A M E R I C A N J O U R N A L O F B OTA N Y 103 (7): 1167 – 1174 , 2016; http://www.amjbot.org/ © 2016 Botanical Society of America • 1167 A M E R I C A N J O U R N A L O F B O T A N Y
R E S E A R C H A R T I C L E
UNVEILING GRASS EVOLUTIONARY HISTORY FROM
ANCESTRAL GRASS KARYOTYPES
Th
e fl owering plants (angiosperms) contain the major clades of
eu-dicots (~275,000 species) and monocots (~65,000 species) and the
smaller groups of magnoliids (~2500 species), Chloranthaceae (~50
species), and Ceratophyllum (5 species) ( Soltis et al., 2008 , 2011 ).
Th
e monocots and eudicots, the two largest and most diverse groups,
accounting respectively for 20% and 75% of modern angiosperm
species, are clades of choice to investigate the forces promoting
species evolution through polyploidization (genome doubling) or
whole-genome duplication (WGD) ( Van de Peer et al., 2009a , b ).
Reconstructing ancestral genomes from the comparison of
modern karyotypes, i.e., the fi eld of paleogenomics research, is
based on the identifi cation of duplications (or any shuffl
ing events)
at orthologous positions between modern species, defi ning
proto-chromosomes (i.e., independent groups of syntenies and paralogies
also referenced as contiguous ancestral regions [CARs]). Th
e
de-duced evolutionary history is then based on the smallest number
of shuffling operations (ancestral chromosome fusion, fission,
inversion, and translocation events) able to account for the
transi-tion from the inferred ancestral karyotypes to the modern genomes
( Salse et al., 2009; Salse, 2012 , 2016 ).
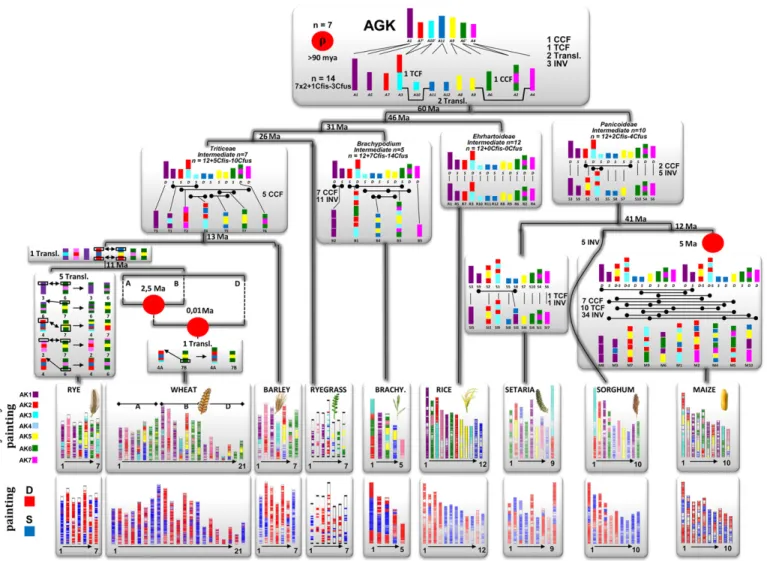
In grasses, for monocots, we inferred an ancestral grass
karyo-type (AGK) structured in seven protochromosomes containing
16,464 protogenes (taking into account gene conservation between
species), with 8581 ordered (taking into account gene adjacency
conservation between species) and with a minimal gene space
physical size of 33 Mb ( Murat et al., 2010 , 2014a , 2014b ). Th
is
an-cestor went through a paleotetraploidization event (consisting of
seven duplicated blocks shared in the modern monocots) dating
back to more than 90 million years ago (Ma) ( Salse et al., 2008 ,
2009 ; Murat et al., 2010 ; Wang et al., 2015 ) and followed by two
symmetric reciprocal translocations, one centromeric (centromeric
chromosome fusion [CCF]) and one telomeric (telomeric
chromo-some fusion [TCF]) and two asymmetric reciprocal translocations
to reach a 12-chromosome intermediate ( Murat et al., 2014a ,
2014b ). A centromeric chromosome fusion (CCF) consists of a
process during which an entire chromosome is inserted (i.e., one
fusion called Cfus in Fig. 1 ) by its telomeres into a break (i.e., one
fi ssion called Cfi s in Fig. 1 ) in the centromeric region of another
chromosome (CCF = 1 fusion + 1 fi ssion and also termed a nested
chromosome fusion NCF; IBI, 2010 ). A telomeric chromosome
fu-sion (TCF) consists of an end-to-end joining of two chromosomes
at their telomeres (CCF = 1 fusion, without fi ssion and also termed
tip-to-tip joining). From the fusion of two ancestral chromosomes,
1 Manuscript received 2 November 2015; revision accepted 1 March 2016.
INRA/UBP UMR 1095 Génétique, Diversité et Ecophysiologie des Céréales, Laboratory of Paleogenomics & Evolution, 5 chemin de Beaulieu 63100 Clermont Ferrand, France
2 E-mail: [email protected]; phone: +33(0)473624380; fax: +33(0)473624453 doi:10.3732/ajb.1500459
I N V I T E D PA P E R
For the Special Issue: The Evolutionary Importance of Polyploidy
Deciphering the evolutionary interplay between
subgenomes following polyploidy: A paleogenomics
approach in grasses
1
Jérôme Salse 2
How did plant species emerge from their most recent common ancestors (MRCAs) 250 million years ago? Modern plant genomes help to address such key questions in unveiling precise species genealogies. The fi eld of paleogenomics is undergoing a paradigm shift for investigating species evolution from the study of ancestral genomes from extinct species to deciphering the evolutionary forces (in terms of duplication, fusion, fi ssion, deletion, and translocation) that drove present-day plant diversity (in terms of chromosome/gene number and genome size). In this review, inferred ancestral karyotype genomes are shown to be powerful tools to (1) unravel the past history of extant species by recovering the variations of ancestral genomic compartments and (2) ac-celerate translational research by facilitating the transfer of genomic information from model systems to species of agronomic interest.
