Diverse RNA Processing Pathways Important for Post-Transcriptional
Gene Regulation
by Calvin H. Jan
B.S., Molecular, Cell and Developmental Biology (2004) University of California, Los Angeles
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY JUNE 2010
© 2010 Massachusetts Institute of Technology All rights reserved
Signature of Author……… Calvin H. Jan Department of Biology May 21, 2010 Certified by……… David P. Bartel Professor of Biology Thesis Supervisor Accepted by………... Stephen P. Bell Professor of Biology Chair, Biology Graduate Committee
Diverse RNA Processing Pathways Important for Post-Transcriptional Gene Regulation by
Calvin H. Jan
Submitted to the Department of Biology on May 21, 2010
In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
ABSTRACT
Cis-acting elements in 3´ untranslated regions (UTRs) of mRNAs are crucial to the regulation of gene expression. Animal microRNAs (miRNAs) each target hundreds of mRNAs, which are recognized by pairing to nucleotides 2-7 of the miRNA. MicroRNAs mature through sequential RNase III cleavage of characteristic stem-loop precursors. Cleavage by Drosha defines the pre-miRNA hairpin, which is then cleaved by Dicer to generate a mature pre-miRNA. This biogenesis pathway ensures high fidelity definition of miRNA 5´ ends, which determine target specificity.
Small RNAs from Caenorhabditis elegans and Drosophila melanogaster are extensively
surveyed here using high-throughput sequencing. Analysis of these libraries led to the discovery of a novel miRNA biogenesis pathway, the mirtron pathway. Unlike canonical miRNAs,
mirtrons are defined by intron splicing. The excised intron lariat is debranched and folds into a pre-miRNA hairpin that is cleaved by Dicer. Because of the accuracy of the spliceosome, the mirtron pathway also allows for high fidelity miRNA maturation.
The trans-acting siRNAs (tasiRNAs) found in plants also reproducibly generate discrete small RNA species. TasiRNAs align to their parent locus (a TAS gene) in a distinctive 21-nt phase. This phasing is crucial; only siRNAs in the appropriate phase have sufficient complementarity to recognize their targets. The register of this phase is established by miRNA cleavage of the TAS transcript. Analysis of siRNAs sequenced from Physcomitrella patens reveals a conserved pathway in which P. patens TAS genes all possess two cleavage sites for miR390, the miRNA that cleaves TAS3 in Arabidopsis. A second miR390 site was found in Arabidopsis TAS3 that is bound by the miRNA but not cleaved. This interaction is important in triggering tasiRNA production from TAS3 transcripts.
A novel approach to mRNA 3´ end identification is applied here to determine 3´ UTRs in C. elegans. C. elegans UTRs are typically 150 nt long and have a higher density of miRNA seed sites than mammals. Ten percent of genes are alternatively polyadenylated. Approximately 1000 convergent gene pairs were found to use bidirectional poly(A) sites. This architecture maximizes gene density and demonstrates the influence of 3´ end formation on the evolution of gene topology.
Thesis Advisor: David P. Bartel Title: Professor
Acknowledgements
None of the work described here would be possible without the support and kindness of my many scientific mentors: Brian Koos, Srinu Reddy, Doug Black and Dave Bartel. I thank them first and foremost for growing my curiosity and capabilities as a scientist. I thank Dave for being so generous with his time, his thoughts, and his encouragement – he has strongly influenced the way I think about biology.
The Bartel lab has been a wonderful place to work. Allison, Uli, Matt, Graham, Sheq, Ed, Mike A., Ramya, I-hung, Soraya, Wendy, Andrew, Sarah, Hervé, Ben, Kyle, Kathy, El Guapo,
Christine, Lena, Alex, Mike N., Igor, Lori, Jinkuk, Sue-Jean, Dave W., Huili, Vincent, Anna, Olivia, Gina, David G., Rosaria, Daehyun, Jinwu, Muhammed and Noah and Robin – you guys are awesome and I am constantly inspired and humbled by your ideas and insights. I am particularly grateful to have overlapped with Graham and Andrew who are excellent scientists and friends. Christine and Lena have been terrific baymates. I thank Sheq for sharing his deep knowledge of RNA biochemistry and pop culture.
I am indebted to my thesis committee members – Phil Sharp, Tania Baker and Chris Burge – for providing focus and perspective throughout my PhD.
