude, and thus enable compensation for permeability losses due to surface functionalization with responsive polymers.[14] In addition, we show here that adoption of smaller diameter SWNTs increases counterintuitively the membrane breathability, a finding that helps resolve this material design dilemma. The polymer layer is designed to provide reversible and rapid response to a specific class of electrophilic reagents, i.e. organophosphate CWAs and structurally related but less toxic CWA simulants. We employ triarylmethanol (TAM)-containing hydrogel polymers that
4
undergo a large colorimetric change and volumetric collapse upon exposure to electrophilic CWA mimics.[15] When anchored to the membrane surface, the CWA-triggered collapse of
the polymer chains blocks molecular transport through the pores and thus provides on-demand protection. Furthermore, the system is chemically reversible as the triarylmethanol-containing polymers can be regenerated to their extended configuration via a base treatment.
The system described herein displays a number of key enabling features including: a) before polymer grafting, large water vapor transport rates that increases with decreasing SWNT diameter up to 11,000 gr m-2 day-1, a trend that transcends the typical selectivity/permeability trade-off for membrane materials;[16] b) rapid responses to reactive CWA mimics and the live agent sarin; c) efficient permeability regulation by the grafted polymer layer in response to environmental triggers; d) performance meeting US Department of Defense (DoD) targets for chemical threat protection.[17]
2. Results and Discussion
2.1. Breathability of SWNT membranes prior to functionalization
Previous reports have shown that fluid flow rates in carbon nanotube (CNT) channels exceed expectations for equally sized pores by orders of magnitude.[14, 18] This large flow
enhancement is typically attributed to the intrinsic atomic smoothness of well-graphitized CNT pore walls.[18-19] Because functionalization tends to disrupt this graphitic order, attachment of chemical moieties on the CNT entrance and, even more so, along the entire CNT length is expected to reduce dramatically the permeability of membranes with CNT pores. For example, Majumder et al.[20] reported that pressure-driven water-flow rates through 7-nm diameter multi-walled CNTs decreased by 2 and 4 orders of magnitude when the CNT tips and inner cores were functionalized, respectively.
Deleted: The responsive polymer chains rest in a native extended conformation, and thus do not obstruct the membrane pores in the absence of CWAs. Exposure to CWAs generates a deep-colored trityl cation with a more planar aromatic structure, which results in a polymer densification by the thermodynamically favorable π-stacking and van der Waals interactions between trityl cations. Commented [FF1]: CUT: “The responsive polymer chains rest in a native extended conformation, and thus do not obstruct the membrane pores in the absence of CWAs. Exposure to CWAs generates a deep-colored trityl cation with a more planar aromatic structure, which results in a polymer densification by the thermodynamically favorable π-stacking and van der Waals interactions between trityl cations.”
Formatted: Highlight Deleted: d
Commented [FF2]: CUT: “, which enables reversible switching between a highly breathable state in safe environments to a protective state in contaminated environments”
Deleted: , which enables reversible switching between a highly breathable state in safe environments to a protective state in contaminated environments
To compensate for a potential permeance loss as a result of functionalization, we sought to maximize the pristine membrane’s breathability by tuning the CNT pore characteristics. CNT forests with tube densities and average diameters in the range of 3.2 – 16.9 × 1011 cm-2 and 1.90 – 3.75 nm (Figure 2.A), respectively, were infiltrated with parylene-N (Figure 2.B) and etched open to form free-standing composite membranes, as described previously.[14] Membrane breathability was then measured at 30 °C and 30% mean relative humidity (RH) under a transmembrane RH gradient of 50%. From these moisture vapor transport rate (MVTR) results, the intrinsic membrane resistance to water vapor diffusion was calculated as
RCNT = ∆C /MVTR - RBL, where RBL = 104.5 s m-1 is the resistance resulting from the air-side
boundary layer at the two membrane surfaces and ∆C is the water vapor concentration difference across the membrane.[14] Figure 2.C shows that the intrinsic membrane resistance decreases and the MVTR increases when smaller diameter (and correspondingly higher density) SWNT arrays are used as moisture-conductive pores. At the smallest SWNT diameters of this study, RCNT dropped by a factor of two and the measured MVTR approached
11,000 gr m-2 day-1. This breathability level matches that of expanded polytetrafluoroethylene (ePTFE)[14] in spite of a much smaller porosity (4.5% vs ~64%) and pore size (1.9 nm vs ~210 nm).[21] Surprisingly, R
CNT and MVTR do not scale with the nominal CNT membrane porosity
(Figure S1). A possible explanation of this unexpected result is that a larger flow enhancement in smaller diameter SWNTs boosts breathability as the pore size decreases. Published experimental[19c] and theoretical findings[22] for pressure-driven water flow in narrow carbon nanotube pores have indeed revealed a rapid increase in flow enhancement with decreasing CNT diameter. To validate this hypothesis, we calculated the water vapor permeability for a single SWNT pore (DCNT) from our experimental breathability values, and
then compared DCNT with the transport diffusivities calculated with Knudsen diffusion theory
(DKn), bulk diffusion (Dbulk = 0.26 cm2 s−1), and transition regime diffusivity (1/Dtrans = 1/DKn
6
flow enhancement factor, E, defined as the ratio between measured SWNT permeability and Knudsen theory prediction. The inset in Figure 2.D shows that E increases sharply at the smallest pore diameters, displays an average four-fold increase when moving from 3.75 nm down to 1.9 nm pores, and exceeds 100 for the most breathable membranes. Remarkably, single pore permeability of water vapor in SWNTs increases steadily with decreasing diameter up to Dbulk (Figure 2.D). This implies that adoption of CNTs as flow channels
enables the simultaneous enhancement of membrane size-sieving selectivity and transport rates. This trend overcomes the traditional permeability/selectivity trade-off both predicted by conventional diffusion theories as well as seen in experiments with established membrane materials.[16]
2.2. Polymer response to nerve agent simulants and live agents
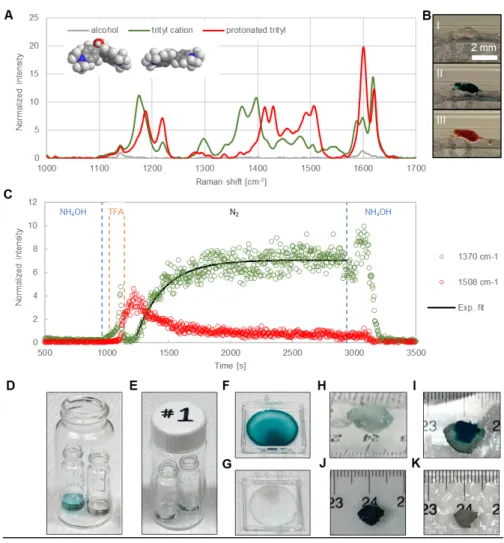
Our rational design of tunable responsive polymers exploits the flexibility provided by ring-opening olefin metathesis polymerization (ROMP) to incorporate multiple repeating units in a single chain in a straightforward manner. Copolymer compositions were previously down-selected for CWA detection applications based on their processability, responsiveness to reactive CWA simulants, and ability to form gels.[15, 23] The polymer system presented herein (Figure 1.C) contains a balance of (1) the CWA-responsive triarylmethanol (TAM) groups, (2) hydrophobic units containing long alkyl chains (CxHy), and (3) hydrophilic hydrogel units (PEG), the last of which facilitate water vapor permeation. In select compositions we have also included an alkyne-containing group (alkyne) as a distinct spectroscopic marker. These copolymers respond very rapidly to vapor simulants and, importantly, exhibit analogous colorimetric and volumetric changes when challenged with live CWA agents.