while CCF delivers a monocentric chromosome, TCF delivers a
di-centric chromosome where one centromere is lost to return to
monocentry through pericentric inversion ( Schubert and Lysak,
2011 ).
All investigated grass genomes can then be reconstructed from
this postpolyploidy ancestral karyotype of 12 protochromosomes
( Fig. 1 ) following CCF, TCF, translocation, or inversion events
( Fig. 1 ). Rice has retained the n = 12 AGK structure and has been
proposed as the slowest-evolving species among the grasses ( Wang
et al., 2015 ), whereas the other species experienced numerous
chro-mosome rearrangements to reach the present-day karyotypes
( Murat et al., 2010 , 2014a , b ). Furthermore, it is possible to represent
the chromosome number for any modern grass species by a
karyo-typic equation refl ecting the evolutionary events, such as duplications
(
× 2), fusions (−Y), fi ssions (+Z), that took place during evolution.
For example in Table 1 , the maize karyotypic equation is [12 − 4 + 2]
× 2 + 7 − 17, refl ecting that the monocot ancestral intermediate
with 12 chromosomes went through 4 fusions (i.e., two CCF) and 2
fi ssions to reach a 10-chromosome Panicoideae intermediate,
fol-lowed by one WGD (to form a 20-chromosome intermediate) and
fi nally 7 CCFs and 10 TCFs (consisting of at least 7 fi ssions and
17 fusions) to reach the modern genome structure of 10
chromo-somes. Such ancestral chromosome fusion events (either TCF or
CCF, Fig. 1 ) that reduce the number of chromosomes in the
mod-ern karyotypes from their postpaleopolyploid ancestors are called
asymmetric and symmetric reciprocal translocations in
chromo-somal mutagenesis approaches and cytogenetic studies ( Schubert
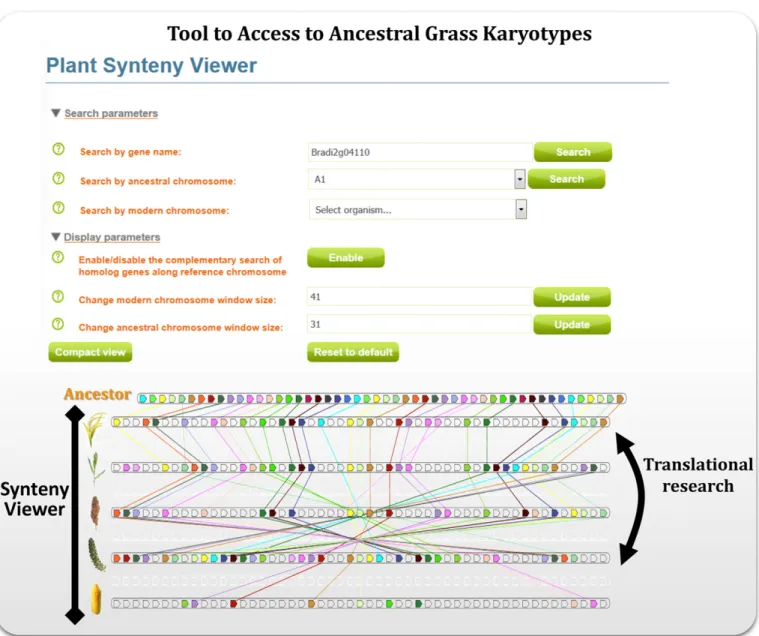
and Lysak, 2011 ). A SyntenyViewer tool ( Fig. 2 ) is available to the
scientifi c community to have access to the inferred AGK to navigate
between grass species. Th
e SyntenyViewer tool delivers a precise
paleogenomics-based repertoire of paralogs/orthologs to perform a
translational research approach in improving (1) complex genome
FIGURE 1 Grass genome evolution from ancestral grass karyotypes (AGKs) (adapted from Murat et al., 2010 , 2014a , b ). Present-day grass genomes (bottom) are represented with color codes to illustrate either (top) the evolution of segments from their founder AGK progenitor with seven proto-chromosomes (referenced as Ax, synteny painting) or (bottom) the mosaic of dominant and sensitive blocks inherited from polyploidization (refer-enced as D in red or S in blue, dominance painting). Whole-genome duplication events that have shaped the structure of the modern grass genomes during their evolution from AGK are indicated as red dots. The geological periods are indicated on the tree branches as millions of years ago (Ma). Evolutionary genome shuffl ing events such as chromosomal fusions, fi ssions, and translocations are abbreviated on the tree branches: CCF, centromeric chromosome fusion; Cfi s, chromosome fi ssion; Cfus, chromosome fusion; INV, inversion; TCF, telomeric chromosome fusion; Transl, translocation.
