Identification of Microsatellite Markers for Plasmopara viticola and Establishment of High throughput Method for SSR Analysis
12
0
0
Texte intégral
Figure


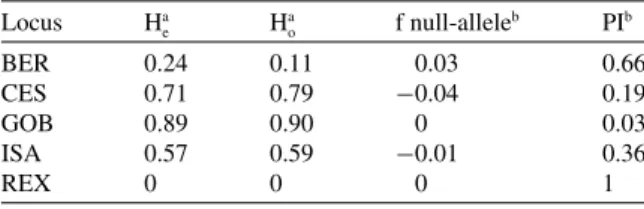
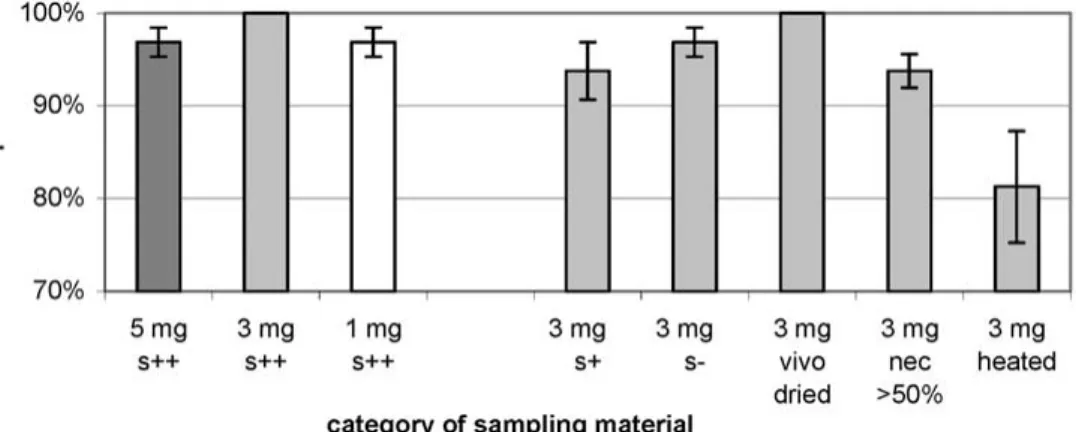
+2
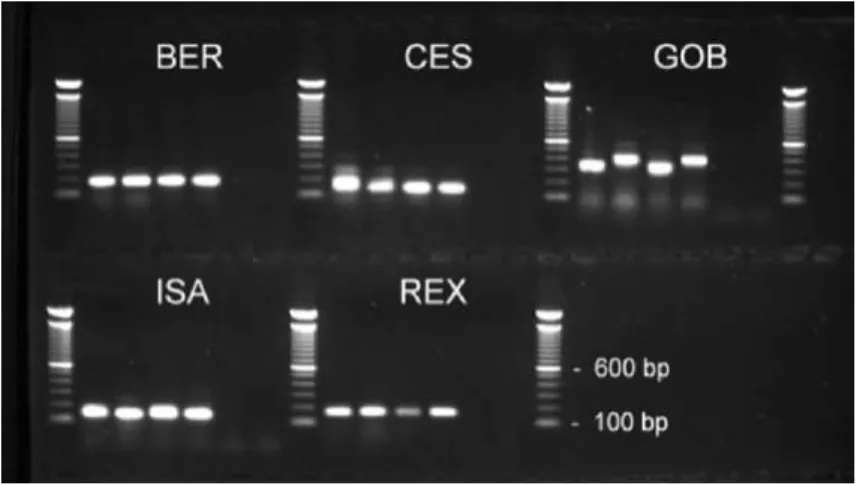
Documents relatifs
