Thermodynamic data provided through the FUNMIG project: Analyses and prospective
Texte intégral
Figure

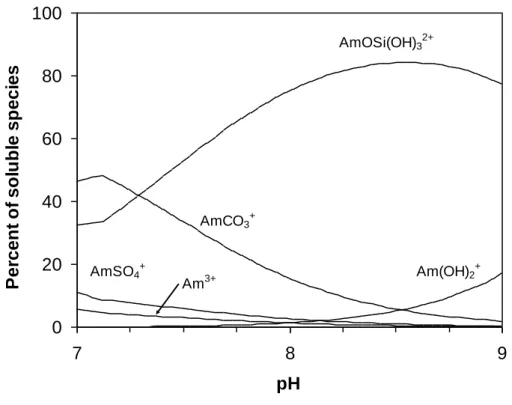
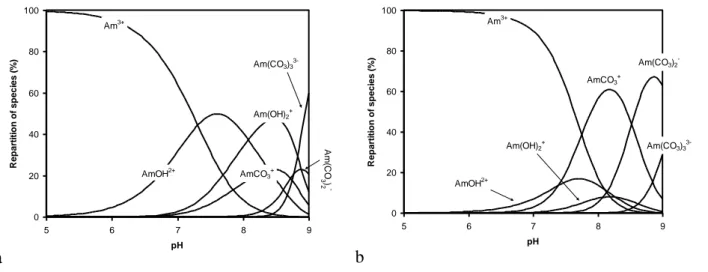
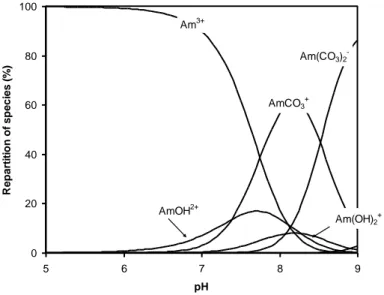
![Figure 4. Solubility of Am(III), [Am], in bicarbonate and carbonate solutions with 4 mol/L NaCl measured (●) in phase 1 (cooling) and (●) in phase 2 (heating) for (a) T = 30°C and (b) T = 70°C](https://thumb-eu.123doks.com/thumbv2/123doknet/13123665.387643/13.892.104.789.191.499/figure-solubility-bicarbonate-carbonate-solutions-measured-cooling-heating.webp)


![Figure 6 Solution speciation of the thorium phosphate system at I = 0.1 mol/L (NaClO 4 ), [PO 4 ] tot = 10 -4 mol/L from the data selected in Rand et al](https://thumb-eu.123doks.com/thumbv2/123doknet/13123665.387643/20.892.99.798.87.411/figure-solution-speciation-thorium-phosphate-naclo-selected-rand.webp)

Documents relatifs
This measurement, combined with a theoretical solubility model, was used to assess the probable order of magnitude of the NH 4 -dawsonite Gibbs free energy value at 25
Abstract Extraction of U(VI), Eu(III) and Am(III) has been performed from acidic aqueous solutions (HNO 3 , HClO 4 ) into the ionic liquid [C 4 mim][Tf 2 N] in which a new
We have measured the optical densities at seven more wavelengths, e.g., 240, 260, 300, 340, 400, 720, and 780 nm, under the same experimental conditions as in the previous
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Novel polydentate phosphonic acids: Protonation and stability constants of complexes with Fe(III) and Cu(II) in aqueous medium... N ovel Polydentate
To offer alternatives to patients while reserving medical resources for the situations that require it, the Greater Paris University Hospitals
[r]
This convergence of different semantic models treating time-related information has recently risen a discussion between their designers about the