J U LY 2016 , V O LU M E 103 • S A L S E — PA L E O G E N O M I C S A N D E V O LU T I O N I N G R A S S E S • 1169 T ABLE 1 .
Plant genome data sets used in paleogenomics studies
. Species C ommon name Ref er enc e C hr omosomes G enome f ea tur es a Annota ted genes E v olutionar y f ea tur es b Siz e (Mbp ) / TE (%) K ar y ot ypic equa tion fr om n = 12 A GK c Rounds (R) of WGD O ryza sativ a R ice IR GSP , 2005 12 372 / 39 40,577 12 = 21 + 0 − 0 [0C CF / 0T CF] 1R Br achypodium distachy on Brach ypodium IBI, 2010 5 271 / 28 25,532 5 = 12 + 7 − 14 [7C CF / 0T CF / 11INV ] 1R Hor deum vulgar e Bar ley M ay er et al ., 2011 7 ~5000 / >80 15,719 7 = 12 + 5 − 10 [5C CF/0T CF] 1R Sec ale c er eale R ye M ar tis et al ., 2013 7 7917 / >80 2940 d 7 = 12+ 5 − 10 [5C CF / 0T CF / 6T ransl .] 1R Triticum aestivum Wheat IWGSC, 2014 21 ~17,000 / >80 99,386 21 = (12+ 5 − 10) × 3 [2WGD / 5C CF / 0T CF / 2T ransl .] 3R Lolium per enne R yeg rass Pf eif er et al ., 2013 7 2600 / >80 762 d 7 = 12 + 5 − 10 [5C CF / 0T CF] 1R Sor ghum bic olor Sor ghum Pat erson et al ., 2009 10 659 / 62 34,496 12+ 2 − 4 [2C CF / 0T CF / 5INV ] 1R Setaria italic a M illet Zhang et al ., 2012 9 490 / 46 38,801 12 + 2 − 5 [2C CF / 1T CF / 6INV ] 1R Z ea mays M aiz e Schnable et al ., 2009 10 2365 / 84 32,540 [12 + 2 − 4] × 2 + 7 − 17 [1WGD / 9C CF / 10T CF / 39INV ] 2R a Genome f eatur es ar e r epor ted accor ding t o the inf or mation a
vailable in the initial genome paper (cf
. Ref er ence column). b Ev olutionar y f eatur es ar e r epor ted accor ding t o r
ecent paleogenomics studies (
Murat et al ., 2014a , b ). c The k ar yot ypic equation in volv es whole -genome duplication ( W GD) ( × 2), fi
ssions (+X) and fusions (−Y
), consisting in centr omer ic chr omosome fusions (C CF), t elomer ic chr omosome fusions ( TCF), in versions (INV ), and translocations (T ransl .) r ef er enced within “[ ]” . d M
apped genes or mar
kers
.
assembly, (2) structural and functional gene annotation, or (3)
can-didate gene or marker selection for traits dissection ( Salse, 2013,
2016 ).
UNVEILING GRASS GENOME PLASTICITY FROM ANCESTRAL
GRASS KARYOTYPES
Th
e number of annotated genes observed in the modern
angio-sperm genomes is lower than what can be predicted based on the
reconstructed ancestral gene pool and the number of
polyploidiza-tion events identifi ed, in theory, doubling the number of genes in
the post-WGD karyotypes. As a case example for the grasses that
underwent one (rye, barley, ryegrass, Brachypodium , rice, sorghum,
and setaria), two (maize) or even three (wheat) polyploidization
events, the maize genome should contain 16,464
× 4 = 65,856
mod-ern genes, instead of the current 32,540 annotated genes ( Fig. 1 ),
indicative that the rate of duplicates deletion is just important as
the rate that new genes are born from duplications through WGDs;
these opposite forces have shaped the gene content of modern plant
genomes.
Th
is phenomenon is explained by the observation that
poly-ploidization events are followed by a dipoly-ploidization (or
fraction-ation) process that consists of gene number reduction aft er WGDs
through the removal of the duplicates until the gene content
re-sembles the diploid progenitor genome plus the retained paralogs
( Langham et al., 2004 ; Th
omas et al., 2006 ; Woodhouse et al., 2010 ;
Schnable et al., 2012 ; Murat et al., 2014b ). However, duplicated
gene deletion is not performed at random because it has been
sug-gested that duplicated gene redundancy is eliminated through a
so-called subgenome dominance in which only one of the duplicated
blocks preferentially retains the majority of ancestral copies of the
duplicates. Th
is diploidization phenomenon then leads, at the
whole chromosome or genome levels, to dominant (D, retention of
duplicated genes, also referenced as LF for least fractionated) and
sensitive (S, loss of duplicated genes, also referenced as MF for most
fractionated) subgenomes in paleo- or neopolyploids ( Salse, 2012,
2016 ).