To characterize the material responsive behavior, we recorded the Raman spectroscopy signatures and the color change of a thin copolymer film (TAM/C16H33/PEG/alkyne = 9.5/42.75/42.75/5) in different gaseous environments (Figure 3.A-B). The alkyne group in this polymer is used as an internal standard with a clear band at 2219 cm-1. Both colorimetric and Raman spectroscopic analysis reveal the presence of three distinct electronic states for the TAM groups (Figure 3.A). After exposure to either acetic acid (AcOH) or diethyl
chlorophosphate (DCP) as CWA simulants,[15] a thin film of this copolymer switches from the colorless neutral state (I - triarylmethanol) to the deep green cationic state (II - trityl cation) (Figure 3.B), as explained in the Supporting Information.[24] This transition is reversible, and the material’s state I can be regenerated with a simple base-vapor treatment (NH4OH). When continuously exposed to a strong acid, namely trifluoroacetic acid (TFA), the material shifts to a third state with a deep red color that is likely caused by the protonation of the
dimethylamino substituted trityl cation (III).[25] This state is unstable in the absence of strong acid, and the material quickly reverts to the green-colored state II once TFA vapor is removed.
The Raman scattering signal intensity in the 1300-1550 cm-1 region of the neutral polymer is orders of magnitude weaker, and thus bands in this region provide a convenient spectroscopic handle for in situ monitoring of the material’s response kinetics. Figure 3.C displays the temporal evolution of Raman scattering upon exposing a thick film of the polymer (c.a. 0.1-0.5 mm) to a sequence of gaseous streams containing NH4OH (10 min), TFA (2 min), N2 (30 min), and NH4OH (10 min). The 1508 cm-1 peak intensity, which is characteristic of the transient protonated dication III, increases during TFA flow, but decays rapidly as soon as the TFA flow is stopped. In contrast, after a transient rise at low TFA doses, the 1370 cm-1 peak intensity (trityl cation II) is nearly undetectable under TFA flow but rises steadily up to a stable plateau with a subsequent N2 purging. The material transition from state III to state II
Formatted: Highlight
Commented [FF3]: CUT: “The ionization of the TAM unit in the copolymer is supported by a Raman spectrum which closely matches that of crystal violet (CV+), a TAM structural analogue
(Figure S3). In particular, the strong Raman bands in the 1350-1400 cm-1 region can be assigned to the vibrational C-C+ stretch modes of
the central positively-charged carbon atom (Table S1).” Deleted: The ionization of the TAM unit in the copolymer is supported by a Raman spectrum which closely matches that of crystal violet (CV+), a TAM structural analogue (Figure S3). In
particular, the strong Raman bands in the 1350-1400 cm-1 region can
be assigned to the vibrational C-C+ stretch modes of the central
positively-charged carbon atom (Table S1).
Commented [FF4]: CUT: “Corresponding Raman scattering bands match indeed those reported for a protonated CV+ dication.”
Deleted: . Corresponding Raman scattering bands match indeed those reported for a protonated CV+ dication
8
can be fit to a single exponential function with a characteristic time t ~ 4-min, and the conversion from neutral (I) to trityl cation (II) occurred within 6 minutes. The regeneration of the polymer with a NH4OH vapor was similarly rapid. In concert with a strong rapid
colorimetric response, 1-cm diameter bulk gels of these polymers were previously shown to display an 8-fold volume collapse in 10 minutes upon contact with TFA vapors.[15]
Organophosphate nerve agents such as sarin (Figure S4) typically display a lower reactivity than their simulants such as DCP. To demonstrate rapid activation of our materials by live agents, we exposed solutions (2 mg/mL in chloroform), thin films, and gels of
TAM/CxHy/PEG terpolymers to both vapor and liquid sarin. Figure 3.D shows that, after spiking with a 25 μL droplet of 100 mM sarin solution, an initially clear polymer solution (225 μL) acquires a strong green color in <1 min (Movie S1). Thin films of the same copolymers respond colorimetrically to sarin vapors in <5 min (Figure 3.F). Polymer gels swollen in dichloromethane displays a nearly instantaneous color transition (Movie S2) and a fast volume collapse (~30 min) when locally challenged with 20 µL of neat sarin (Figure 3.I and J). Finally, polymer solutions, films, and gels could be fully regenerated to their colorless state in <10 min by exposure to NH4OH vapor (Figure 3.E, G, and K, respectively).
The dimensions of the gels in this volumetric change study using sarin and in a previous study with simulants[15] are several orders of magnitude larger than the length of the polymer chains we grafted on a SWNT membrane surface. Thus, as a result of much shorter diffusion times, these results suggest that functionalized SWNT membranes will be able to transition nearly instantaneously to a protective state when in contact with both simulants and live agents.
Integration of the responsive polymer layer must preserve the high breathability of the SWNT membranes and still enable efficient threat-triggered pore blocking. To provide the highest level of protection against CWAs, we aimed to maximize the density of polymer chains and ensure grafting near or at the pore entrances through a robust linkage. We also selected polymer chains short enough to provide low resistance to water vapor diffusion.
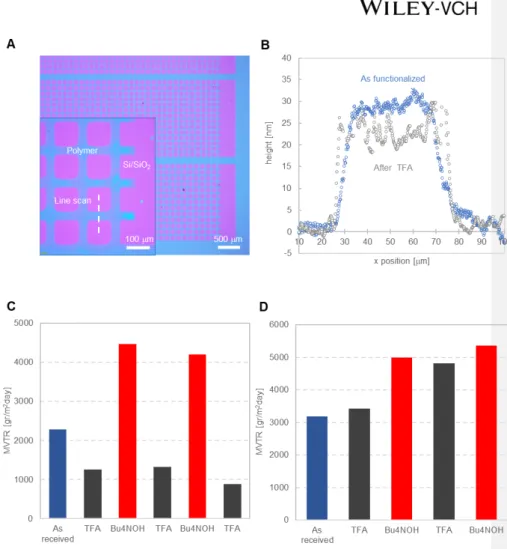
To this end, we adapted a functionalization method previously developed to covalently tether triarylmethanol-containing polymers to SiO2 nanoparticles by surface-initiated ROMP.[23] Specifically, we used carboxylic acid groups generated by air-plasma etching on the exposed tips of open nanotubes and on the surface of the polymer matrix as anchor points. Following the synthetic scheme outlined in the Experimental section, SWNT membranes were first functionalized with norbornene groups via amide coupling[26] and then exposed to a solution of the ruthenium olefin metathesis catalyst (Grubbs second generation). This catalyst-primed surface was finally treated with a solution of norbornene-based monomers (1, 2, and 3) to promote surface-initiated growth of the responsive polymers (Figure 1.C).[23, 27] This process produces grafted polymer chains with a large area density (about 1.25 molecules/nm2) and a molecular weight of 47.5 kDa (see Supporting Information).
