In vitro imaging of primary neural cell culture from Drosophila
8
0
0
Texte intégral
Figure
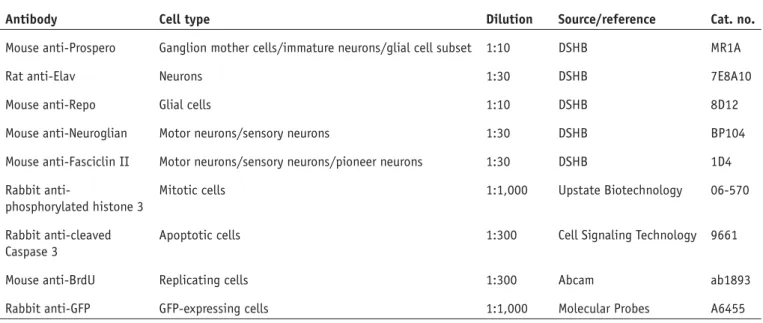
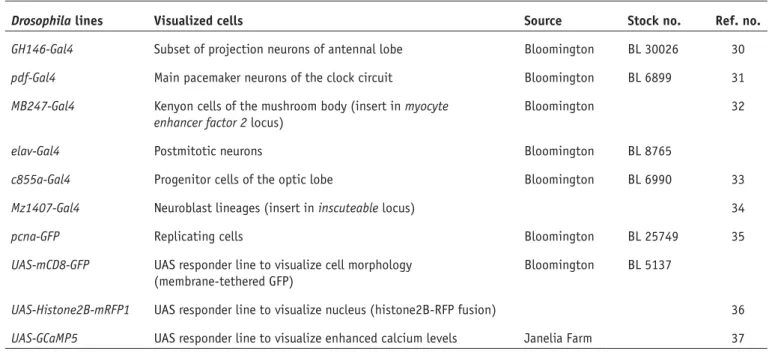

Documents relatifs
Effects of Silver Nanoparticles on Primary Mixed Neural Cell Cultures: Uptake, Oxidative Stress and Acute Calcium Responses.. Andrea Haase,* ,1 Stephanie Rott,† Alexandre
LIF broadly promotes survival in sphere-derived cultures An additional mechanism by which LIF could increase the number of neural and glial cells in spiral ganglion stem