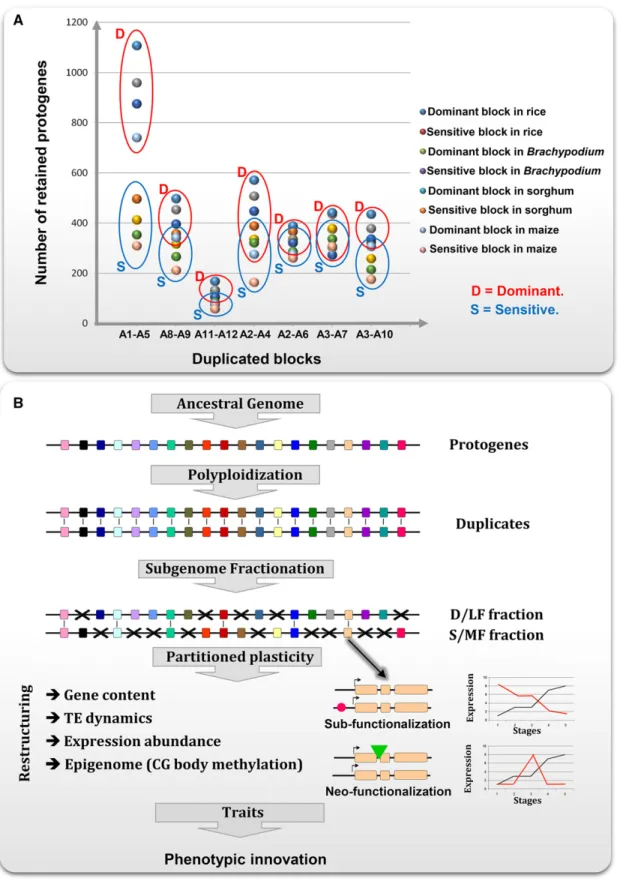
Bias in ancestral gene retention between duplicated blocks
aris-ing from lineage-specifi c polyploidizations has been documented
in Arabidopsis ( Th
omas et al., 2006 ), maize ( Schnable et al., 2011 ),
Brassica ( Cheng et al., 2012 ; Murat et al., 2015 ), and wheat ( Pont
et al., 2013 ). In grasses, this genome fractionation process is clearly
ancestral, as shown in Fig. 3A , in response to shared paleo-WGD
events between ancestral chromosomes A1 and A5, A8 and A9, A2
and A4, A2 and A6, A3 and A7, A3 and A10 ( Schnable et al., 2012 ;
Murat et al., 2014a ). However, an exception to this general
subge-nome dominance pattern has been characterized in cereals where a
highly conserved region has been retained during 50–70 Myr of
evolution between ancestral chromosomes A11–A12 by an active
and recurrent gene conversion phenomenon ( Fig. 3A ) ( Jacquemin
et al., 2011 ; Wang et al., 2011 ).
Investigations of the expressional dynamics of grass duplicates,
deriving form a 90 Myr paleotetraploidization event, suggests that
57.4% ( Yim et al., 2009 ) up to 85% ( Th
roude et al., 2009 ) of the rice
paleoduplicates have diverged in expression. In rice, retained
an-cient gene duplicates associated with high expression tended to
have higher CG body methylation ( Wang et al., 2013 ), suggesting a
direct role of epigenetic regulation in structural and expressional
maintenance of duplicates preventing pseudogenization, silencing
FIGURE 2 Grass SyntenyViewer tool (adapted from Murat et al., 2014a , b ). Screen capture of the SyntenyViewer web tool (http://urgi.versailles.inra.fr/ synteny) visualizing the synteny between inferred AGK s and modern grass species for applied translational research from model species ( Brachypo-dium ) to crops (wheat, rice, sorghum, maize) or between crops.
and deletion, and ultimately retaining WGD-derived genes. In wheat,
only 28% of the homoeologous triplets, deriving from a
hexaploidi-zation event 0.01–2.5 Ma, have the same expression pattern during
grain development ( Pont et al., 2011 ; Pfeifer et al., 2014 ). Such
mas-sive divergence in expression between duplicates has been
pro-posed as a source of subfunctionalization (partitioning of ancestral
functions between the duplicates, mainly for younger duplicates)
and neofunctionalization (gain of a new nonancestral function of
one duplicate, mainly for old duplicates) of genes during evolution,
both being key forces of evolutionary plasticity in plants ( Zou et al.,
2009 ). Bias in expression abundance between subgenomes
follow-ing duplication has been reported where the D/LF genes harbor
the highest level of expression compared with the S/MF
compart-ment ( Schnable et al., 2011 ; Freeling et al., 2012 ).
Bias in transposable element (TE) content has been also
re-ported between subgenomes in synthetic or naturally occurring
allopolyploids ( Renny-Byfi eld et al., 2011 , 2012 ; Buggs et al., 2012 ),
with the paternal subgenome subject to massive removal of
repeti-tive and nongenic DNA aft er hybridization. Bias in transposable
element (TE) content is not yet confi rmed in paleopolyploids
be-cause the rapid turnover of intergenic repeats makes it diffi
cult
(and in most cases impossible) to detect ancient elements that may
have diff erentially targeted subgenomes that were inherited from
polyploidizations that date back to more than 10 Ma. Transposable
element insertional dynamics may have then participated in the
subgenome restructuring aft er polyploidization so that such
subge-nomes become totally indistinguishable aft er million years of
evo-lution. Th
e insertional activity of TEs may not only drive subgenome
structural diff erentiation, but also the observed epigenetic changes
at the genome-wide and gene-based scales, as one of the
mecha-nisms driving duplicated genes expression partitioning ( Rodin and
Riggs, 2003 ; Rapp and Wendel, 2005 ). It has been reported that
J U LY 2016 , V O LU M E 103 • S A L S E — PA L E O G E N O M I C S A N D E V O LU T I O N I N G R A S S E S • 1171
FIGURE 3 Evolutionary plasticity following polyploidization (adapted from Murat et al., 2014a ; Salse, 2012 ). (A) Illustration of the subgenome domi-nance after whole-genome duplication in grasses with the average number of ancestral genes (protogenes) retained ( y -axis) in the investigated genomes (species illuminated with a specifi c color code) between duplicated blocks (A1–A5, A8–A9, A11–A12, A2–A4, A2–A6, A3–A7, A3A–10, x -axis), defi ning dominant (D, red circle) and sensitive (S, blue circle) blocks. (B) Schematic illustration of the biased structural and expressional genome par-titioning following polyploidization in plants acting as a source of genomic plasticity in modern species. Genes are illustrated as colored boxes, deleted genes with red crosses, and expression modifi cation as a theoretical expression pattern during development. TE, transposable element.