To measure the length of these polymer chains in their native and collapsed states, a responsive polymer layer grafted from a silicon wafer was first patterned through a shadow mask by plasma etching (Figure 4.A) and then exposed to TFA (Figure S8). Profilometry line scans in Figure 4.B before and after the chemical challenge show that the thickness of the responsive polymer layer is only 28 ± 3 nm in the native state, thus providing a relatively low barrier to moisture permeation. The polymer densifies to 22 ± 5 nm in the protective state triggered by TFA and, as shown next, this densification is large enough to efficiently block the pore entrances.
10
2.4. Breathability of responsive SWNT membranes
To demonstrate that SWNT membranes functionalized with triarylmethanol-containing polymers are capable of responding to environmental cues, we quantified the trans-membrane water vapor flux after exposure to chemicals that trigger either a polymer chain collapse (TFA vapor) or extension (2 M Bu4NOH). Membranes grafted with polymers lacking the responsive groups (but otherwise identical to the responsive polymers) were used as controls and subjected to the same chemical challenges.
The permeability data in Figure 4.C indicates that the responsive membranes switch reversibly between two well-defined states triggered by environmental stimuli. Specifically, TFA vapor exposure induces a transition to a protective state with a low MVTR of ~1,000 gr m-2 day-1, while Bu4NOH restores the breathable state with a water vapor transport rate exceeding 4,000 gr m-2 day-1. Control membranes functionalized with polymer lacking the TAM responsive units did not display significant changes in permeability across multiple cycles of chemical challenges. Thus, the TAM group is critical to achieve the chemically-induced actuation of the polymer chains to modulate the membrane transport properties. Close inspection of Figure 4.C-D reveals also that both responsive and control membranes exhibit larger MVTR values after the first reset treatment with Bu4NOH. This is the result of selective hydrolysis of the PEG ester and imide portions of the polymer, yielding carboxylate groups which augment the polymer hydrophilicity. FTIR studies confirmed that the ester groups of monomer 3 and the imide groups of monomer 2 hydrolyze completely in less than 5 mins in the presence of Bu4NOH (Figure S9).
By comparing the water vapor permeabilities of SWNT membranes before and after functionalization, we conclude that the responsive polymer layer provides the largest resistance to water vapor transport despite constituting a tiny fraction of the entire membranes thickness (~30 nm of a 30-50 μm total thickness). Anchoring the responsive polymers on the
membrane surface reduces the membrane breathability by a factor of 2 (7.5) and increases the membrane resistance to water vapor diffusion by 5 (25) fold in the breathable (protective) state.
2.5. CWA protection with responsive SWNT membranes
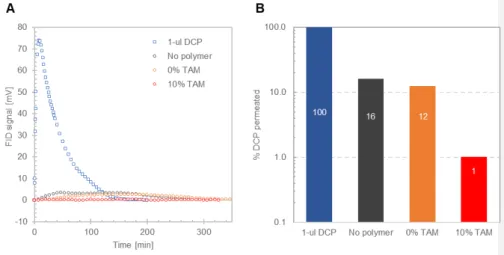
The large resistance to diffusion of a small molecule like water suggests that, in the protective state, the collapse of the polymeric chains could greatly hinder the permeation of larger chemical agents. To validate this hypothesis, we quantified the fraction of nerve agent simulant (DCP) that permeates through functionalized and unfunctionalized SWNT membranes within 5-6 hours. In these tests, a 1 μL DCP droplet was placed on the surface of glass filter paper resting on top of a ~1 cm2 SWNT membrane, and a nitrogen sweep on the opposite side of the membrane was used to carry the permeated simulant to a flame ionization detector (FID). Figures 5.A and B respectively display the raw FID signal and the
corresponding fraction of the 1 μL DCP droplet that permeated across three types of SWNT membranes: a) unfunctionalized; b) functionalized with a control polymer that does not contain the responsive unit (0% TAM) and thus cannot collapse in the presence of DCP; and c) functionalized with the polymer containing 10% TAM (10% TAM). The unfunctionalized SWNT membrane permits the permeation of about 16% of the chemical challenge in a 6 hr run, and the addition of a control polymer layer further reduces the threat passage to 12%. The FID signal recorded for the membrane functionalized with 10% TAM-polymer barely deviates from the baseline, indicating that DCP permeation was restricted to only ~1%. Even if the entire protective suit area is hypothetically exposed to a 1 μL cm-2 dose, this strong permeance attenuation in the protective state would maintain the skin chemical exposure level one order of magnitude below sarin’s LD50.[28] Thus, in CWA-contaminated environments, the autonomous, threat-triggered collapse of the TAM-containing polymer chains
12
dramatically reduces the permeation rate of nerve agent simulants across the membrane, thereby providing adequate levels of protection for several hours.