dominantly expressed genes in D/LF subgenomes have fewer 24-bp
RNA-targeted TEs in their 1-kb fl anking regions compared with
their S/MF paralogs ( Woodhouse et al., 2014 ), with small
RNA-targeted TEs shown to be subject to methylation, potentially
sup-pressing nearby gene expression ( Hollister and Gaut, 2009 ). All
these observations may suggest that diff erences in TE content and
insertional dynamics either between prepolyploidization progenitors
(in the case of allopolyploidy, merging paternal/maternal genomes
from distinct species) and/or between the postpolyploidization
subgenomes (in the case of autopolyploidy, doubling genomes from
the same species) may drive (small RNA-targeted) TE-mediated
epigenetic changes in promoting gene neo- or
subfunctionaliza-tion, silencing and ultimately deletion following polyploidizasubfunctionaliza-tion,
leading to the observed biased fractionation between D/LF and S/
MF compartments. However, this postpolyploidy structural and
functional plasticity driven by subgenomic dominance seems to
re-quire some time to “evolve” and “stabilize” as proposed from
stud-ies on resynthesized polyploids ( Renny-Byfi eld et al., 2015 ).
Despite this general phenomenon of massive and biased DNA
loss (including duplicates) following WGDs, ancestral genes can be
retained as pairs (referred to as Ohnologs; Ohno, 1970 ) during
evo-lution. Th
ese genes that survive diploidization (or
“diploidization-resistant” genes) are enriched in functional annotations such as
transcription factor, transcription regulator, and ribosomal protein
gene ( Freeling, 2009 ). Several hypotheses (probably interconnected
ones) have been proposed to explain duplicated gene retention such
as at (1) the function level with the acquisition of novel function
(neo- and subfunctionalization; Lynch and Conery, 2000 ; Wang
et al., 2012 ), (2) the network level with the maintenance of a
stoi-chiometric balance of gene product interactions (or connectivity)
in macromolecular complexes ( Birchler and Veitia, 2011 , 2014 ),
(3) the phenotypic level with a heterotic eff ect with transgressive
performances ( Birchler et al., 2010 ), (4) the allelic level in masking
deleterious recessive mutations ( Gu, 2003 ), (5) the adaptation level
with escape from adaptative confl icts ( Des Marais and Rausher,
2008 ), (6) the expression level with a higher level of expression
as-sociated with higher CG gene body methylation (
Seoighe and
Wolfe, 1999 ; Aury et al., 2006 ; Yang and Gaut, 2011 ), all potentially
delivering to the newly formed polyploid a machinery absent from
the diploid progenitors. It has been reported that such retained
du-plicated genes in rice are enriched in single nucleotide
polymor-phisms (SNPs) encoding less radical amino acid changes, suggesting
that such “advantageous” material/genes inherited from WGDs are
highly stable (i.e., slow rate of evolution) over the time ( Chapman
et al., 2006 ).
Overall, such structural (chromosome fusion/fi ssion/translocation,
gene/DNA loss, TE dynamics) and functional (neo- and
subfunc-tionalization, expression, network connectivity) partitioning of the
subgenomes following polyploidization participate in
homoeolo-gous chromosome (subgenomes) diff erentiation, then (1)
stabiliz-ing meiotic pairstabiliz-ing by preventstabiliz-ing chromosome pairstabiliz-ing while
increasing fertility of the nascent postpolyploidy lineages distinctly
diff erent from their ancestral progenitors and (2) ultimately
pro-vide genetic material to be “specialized” for key innovations such as
phenotypic novelty, altogether being potentially responsible for the
evolutionary success of polyploidy plants and novel desirable traits
selected during domestication by humans ( Fig. 3B ). For example,
nascent hexaploid wheat has been shown to exhibit hybrid vigor
and adaptive traits (such as robust seedling growth, larger spikes
with longer rachis internodes, and salt tolerance) when compared
with their parents ( He et al., 2003 ; Colmer et al., 2006 ; Yang et al.,
2014 ). Beside the knowledge gained during the last decade on the
role of polyploidy in plant species evolution and adaptation, still a
lot needs to be achieved and investigated to precisely characterize
the driving molecular mechanisms and so that they can be used in
breeding.