3. Conclusion
To summarize, we have demonstrated a responsive SWNT membrane system that transitions locally, rapidly, and reversibly from a highly breathable state to a protective activated form upon exposure to nerve agents such as sarin. Response to external chemical stimuli is achieved through CWA-responsive polymers covalently anchored to the membrane surface, which undergo a 20-25% chain collapse triggered by organophosphate agents. This physical change efficiently limits threat-vapor permeation through the SWNT pores to only 1% of a 1 μL cm-2 challenge during a six-hour exposure. In safe environments, the hydrophilicity and thinness of the functional layer as well as the extended configuration of the polymer chains confer high breathability (MVTR > 4,000 gr m-2 day-1) to the functionalized SWNT membranes. These protection and breathability levels compare well with DoD targets for novel protective materials.[17]
Adoption of nm-wide, vertically-aligned SWNTs as moisture conductive pores enabled us to overcome a limiting breathability/protection trade-off predicted by conventional diffusion theories, and simultaneously increase size-sieving selectivity and water-vapor permeability by decreasing SWNT diameters. Integration of these types of responsive materials systems into next-generation protective garments is foreseen to greatly extend the time that first responders, medical, or military personnel can operate in a chemically threatening
environment. Actuating materials like those adopted here offer an intriguing toolbox to create dynamically responsive membranes outperforming state-of-the-art products in other
4. Experimental Section
SWNT growth: Carbon nanotube forests were synthesized from either Fe/Al2O3 or
Fe/Mo/Al2O3 multilayer, thin-film catalysts. Each layer was sequentially deposited onto 100 mm Si (100) wafers by electron-beam evaporation without breaking vacuum (base pressure ≤ 1.6 × 10-6 mbar). Nominal thicknesses of the catalyst layers used in this study (Fe/Al2O3 = 5.5/300 Å or Fe/Mo/Al2O3 = 5.5/0.5/300 Å) were recorded in situ by a quartz crystal monitor during deposition. SWNTs with average diameter > 3.2 nm were grown in an atmospheric-pressure, hot-wall chemical-vapor-deposition (CVD) furnace (Lindberg Blue M Mini-Mite, Thermo Scientific) following the recipe reported in a previous publication.[14] Synthesis of smaller diameter SWNTs was performed at a low-pressure in cold-wall furnace (AIXTRON® Black Magic), featuring a wafer-scale heater stage and gas showerhead. The chamber was pumped down below 0.2 mbar prior to inititating the growth recipe, which began with a thermal annealing step in a reducing environment before introducing the hydrocarbon feedstock growth gas (C2H2). For this study, we utilized two different recipes: 1) ramp with top and bottom heaters at 300 °C min-1 to 750 °C at 3.2 mbar in H2 = 400 sccm and holding for 5 min before switching the gas mixture to C2H2/H2/Ar = 4/0/400 sccm at 1.7 mbar for 30 min; and 2) ramp with top heater at 300 °C min-1 to 700 °C and bottom heater at 300 °C min-1 to 800 °C at 80 mbar in H2/Ar = 700/200 sccm and holding for 2 min at 800 °C before switching the gas mixture to C2H2/H2/Ar = 4/700/380 sccm at 80 mbar for 13 min (with addition of 20 sccm Ar through a bubbler containing H2O, resulting in ~170 ppmv). These low-pressure recipes produced forests with 30-50 µm thickness and number density > 1012 cm-2.
SWNT characterization: High-resolution transmission electron microscopy (HRTEM) was
14
number N ≥ 65 (image processing performed using custom MATLAB script). We used a JEOL 2100-F field-emission analytical TEM, operating at 120 kV or 200 kV, ≥ 150 kx magnification, with a pixel resolution of ≤ 0.05 nm. We prepared samples by dispersing CNT forests in ethanol with ultrasonication and subsequently dropcasting the dispersion onto Cu TEM grids coated with Formvar. Scanning electron microscopy (SEM) was performed with a Zeiss Gemini Ultra-55 analytical field emission SEM, operated at 5 kV accelerating voltage. The mean number density ρn (cm-2) was quantified from the mass increase of the Si substrate after CNT growth and from the forest height with the equation[29]
𝜌!= "!
$%/''(" , (1)
where ρm is the CNT volumetric mass density, d is the mean SWNT diameter measured by TEM, and SSAG is the specific surface area of graphene, 1315 m2 g-1.
Micro-Raman spectroscopy of CNT forests was performed using a Renishaw InVia Qontor Raman spectrometer with excitation wavelength λ = 633 nm, a grating of 1200 lines/mm, and a 50× long working-distance objective. Final spectra used for analysis were generated by averaging over five 5-s collections. A representative spectrum for each average forest diameter shown in Figure 2 is reported in Figure S2.
Membrane fabrication: The CNT-parylene membranes were fabricated according to a
previously reported process[14] with minor modifications. To form the CNT-parylene composite, the inter-tube spacing of the CNT arrays on Si wafers was infiltrated with parylene-N with a target coating thickness of 12000 Å. The excess polymer on top of the composite wafer was then removed by reactive ion etching using a PETS etcher with CF4/O2 flow rates of 40/10 sccm, a Rf power of 150 W and a total etching time between 10.5 and 13.5 minutes. The etched composite wafers were then immersed in 37% hydrochloric acid (Sigma-Aldrich, product # 320331) for 5 to 24 hr to dissolve the alumina and catalyst layer between
the CNTs and the Si-wafers and delaminate the CNT-parylene composite. The free-standing composites were cut into smaller coupons and glued to 0.005-inch-thick Kapton-HN frames (Hardman Double/Bubble epoxy, product # 04007). Lastly, the framed composite was etched in a Harrick PDC-001 plasma cleaner with a 30 W Rf power to open the pores until the target N2 permeance (1-2.5 × 10-5 mol m-2 s-1 pa-1) was obtained.
Synthesis of responsive polymer: All air- and water-sensitive syntheses were performed in
flame-dried flasks under an inert atmosphere with dry nitrogen using standard Schlenk techniques. Diethyl ether, tetrahydrofuran, dichloromethane, and toluene were taken from an Innovative Technologies solvent purification system containing activated alumina columns and stored under argon over 3 Å or 4 Å molecular sieves. All chemicals were purchased from Acros, Aldrich, Fluka, VWR, Aplichem or Merck and used as such unless stated otherwise. Reaction progress was monitored by thin layer chromatography (TLC) (Merck silica gel 60 F254 plates), and column chromatography was performed using silica gel (60 Å pore size, 230–500 mesh, Aldrich). 1H and 13C NMR spectra were recorded at 400 MHz (100 MHz) using Bruker AVANCE-400 NMR spectrometers. Chemical shifts are reported in ppm and referenced to residual NMR solvent peaks (CDCl3: δ 7.26 for 1H and δ 77.2 for 13C). High resolution mass spectra were determined with a Bruker Daltonics APEXIV 4.7 Tesla FT– ICR–MS using ESI or DART ionization. Gel permeation chromatography (GPC)
measurements were performed in tetrahydrofuran using an Agilent 1260 Infinity system and calibrated with a polystyrene standard. Diethyl chlorophosphate was purchased from Sigma-Aldrich and filtered over K2CO3 before being used, thereby removing any acid impurities that might have formed during storage.
Preparation of reagents: Monomers (4-(Bicyclo[2.2.1]hept-5-en-2-yl)phenyl)bis(4-(dimethylamino)phenyl)methanol (1), Di(2,5,8,11-tetraoxatridecan-13-yl)
7-Deleted: ( Deleted: )
16
oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate (2), 2-Hexadecyl-3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione (3a), the corresponding octyl analog (3b), 1-Bromo-4-(phenylethynyl)benzene (4) and 2,2-Dimethyl-4,7-dihydro-1,3-dioxepine (5) were synthesized according to literature procedures.[15, 30]
Representative polymer synthesis protocol: Soluble copolymers and gels for live agent testing were prepared following similar procedures. A representative example is given here for gel TAM/C8H17/PEG= 5/30/65. 0.01 g (0.0228 mmol, 5 eq.) of component 1, 0.188 g (0.335 mol, 65 eq.) of 2, and 0.044 g (0.159 mmol, 30 eq.) of 3b were placed in a 20 mL vial containing no solvent and flushed with nitrogen. 0.5 mL of Grubbs thirdgeneration catalyst stock solution (C = 1.0 mg mL-1, catalyst/monomer ratio = 1:1034) was then injected into the vial, and the reaction was allowed to proceed for 4 hr under stirring before adding the quenching agent ethyl vinyl ether. Obtained polymer gels were washed with DCM (~ 5 mL) to remove soluble monomers and oligomers. DCM-soluble copolymers were synthesized using a
catalyst/monomer ratio = 1:272, precipitated by dropwise addition of the solution to vigorously stirred hexane, and recovered by filtration.
Membrane functionalization procedure (MFP)
Step 1: Norbornene substitution: A CNT/PaN membrane was immersed in a solution of EDC-HCl (120 mg), catalytic DMAP (7 mg), and norbornene methyl amine (80 μL) in water (3.5 mL). After sitting in a covered beaker overnight, the membrane was removed from the solution, and washed with water (×3) and ethanol (×1). The membrane was air dried and used in the next step.
Step 2: Ruthenium substitution: Grubbs second generation catalyst (42 mg) was solubilized in THF (3.3 mL, 17 mM solution). The norbornene-substituted membrane was added to this solution and was allowed to react for 20 minutes before it was removed and washed with THF (×3). The membrane was then air dried.
Step 3: Monomer treatment: Next the Ru-substituted membrane was added to a solution of monomers in N2-degassed EtOH (21 mM for monomers 2 and 3a, the concentration of monomer 1 was adjusted according to different polymer composition) to initiate the polymerization reaction. After 30 min, the membrane was transferred to a solution of ethyl vinyl ether in EtOH and allowed to sit for 2 min. Finally, the membrane was washed with EtOH (×3) and dried in air.
Determination of grafted polymer density: The Ru content in the ethyl vinyl bath from the last
functionalization step was used as a measure of the number of polymer chains grown from the membrane surface, and was quantify by Inductively Coupled Plasma Mass Spectrometry (ICP-MS, Agilent 7900) using indium as internal standard (added at 2 μg L-1 to all solutions). After performing the membrane functionalization procedure (MFP) on a CNT membrane supported on a Si chip (surface area of ~ 1.7 cm2), the ethyl vinyl ether solution was collected,
18
transfered to a plastic centrifugal tube, and dried. 2 mL of 65% ultrapure HNO3 (Sigma-Aldrich) was added to the residue in order to convert organic ruthenium into ruthenium salt (Scheme S1). The mixture was kept at room temperature overnight. 0.2 mL of this mixture was diluted to 5 mL, and 0.4 mL aliquotes of this solution were then used to prepare the ICP-MS samples. Ruthenium ICP standards (Certipur®, RuCl₃ in 7% HCl, 1000 mg L-1 Ru) with concentrations of 10 nM, 100 nM, 1 μM, 10 μM, and 100 μM were used for calibration. The obtained Ru density and, hence, the density of the living polymer chains was 0.21 nM/cm2, which corresponds to 1.25 molecule nm-2.
Determination of polymer molecule weight: Following a modified version of the membrane
functionalization procedure MFP, responsive polymers were grafted from the membrane surface via an acid-labile linkage. After MFP step 2, a CNT membrane was placed into a bath of molecule 5 (10 nM in EtOH) for 1 minute, removed from the bath and washed with EtOH. Then, the membrane was treated according to step 3 of the MFP and kept in an HCl bath (1.0 M in MeOH) overnight to cleave the polymer chains from the membrane surface (Scheme S2). After concentrating the polymer solution, the resulting residue was dissolved in 0.5 mL of DMF and analyzed by GPC (Watty Technology) using a dynamic light scattering detector and DMF as the eluant. The polymer molecular weight was calculated from a calibration curve based on external polystyrene standards (Easical PS-2, Agilent). The recorded retention time of the cleaved polymer sample at the peak maximum was 12.886 min, corresponding to a molecular weight of 47.5 KDa.
Grafted polymer layer thickness: The thickness of the responsive polymer layer was measured
in air on a model, functionalized silicon wafer surface using a profilometer with a 12.5 µm radius diamond tip stylus (Dektak 6 M, VEECO; line profiles: 1500 µm scan length, 50 µm s -1 scan speed, 0.3 mg tracking force). To create the step feature necessary to quantify the
thickness changes on a uniformly functionalized silicon chip, the polymer layer was patterned into a grid by plasma etching (ULVAC NE-550EXa etcher; antenna power = 125 W, bias power = 125 W, chiller temperature = 0 ºC, pressure = 0.5 Pa, O2 flow rate = 99 sccm) through a shadow mask. Representative line profiles across 50 µm wide polymer gridlines were used to measure the height of the responsive polymer brush and detect subsequent changes at the same location after exposure to TFA. Reported heights are averages of three or more closely located line profiles. Sample preparation and measurement conditions are described in detail in the Supporting Information.
Membrane transport measurements: Fluid transport measurements followed previously
reported protocolswith minor modifications.[14]
Gas transport measurement: N2 permeance of SWNT membranes was measured with a mass flow meter located downstream of a dead-end filtration cell. Typically, the mass flow rate was recorded at 5 pressure points up to 4 psi. N2 permeance measurements were used to gauge the degree of membrane pore opening and as a screening tool to qualitatively identify the presence of large defects. Since transport through a few nanometer wide SWNT pores is expected to be in the Knudsen regime and thus independent of the applied feed pressures, only membranes with N2 permeance independent of pressure were used in this study. Water permeance and selectivity measurement: To further test for the selectivity properties of our membranes, we measured the rejection coefficient for a negatively charged dye, Direct Blue 71 (DB71, Sigma Aldrich, product # 212407), with size of 3 × 1.5 × 1-nm. 10 µM DB71 aqueous feed solution was pressurized at 2 psi with a controlled in-house N2 line in a dead-end filtration cell. Water permeance was quantified from the reduction of feed solution volume or from the increase of the permeate weight in a collection vial over time. After filtration, the solution permeated through the membrane was analyzed by UV-vis
20
spectroscopy (Varian Cary 100 UV-Vis Spectrophotometer) to determine the analyte concentration and then calculate the rejection coefficient (R) using the equation: 𝑅 = 1 −(#
($ (2)
where, AP and AF are the absorbance of the permeate and feed solution, respectively, at the peak wavelength l = 587 nm. Only membranes with a R above 99% were considered defect-free and used in this study.[14]
Moisture vapor transport rate: SWNT membranes were housed in a cross-flow Dynamic Moisture Permeation Cell[31] and exposed on both sides to 1000-sccm (Q) N2 gas streams with controlled RH. During a typical test, a 50% RH difference was maintained across the membrane, the pressure gradient (monitored by an Omega PX409-001DWUI differential pressure transmitters) was set to zero with a back-pressure regulator, and the temperature was maintained at 30˚C by enclosing the entire system in a thermostated box. RH of the gas stream entering the high-humidity cell side was monitored with an Omega RH-USB humidity sensor, while RHs of the gas stream flowing through the low-humidity side were monitored by two Vaisala HM70 RH sensors before and after the cell. Once the desired flowrates and relative humidity were established, the system was left to stabilize for > 30 min before recording the water concentration readings from the two Vaisala RH sensors. The difference between these two water concentrations (∆C) was used to calculate the mass flow rate (m) of water vapor diffusing across the membrane, 𝑚 = 𝑄 × ∆𝐶. Measurements were performed at two mean RH (30% and 40%), which is defined as the RH average of the two incoming gas streams.