CONCLUSION
Overall, the consequences of polyploidization (reshaping gene
con-tent, expression, functions, TE compartment, CG gene body
meth-ylation) could explain how WGDs may have favored the emergence
of new plant species during the last 300 Myr of evolution. Such
evo-lutionary plasticity gained from polyploidization events provides
the basis for functional and phenotypic novelty in angiosperms.
Such novelty may fi nally be achieved through duplicate-based
functional divergence, gene conversion, changes in expression,
dosage eff ects, and network specialization and may ultimately
un-derlie the observed evolutionary success of angiosperms. However,
the continuum and interplay between the reported structural and
functional reprogramming aft er polyploidization are still poorly
understood. Synthetic polyploids may further expand our nascent
knowledge about this major phenomenon driving plant
evolution-ary dynamics.
ACKNOWLEDGEMENTS
Th
is work has been supported by grants from the Agence Nationale
de la Recherche (ANR Blanc-PAGE, ref : ANR-2011-BSV6-00801).
Th
e author thanks Caroline Pont and Florent Murat for their
participation in formatting the illustrations.
LITERATURE CITED
Aury , J. M. , O. Jaillon , L. Duret , B. Noel , C. Jubin , B. M. Porcel , B. Ségurens , et al . 2006 . Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia . Nature 444 : 171 – 178 .
Birchler , J. A. , and R. A. Veitia . 2011 . Protein–protein and protein–DNA dos-age balance and diff erential paralog transcription factor retention in poly-ploids. Frontiers in Plant Science 2 : 64 .
Birchler , J. A. , and R. A. Veitia . 2014 . Th e gene balance hypothesis: Dosage ef-fects in plants. Methods in Molecular Biology 1112 : 25 – 32 .
Birchler , J. A. , H. Yao , S. Chudalayandi , D. Vaiman , and R. A. Veitia . 2010 . Heterosis. Plant Cell 22 : 2105 – 2112 .
Buggs , R. J. , S. Renny-Byfi eld , M. Chester , I. E. Jordon-Th aden , L. F. Viccini , S. Chamala , and A. R. Leitch . 2012 . Next-generation sequencing and genome evolution in allopolyploids. American Journal of Botany 99 : 372 – 382 . Chapman , B. A. , J. E. Bowers , F. A. Feltus , and A. H. Paterson . 2006 . Buff ering
of crucial functions by paleologous duplicated genes may contribute cy-clicality to angiosperm genome duplication. Proceedings of the National Academy of Sciences, USA 103 : 2730 – 2735 .
Cheng , F. , J. Wu , L. Fang , S. Sun , B. Liu , K. Lin , G. Bonnema , and X. Wang . 2012 . Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS One 7 : e36442 .
Colmer , T. D. , T. J. Flowers , and R. Munns . 2006 . Use of wild relatives to im-prove salt tolerance in wheat. Journal of Experimental Botany 57 : 1059 – 1078 . Des Marais , D. L. , and M. D. Rausher . 2008 . Escape from adaptive confl ict aft er
duplication in an anthocyanin pathway gene. Nature 454 : 762 – 765 . Freeling , M. 2009 . Bias in plant gene content following diff erent sorts of
du-plication: Tandem, whole-genome, segmental, or by transposition. Annual Review of Plant Biology 60 : 433 – 453 .
J U LY 2016 , V O LU M E 103 • S A L S E — PA L E O G E N O M I C S A N D E V O LU T I O N I N G R A S S E S • 1173
Freeling , M. , M. R. Woodhouse , S. Subramaniam , G. Turco , D. Lisch , and J. C. Schnable . 2012 . Fractionation mutagenesis and similar consequences of mechanisms removing dispensable or less-expressed DNA in plants. Current Opinion in Plant Biology 15 : 131 – 139 .
Gu , X. 2003 . Evolution of duplicate genes versus genetic robustness against null mutations. Trends in Genetics 19 : 354 – 356 .
He , P. , B. R. Friebe , B. S. Gill , and J. M. Zhou . 2003 . Allopolyploidy alters gene expression in the highly stable hexaploid wheat. Plant Molecular Biology 52 : 401 – 414 .
Hollister , J. D. , and B. S. Gaut . 2009 . Epigenetic silencing of transposable ele-ments: A trade-off between reduced transposition and deleterious eff ects on neighboring gene expression. Genome Research 19 : 1419 – 1428 .
IBI [International Brachypodium Initiative] . 2010 . Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463 : 763 – 768 . IRGSP [International Rice Genome Sequencing Project] . 2005 . Th e map-based
sequence of the rice genome. Nature 436 : 793 – 800 .
IWGSC [International Wheat Genome Sequencing Consortium] . 2014 . A chromosome-based draft sequence of the hexaploid bread wheat ( Triticum aestivum ) genome. Science 345 : 1251788 .
Jacquemin , J. , C. Chaparro , M. Laudié , A. Berger , F. Gavory , J. L. Goicoechea , R. A. Wing , and R. Cooke . 2011 . Long-range and targeted ectopic recom-bination between the two homeologous chromosomes 11 and 12 in Oryza species. Molecular Biology and Evolution 28 : 3139 – 3150 .
Langham , R. J. , J. Walsh , M. Dunn , C. Ko , S. A. Goff , and M. Freeling . 2004 . Genomic duplication, fractionation and the origin of regulatory novelty. Genetics 166 : 935 – 945 .
Lynch , M. , and J. S. Conery . 2000 . Th e evolutionary fate and consequences of duplicate genes. Science 290 : 1151 – 1155 .