DCP permeation: To quantify the permeation of diethyl chlorophosphate (CAS [814-49-3]) through SWNT membranes, we adopted a previously described method[31] summarized below for completeness. The membrane was placed into a diffusion cell having a gas inlet and outlet
in the base semicell. On top of the SWNT membrane, glass filter paper supported by a metal mesh was used to wick and spread evenly a 1-μL droplet of the DCP challenge, and ensure that the membrane surface is uniformly exposed to DCP vapor. After dosing the DCP droplet with a Hamilton syringe, the top semicell was sealed with a rubber cap covered with plastic wrap to prevent absorption of analyte, so that the stagnant headspace above the sample surface became saturated with the evaporated simulant. A high purity dry N2 sweep (110 cm3/min) below the sample was then used to carry any permeated DCP to a flame ionization detector (SRI Instruments, Torrance, CA, USA). Before each experiment, the FID signal was recorded in the absence of any chemical until a stable baseline was achieved. Data were collected at 1-point/s using PeakSimple software (SRI Instruments) and baseline corrected before quantifying the permeated amount over the duration of the experiment (Figure 5).
Raman spectroscopy of responsive polymers: Micro-Raman spectroscopy of the responsive
materials was performed in situ using a Renishaw InVia Qontor Raman spectrometer with excitation wavelength λ = 785 nm, a grating of 1200 lines mm-1, and a 50× long working-distance objective. Final spectra for static tests were generated by averaging over five 10-s acquisitions at 1, 5, or 100% power, whereas time-resolved spectra were continuously collected over the duration of the experiment with 4-s acquisitions at 10% power. We applied an 11th-order polynomial fit to subtract the resultant baseline from all spectra. The materials were encapsulated in quartz capillary tubes that had 1.5 mm diameter and 0.01 mm wall thickness (Charles Supper, Part# 15-OQZ), which were sealed and connected to a custom-built gas manifold for selectively delivering acidic or basic vapors with a pure N2 carrier gas feed at ~100 sccm. The laser was focused on a side of the capillary tube, perpendicular to its long axis, such that we probed the inner surface of the capillary wall where the responsive materials were fixed.
22
Live agent testing of responsive polymers: Sarin (GB) employed in the following experiments
was synthesized and vacuum distilled at Lawrence Livermore National Laboratory (LLNL) and has a purity of >98% as measured by 1H and 31P NMR in a Bruker 600 MHz Avance Spectrometer.
Safety note: GB, as well as other nerve agents, is a highly toxic, schedule 1 classified-compound that can cause severe harm to exposed individuals at extremely low doses. Only properly trained personnel, in a certified laboratory possessing the adequate equipment to carry out their synthesis and subsequent purification, should handle these highly toxic chemical warfare agents. The Forensic Science Center at LLNL has the authority and capability to synthesize and handle dilute and neat quantities of GB through its accreditation as a United States laboratory for the Organization for the Prohibition of Chemical Weapons (OPCW). Proper protective personal equipment (PPE) should be worn at all times which include lab coats, safety glasses, butyl-based gloves with nitrile gloves underneath to provide further protection and a face shield. All the experiments described herein were performed on a well-ventilated chemical surety fume hood.
Polymer solutions in CHCl3: The composition of the copolymer tested in CHCl3 solution was TAM/C16H33/PEG = 5/30/65. Solution volume employed in these experiments was 225 µL in a glass autosampler vial. The colorless solutions were treated with 25 μL of a 100 mM GB solution in CHCl3 at ambient temperature (RT). Each experiment was done in duplicate. The first attempt was to record still pictures of the color change of the polymer solutions, while during the second attempt we recorded a real-time video. After activation with GB, the polymer solutions were regenerated in a wide mouth jar by opening and exposing their vials to NH4OH vapors coming from two open 20 mL scintillation vials. For all polymer solutions, the time for their complete regeneration was ~10 min at RT.
Polymer films: To form thin films, the random copolymer TAM/C16H33/PEG = 5/30/65 was drop-casted from a CHCl3 solution onto glass slides. 20 μL of GB was introduced in a glass autosampler vial via pipette. The vial was then inserted in a wide mouth jar together with the polymer film, the lid was capped tightly, and the film color change was monitored from colorless to dark green. After use, the polymer films were regenerated by contact with NH4OH vapor from two open 20 mL scintillation vials, all enclosed in a wide mouth jar. As in the case of the polymer solutions, the time for the films’ complete regeneration was ~10 min at RT.
Polymer gels: A piece of a high-molecular-weight copolymers gel (TAM/C8H17/PEG = 5/30/65) swollen in dichloromethane (DCM) was placed on top of a small ruler to measure volume changes. The gel sample on the ruler was then sealed inside a wide mouth jar hosting an autosampler vial containing 20 µL of neat liquid GB. After tightly sealing the jar, the change in color and morphology of the gel piece was monitored over 16 min at RT. After this time, the gel along with the ruler were removed from the jar and any changes in the polymer size were noted. Lastly, regeneration of the gel pieces was accomplished following the same procedure used for the polymer films and the solutions.
Two videos associated with the experiments described above for polymer solutions (Movie S1) and gels (Movies S2) are included in the Supporting Information.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Acknowledgements
This work was enabled by financial support from the Chemical and Biological Technologies Department of the Defense Threat Reduction Agency (DTRA-CB) via grant BA12PHM123 in
24
the “Dynamic Multifunctional Materials for a Second Skin D[MS]2” program. Work at LLNL was performed under the auspices of the US Department of Energy under contract DE-AC52-07NA27344. A portion of this work was performed at the Molecular Foundry, which was supported by the Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under contract DE-AC02-05CH11231.
Received: ((will be filled in by the editorial staff)) Revised: ((will be filled in by the editorial staff)) Published online: ((will be filled in by the editorial staff))
References
[1] M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov, S. Minko, Nature Materials 2010, 9, 101.
[2] S. Mura, J. Nicolas, P. Couvreur, Nature Materials 2013, 12, 991.
[3] a) E. Aznar, M. Oroval, L. Pascual, J. R. Murguía, R. Martínez-Máñez, F. Sancenón,
Chemical Reviews 2016, 116, 561; b) J. Liu, N. Wang, L.-J. Yu, A. Karton, W. Li, W. Zhang,
F. Guo, L. Hou, Q. Cheng, L. Jiang, D. A. Weitz, Y. Zhao, Nature Communications 2017, 8, 2011.
[4] S. Pan, R. Guo, M. Björnmalm, J. J. Richardson, L. Li, C. Peng, N. Bertleff-Zieschang, W. Xu, J. Jiang, F. Caruso, Nature Materials 2018.