Martis , M. M. , R. Zhou , G. Haseneyer , T. Schmutzer , J. Vrána , M. Kubaláková , S. König , et al . 2013 . Reticulate evolution of the rye genome. Plant Cell 25 : 3685 – 3698 .
Mayer , K. F. , M. Martis , P. E. Hedley , H. Simková , H. Liu , J. A. Morris , B. Steuernagel , et al . 2011 . Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 23 : 1249 – 1263 .
Murat , F. , A. Louis , F. Maumus , A. Armero , R. Cooke , H. Quesneville , H. Roest Crollius , and J. Salse . 2015 . Understanding Brassicaceae evolution through ancestral genome reconstruction. Genome Biology .
Murat , F. , C. Pont , and J. Salse . 2014a . Paleogenomics in Triticeae for transla-tional research. Current Plant Biol 1 : 34 – 39 .
Murat , F. , J. H. Xu , E. Tannier , M. Abrouk , N. Guilhot , C. Pont , J. Messing , and J. Salse . 2010 . Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffl ing as a source of plant evolution. Genome Research 20 : 1545 – 1557 .
Murat , F. , R. Zhang , S. Guizard , R. Flores , A. Armero , C. Pont , D. Steinbach , et al . 2014b . Shared subgenome dominance following polyploidization ex-plains grass genome evolutionary plasticity from a seven protochromosome ancestor with 16K protogenes. Genome Biology and Evolution 6 : 12 – 33 . Ohno , S. 1970 . Evolution by gene duplication . Springer, Berlin, Germany. Paterson, A. H. , J. E. Bowers , R. Bruggmann , I. Dubchak , J. Grimwood , H.
Gundlach , G. Haberer , et al . 2009 . Th e Sorghum bicolor genome and the diversifi cation of grasses. Nature 457 : 551 – 556 .
Pfeifer , M. , M. Martis , T. Asp , K. F. Mayer , T. Lübberstedt , S. Byrne , U. Frei , and B. Studer . 2013 . Th e perennial ryegrass GenomeZipper: Targeted use of genome resources for comparative grass genomics. Plant Physiology 161 : 571 – 582 .
Pfeifer , M. , K. G. Kugler , S. R. Sandve , B. Zhan , H. Rudi , T. R. Hvidsten , International Wheat Genome Sequencing Consortium , et al. 2014 . Genome interplay in the grain transcriptome of hexaploid bread wheat. Science 345 : 1250091 .
Pont , C. , F. Murat , C. Confolent , S. Balzergue , and J. Salse . 2011 . RNA-seq in grain unveils fate of neo- and paleopolyploidization events in bread wheat ( Triticum aestivum L.). Genome Biology 12 : R119 .
Pont , C. , F. Murat , S. Guizard , R. Flores , S. Foucrier , Y. Bidet , U. M. Quraishi , et al . 2013 . Wheat syntenome unveils new evidences of contrasted evo-lutionary plasticity between paleo- and neoduplicated subgenomes. Plant Journal 76 : 1030 – 1044 .
Renny-Byfi eld , S. , M. Chester , A. Kovařík , S. C. Le Comber , M. A. Grandbastien , M. Deloger , R. A. Nichols , et al. 2011 . Next generation sequencing reveals genome downsizing in allotetraploid Nicotiana tabacum , predominantly through the elimination of paternally derived repetitive DNAs. Molecular Biology and Evolution 28 : 2843 – 2854 .
Renny-Byfi eld , S. , A. Kovařík , M. Chester , R. A. Nichols , J. Macas , P. Novák , and A. R. Leitch . 2012 . Independent, rapid and targeted loss of highly re-petitive DNA in natural and synthetic allopolyploids of Nicotiana tabacum . PLoS One 7 : e36963 .
Rapp , R. A. , and J. F. Wendel . 2005 . Epigenetics and plant evolution . New Phytologist 168 : 81 – 91 .
Renny-Byfi eld , S. , L. Gong , J. P. Gallagher , and J. F. Wendel . 2015 . Persistence of subgenomes in paleopolyploid cotton aft er 60 My of evolution . Molecular Biology and Evolution 32 : 1063 – 1071 .
Rodin , S. N ., and A. D. Riggs . 2003 . Epigenetic silencing may aid evolution by gene duplication . Journal of Molecular Evolution 56 : 718 – 729 .
Salse , J. 2012 . In silico archeogenomics unveils modern plant genome or-ganisation, regulation and evolution. Current Opinion in Plant Biology 15 : 122 – 130 .
Salse , J. 2013 . Paleogenomics as a guide for traits improvement . In R. Tuberosa, A. Graner, and R. Frison [eds.], Genomics of plant genetic resources, vol. 1, Genomics platforms, crop domestication and allele mining, 131–172. Springer Verlag, Dordrecht, Netherlands.
Salse , J. 2016 . Ancestors of modern plant crops . Current Opinions in Plant Biology 30 : 134 – 142 .
Salse , J. , M. Abrouk , S. Bolot , N. Guilhot , E. Courcelle , T. Faraut , R. Waugh , et al . 2009 . Reconstruction of monocotelydoneous proto-chromosomes reveals faster evolution in plants than in animals. Proceedings of the National Academy of Sciences, USA 106 : 14908 – 14913 .
Salse , J. , S. Bolot , M. Th roude , V. Jouff e , B. Piegu , U. Masood , T. Calcagno , et al . 2008 . Identifi cation and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell 20 : 11 – 24 .
Schnable , J. C. , N. M. Springer , and M. Freeling . 2011 . Diff erentiation of the maize subgenomes by genome dominance and both ancient and ongo-ing gene loss. Proceedongo-ings of the National Academy of Sciences, USA 108 : 4069 – 4074 .
Schnable , J. C. , X. Wang , J. C. Pires , and M. Freeling . 2012 . Escape from pref-erential retention following repeated whole genome duplications in plants. Frontiers in Plant Science 3 : 94 .
Schnable , P. S. , D. Ware , R. S. Fulton , J. C. Stein , F. Wei , S. Pasternak , C. Liang , et al . 2009 . Th e B73 maize genome: Complexity, diversity, and dynamics. Science 326 : 1112 – 1115 .
Schubert , I. , and M. A. Lysak . 2011 . Interpretation of karyotype evolution should consider chromosome structural constraints. Trends in Genetics 27 : 207 – 216 .
Soltis , D. E. , C. D. Bell , S. Kim , and P. S. Soltis . 2008 . Origin and early evolution of angiosperms . Annals of the New York Academy of Sciences 1133 : 3 – 25 . Soltis , D. E. , S. A. Smith , N. Cellinese , K. J. Wurdack , D. C. Tank , S. F. Brockington ,
N. F. Refulio-Rodriguez , et al . 2011 . Angiosperm phylogeny: 17 genes, 640 taxa. American Journal of Botany 98 ( 4 ): 704 – 730 .
Seoighe , C. , and K. H. Wolfe . 1999 . Yeast genome evolution in the post-genome era. Current Opinion in Microbiology 2 : 548 – 554 .
Th omas , B. C. , B. Pedersen , and M. Freeling . 2006 . Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeo-log leaving clusters enriched in dose-sensitive genes. Genome Research 16 : 934 – 946 .
Th roude , M. , S. Bolot , M. Bosio , C. Pont , X. Sarda , U. M. Quraishi , F. Bourgis , et al . 2009 . Structure and expression analysis of rice paleo duplications. Nucleic Acids Research 37 : 1248 – 1259 .
Van de Peer , Y. , J. A. Fawcett , S. Proost , L. Sterck , and K. Vandepoele . 2009a . The flowering world: A tale of duplications. Trends in Plant Science 14 : 680 – 688 .
Van de Peer , Y. , S. Maere , and A. Meyer . 2009b . The evolutionary sig-nificance of ancient genome duplications. Nature Reviews. Genetics 10 : 725 – 732 .
Wang , X. , H. Tang , and A. H. Paterson . 2011 . Seventy million years of con-certed evolution of a homoeologous chromosome pair, in parallel, in major Poaceae lineages. Plant Cell 23 : 27 – 37 .
Wang , X. , J. Wang , D. Jin , H. Guo , T. H. Lee , T. Liu , and A. H. Paterson . 2015 . Genome Alignment spanning major Poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events. Molecular Plant 8 : 885 – 898 .
Wang , Y. , X. Wang , T. H. Lee , S. Mansoor , and A. H. Paterson . 2013 . Gene body methylation shows distinct patterns associated with different gene origins and duplication modes and has a heterogeneous relation-ship with gene expression in Oryza sativa (rice). New Phytologist 198 : 274 – 283 .
Wang , Y. , X. Wang , and A. H. Paterson . 2012 . Genome and gene duplications and gene expression divergence: A view from plants. Annals of the New York Academy of Sciences 1256 : 1 – 14 .
Woodhouse , M. R. , J. C. Schnable , B. S. Pedersen , E. Lyons , D. Lisch , S. Subramaniam , and M. Freeling . 2010 . Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biology 8 : e1000409 .
Woodhouse , M. R. , F. Cheng , J. C. Pires , D. Lisch , M. Freeling , and X. Wang . 2014 . Origin, inheritance, and gene regulatory consequences of genome dominance in polyploids . Proceedings of the National Academy of Sciences, USA 111 : 5283 – 5288 .
Yang , C. , L. Zhao , H. Zhang , Z. Yang , H. Wang , S. Wen , C. Zhang , S. Rustgi , D. von Wettstein , and B. Liu . 2014 . Evolution of physiological responses to salt stress in hexaploid wheat. Proceedings of the National Academy of Sciences, USA 111 : 11882 – 11887 .
Yang , L. , and B. S. Gaut . 2011 . Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Molecular Biology and Evolution 28 : 2359 – 2369 . Yim , W. C. , B. M. Lee , and C. S. Jang . 2009 . Expression diversity and evolution-ary dynamics of rice duplicate genes. Molecular Genetics and Genomics 281 : 483 – 493 .
Zhang , G. , X. Liu , Z. Quan , S. Cheng , X. Xu , S. Pan , M. Xie , et al . 2012 . Genome sequence of foxtail millet ( Setaria italica ) provides insights into grass evolu-tion and biofuel potential. Nature Biotechnology 30 : 549 – 554 .
Zou , C. , M. D. Lehti-Shiu , M. Th omashow , and S. H. Shiu . 2009 . Evolution of stress-regulated gene expression in duplicate genes of Arabidopsis thaliana . PLOS Genetics 5 : e1000581 .