[5] T. J. White, M. E. McConney, T. J. Bunning, J. Mater. Chem. 2010, 20, 9832. [6] X. Chen, L. Mahadevan, A. Driks, O. Sahin, Nat. Nanotechnol. 2014, 9, 137. [7] Z. J. Yang, J. J. Wei, Y. I. Sobolev, B. A. Grzybowski, Nature 2018, 553, 313. [8] X. Hou, W. Guo, L. Jiang, Chem. Soc. Rev. 2011, 40, 2385.
[9] L. Ionov, Adv. Funct. Mater. 2013, 23, 4555.
[10] A. K. Yetisen, H. Butt, L. R. Volpatti, I. Pavlichenko, M. Humar, S. J. J. Kwok, H. Koo, K. S. Kim, I. Naydenova, A. Khademhosseini, S. K. Hahn, S. H. Yun, Biotechnology
Advances 2016, 34, 250.
[11] a) R. Spitz Steinberg, M. Cruz, N. G. A. Mahfouz, Y. Qiu, R. H. Hurt, ACS Nano
2017, 11, 5670; b) B. D. Martin, G. A. Justin, M. H. Moore, J. Naciri, T. Mazure, B. J. Melde,
R. M. Stroud, B. Ratna, Adv. Funct. Mater. 2012, 22, 3116; c) J. L. Poole, S. Donahue, D. Wilson, Y. M. Li, Q. Zhang, Y. Gu, R. Ferebee, Z. Lu, R. M. Dorin, L. F. Hancock, L. Takiff, I. F. Hakem, M. R. Bockstaller, U. Wiesner, J. Walker, Macromolecular Rapid
Communications 2017, 38, 1700364; d) A. Gugliuzza, E. Drioli, J. Membr. Sci. 2013, 446,
26
[12] a) S. A. Brewer, Recent Patents on Materials Science 2011, 4, 1; b) M. A. Wartell, M. T. Kleinman, B. M. Huey, L. M. Duffy, Strategies to Protect the Health of Deployed U.S.
Forces: Force Protection and Decontamination, National Academy Press, Washington, D.C.
1999; c) G. R. Lomax, J. Mater. Chem. 2007, 17, 2775.
[13] M. Cacas, in Signal, AFCEA International, 2013.
[14] N. Bui, E. R. Meshot, S. Kim, J. Peña, P. W. Gibson, K. J. Wu, F. Fornasiero, Adv.
Mater. 2016, 28, 5871.
[15] C. Belger, J. G. Weis, E. Egap, T. M. Swager, Macromolecules 2015, 48, 7990. [16] H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech, B. D. Freeman, Science 2017,
356.
[17] https://arpa-e.energy.gov/sites/default/files/12_Whitfield - DMMSS Overview - DELTA Kick-off.pdf 2015
[18] a) M. Majumder, N. Chopra, R. Andrews, B. J. Hinds, Nature 2005, 438, 44; b) J. K. Holt, H. G. Park, Y. M. Wang, M. Stadermann, A. B. Artyukhin, C. P. Grigoropoulos, A. Noy, O. Bakajin, Science 2006, 312, 1034.
[19] a) G. Hummer, J. C. Rasaiah, J. P. Noworyta, Nature 2001, 414, 188; b) A. Kalra, S. Garde, G. Hummer, Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 10175; c) E. Secchi, S. Marbach, A. Nigues, D. Stein, A. Siria, L. Bocquet, Nature 2016, 537, 210.
[20] M. Majumder, N. Chopra, B. J. Hinds, ACS Nano 2011, 5, 3867.
[21] H. Schreuder-Gibson, P. Gibson, K. Senecal, M. Sennett, J. Walker, W. Yeomans, D. Ziegler, P. P. Tsai, Journal of Advanced Materials 2002, 34, 44.
[22] a) J. A. Thomas, A. J. H. McGaughey, Phys. Rev. Lett. 2009, 102; b) J. A. Thomas, A. J. H. McGaughey, Nano Lett. 2008, 8, 2788.
[23] S.-C. Sha, R. Zhu, M. B. Herbert, J. A. Kalow, T. M. Swager, Journal of Polymer
Science Part A: Polymer Chemistry 2017, n/a.
[25] L. Angeloni, G. Smulevich, M. P. Marzocchi, J. Raman Spectrosc. 1979, 8, 305. [26] M. Majumder, N. Chopra, B. J. Hinds, J. Am. Chem. Soc. 2005, 127, 9062. [27] a) N. Y. Kim, N. L. Jeon, I. S. Choi, S. Takami, Y. Harada, K. R. Finnie, G. S. Girolami, R. G. Nuzzo, G. M. Whitesides, P. E. Laibinis, Macromolecules 2000, 33, 2793; b) I. Njoroge, P. A. Kempler, X. Deng, S. T. Arnold, G. K. Jennings, Langmuir 2017, 33, 13903. [28] F. Worek, J. Jenner, H. Thiermann, in Chemical Warfare Toxicology, Volume 1 -
Fundamental Aspects, Royal Society of Chemistry, 2016, 81.
[29] S. Esconjauregui, R. S. Xie, M. Fouquet, R. Cartwright, D. Hardeman, J. W. Yang, J. Robertson, J. Appl. Phys. 2013, 113.
[30] a) P. Li, L. Wang, M. Wang, F. You, European Journal of Organic Chemistry 2008,
2008, 5946; b) J. A. Kalow, A. G. Doyle, J. Am. Chem. Soc. 2010, 132, 3268.
[31] P. W. Gibson, H. L. Schreuder-Gibson, Journal of Engineered Fibers and Fabrics
2009, 4, 11.
[32] G. F. Zhong, J. H. Warner, M. Fouquet, A. W. Robertson, B. A. Chen, J. Robertson,
28
Figure 1. A CNT membrane functionalized with copolymers responsive to nerve agents.
(A) Example of tri-layer laminate mimicking a protective military garment and consisting of a nylon/cotton outer-shell fabric with a camouflage pattern, an intermediate protective CNT membrane layer, and a cotton comfort liner. (B) Schematic representation of the membrane response mechanism to environmental chemical stimuli, in which the collapse of actuating polymer chains grafted on the membrane surface prevents nerve agents like sarin from entering the SWNT membrane pores. In a safe environment, the responsive polymer chains remain extended and allow rapid transport of water vapor. (C) Surface-initiated ROMP
functionalization scheme adopted to covalently link responsive copolymers to the carboxylic groups on the membrane surface and SWNT openings. The responsive copolymers consist of three monomers: 1 an organophosphate responsive unit based on triarylmethanol (TAM) derivatives; 2 a hydrophobic monomer containing long alkyl chains (CxHy); and 3 a hydrophilic pegylated hydrogel unit (PEG).
30
Figure 2. Unfunctionalized SWNT membranes. (A) Area number densities and average
nanotube diameters, dCNT, of the SWNT forests employed in membrane fabrication, and comparison with the theoretical limit from close-packing (solid line) calculations[32]. (B) SEM image of the cross-section of a parylene-N/SWNT composite before surface plasma etching. Inset: higher magnification highlighting the conformal parylene-N coating on the nanotubes. (C) Moisture vapor transport rate (MVTR, blue points) and corresponding SWNT-membrane resistance (red points) to water vapor diffusion as a function of the average CNT pore diameter. Lines are to guide the eye. (D) Dependence of single-pore water-vapor
permeability on pore diameter. Experimental results for several SWNT diameters (red points) were obtained at 30 °C with average RH = 30% and an incoming gas stream humidity difference of 50%. The pore size dependences predicted by bulk, transition, and Knudsen diffusion equations are represented by the black lines. Inset: Enhancement factor (defined as the ratio of measured to predicted permeability) for water vapor diffusion through the SWNT diameters considered in this study. All error bars are standard deviations across several membranes with the same average nanotube diameter (total number of membranes = 31).
32
Figure 3. Spectroscopic, colorimetric, and volumetric changes induced on the responsive copolymers by chemical stimuli. (A) Raman spectra (λ = 785 nm, 5×10 s acquisitions)
collected at three different vapor conditions for a responsive copolymer with percent composition TAM/C16H33 /PEG/alkyne = 9.5/42.75/42.75/5. Raman intensity on the y-axis is normalized to that of the alkyne internal standard at 2219 cm-1. The gray/green/red line shows the characteristic peaks of the alcohol form of the TAM unit (I)/ trityl cation (II)/ protonated
trityl cation (III) under NH4OH/ acetic acid/ TFA vapor flow collected with a 1/100/5% laser power, respectively. Inset: space filling models of the 3D structure of the TAM alcohol and of the more planar trityl cation. (B) Photographs showing the color of the responsive copolymer inside a quartz capillary flushed with NH4OH (top: colorless state I), acetic acid (middle: green state II), and TFA vapors (bottom: red state III). (C) Polymer response kinetics monitored by in-situ micro-Raman spectroscopy. A thin layer of the copolymer inside a glass capillary was exposed to the following sequence of gases: NH4OH (10 min), TFA (2 min), N2 (30 min), and NH4OH (10 min). Time interval for exposure to each gas stream is bracketed by the vertical dashed lines. The Raman peak intensities at 1370 cm-1 and 1508 cm-1 (λ = 785 nm, 10% power, 4 s collection time) were normalized to that of the alkyne internal standard at 2219 cm-1. The black line represents a fit of the 1370 cm-1 peak intensity rise after
introduction of the N2 stream with the function y = y0 + A×exp (-t/t). (D) Photographs of a 2 mg/mL chloroform solution of polymer TAM/C16H33/PEG = 5/30/65 as prepared (right), 1 min after spiking with 25 μL of 100 mM sarin solution (left), and (E) after 10 min
regeneration with NH4OH vapors. (F) Photographs of a polymer thin film (TAM/C16H33/PEG = 5/30/65) after 5 min exposure to sarin vapors and (G) after 10 min regeneration with NH4OH vapors. (H) Photographs of a DCM-swollen gel (TAM/C8H17/PEG = 5/30/65) as prepared, (I) 15 min and (J) 30 min after spiking with 20 μL of neat sarin, and (K) after 10 min regeneration with NH4OH vapors.
34
Figure 4. Chemically-triggered collapse and extension of the responsive polymer chains and corresponding MVTR modulation when grafted on a SWNT membrane surface. (A)
Optical image of the responsive polymer layer (blue) grafted from a Si/SiO2 surface (pink) and patterned by plasma etching through a shadow mask. Inset: magnification of the pattern edge showing a line scan across a 50 μm wide grid line. (B) Profilometry measurements of the thickness of a responsive polymer layer (TAM/C16H33/PEG = 20/40/40) before and after exposure to TFA vapor (1 min). For the profilometry experiments, the responsive polymer
was grown from a model Si/SiO2 surface primed with norbornenyl trichlorosilane using a functionalization method analogous to that displayed in Figure 1.C (see Figure S8 for details). (C) Water vapor transport rate across a SWNT membrane functionalized with the responsive polymer TAM/C16H33/PEG = 20/40/40, and (D) with a TAM-free control polymer
(TAM/C16H33/PEG = 0/50/50). Breathability data were collected with membranes having SWNT average diameter < 2.2 nm at 30 °C under a transmembrane RH gradient of 50% (RH = 65% on one side and 15% on the opposite side of the membrane). In both panels C and D, the blue bar represents the functionalized membrane before chemical exposure, the dark gray bars are data collected after TFA (2 min) exposure, and the red bars are for membranes that were regenerated in a 2 M Bu4NOH aqueous solution (2 hr).
36
Figure 5. DCP permeation through SWNT membranes. (A) Time evolution of the raw
signal at the flame ionization detector (FID), which is proportional to the instantaneous DCP vapor concentration permeating through the membrane. (B) Percent of a 1 μL DCP challenge permeated through SWNT membranes in ~6 hr, calculated by integrating the area under the FID signal over time. In both panels A and B, the color blue indicates data for permeation experiments conducted without membranes in the diffusion cell; dark gray is for permeation through an unfunctionalized SWNT membrane (average dCNT = 2.57 nm); orange is for a SWNT membrane (dCNT = 3.16 nm) functionalized with a polymer lacking the responsive unit (TAM/C16H33/PEG = 0/50/50); and red is for a SWNT membrane (dCNT = 1.8 nm)
functionalized with a responsive polymer (TAM/C16H33/PEG = 10/45/45). All experiments were conducted at room temperature.
Table of contents entry
A bistable membrane is demonstrated that rapidly and reversibly transitions from a highly
breathable state to a chemically protective state when exposed to organophosphates. The threat-triggered physical collapse of an ultrathin copolymer layer on the membrane surface efficiently gates transport through membrane pores made of single-walled carbon nanotubes (SWNT). SWNT ultrafast moisture conduction enables overcoming the
breathability/protection trade-off that limits conventional membrane materials.
Keyword: Stimuli-Responsive Materials
Yifan Li†, Chiatai Chen†, Eric R. Meshot, Steven F. Buchsbaum, Myles Herbert, Rong Zhu, Oleg Kulikov, Ben McDonald, Ngoc Bui, Melinda L. Jue, Sei Jin Park, Carlos A. Valdez, Saphon Hok, Qilin He, Christopher J. Doona, Kuang Jen Wu, Timothy M. Swager*, Francesco Fornasiero*
![Figure 2. Unfunctionalized SWNT membranes. (A) Area number densities and average nanotube diameters, d CNT , of the SWNT forests employed in membrane fabrication, and comparison with the theoretical limit from close-packing (solid line) calculations [32]](https://thumb-eu.123doks.com/thumbv2/123doknet/14137713.469926/31.892.86.592.257.736/unfunctionalized-membranes-densities-diameters-fabrication-comparison-theoretical-calculations.webp)