I thank my classmates, especially James and Steve for sharing the grad school experience with me, scientific and otherwise. I thank JP, Vincent, David G. and Crazy Dave K. for helping me escape from Cambridge via bike, and Vincent and K. Wang for organizing IM tennis.
I am grateful to Ellie for starting the bioREFS program. Ellie, Lourdes, Kevin, Brian, Shankar, Josh and Michael are tremendously dedicated and thoughtful peers; they have made the
department a better place.
I thank Karen for her tireless understanding, love and encouragement. You are the best! Lastly I thank my family for their support. I am grateful to my father for teaching me to leave things in better condition than I found them (I have tried to apply this philosophy to the lab and to the literature). I thank my mother for helping me find my first research experience and encouraging me ever since.
Table of Contents
Abstract 3
Acknowledgements 5
Table of Contents 7
Chapter 1. Introduction 9-46
Chapter 2. A Two-Hit Trigger for siRNA Biogenesis in Plants 47-95
Chapter 3. Intronic MicroRNA Precursors that Bypass Drosha Processing 97-129
Chapter 4. 3´ UTRs and Their Regulation by MicroRNAs in C. elegans 131-205
Chapter 5. Future Directions 207-213
Appendix A. The Widespread Impact of Mammalian MicroRNAs on
mRNA Repression and Evolution 215-220
Appendix B. Large-Scale Sequencing Reveals 21U-RNAs and Additional
MicroRNAs and Endogenous siRNAs in C. elegans 221-236
Appendix C. A Single Hox Locus in Drosophila Produces Functional
MicroRNAs from Opposite DNA Strands 237-243
Curriculum vitae 245-246
Additional electronic materials are included on the accompanying CD-ROM. Electronic materials are organized into directories by chapter affiliations.
Chapter 1 Introduction
Post-transcriptional gene regulation
All known organisms exert temporal and spatial control over the gene products they express. Gene regulation is achieved by trans-acting regulatory factors operating on genes as specified by genetic information encoded in cis. Comparative genomic studies indicate that the evolution of cis-regulatory systems is a primary driver of morphological diversity (Carroll, 2000). It is thus crucial to understand not only the biological function of gene products, but also how their activity is regulated. From a biochemical perspective, activity is directly related to concentration, as the concentrations of molecules determine reaction rates and the formation of intermolecular complexes. This implies that regulation of expression level is the most universal way to regulate gene activity.
Gene regulation relies on multiple processes that can be classified as transcriptional or post-transcriptional. The earliest studies of gene regulatory mechanisms provided conceptual frameworks for both transcriptional and post-transcriptional modalities. In their landmark study of lactose metabolism in Escherichia coli, Jacob and Monod proposed two generalized models to explain negative gene regulation (Jacob and Monod, 1961). Though lacking in molecular detail, Model I postulates that the regulatory gene product acts on the promoter of the target gene to inhibit transcription (transcriptional regulation). Model II posits that the repressive gene product is an RNA that interacts with a regulatory region in the target mRNA (post-transcriptional regulation).
In the time since Jacob and Monod proposed these mechanisms of gene regulation, we have come to appreciate the multitude of regulatory mechanisms that synergize to control the expression of genes. Transcription represents the first committed step to gene expression and, as such, is often a key regulated step. However, there are significant limits to the control that can be exerted by transcription alone. Although transcriptional regulation can determine the rate at which RNA is made from a genetic locus and is absolutely critical for determining whether a gene should be expressed, this alone cannot dictate the duration of expression or cellular location of the gene products. In order to control these properties, cells must rely on post-transcriptional mechanisms that act on RNA and protein, such as regulation of localization, translation and decay.
The extent to which genes are post-transcriptionally regulated is unclear. However, several recent discoveries – highlighted below – suggest that post-transcriptional mechanisms broadly influence the evolution of genes and their regulatory systems. The synthesis of detailed biochemical and genetic studies with genome-wide and multi-genome studies will be
indispensable for understanding the logic of gene regulation.
The discovery of small non-coding RNAs in plants and animals
The discovery of small regulatory RNAs first in Caenorhabditis elegans and
subsequently across Bilateria hinted at a much larger realm of post-transcriptional regulation. Small non-coding RNAs have been identified in members of all eukaryotic kingdoms (Cerutti and Casas-Mollano, 2006; Ghildiyal and Zamore, 2009). In plants and animals, these RNAs range in size from 20~30 nucleotides and have demonstrated roles as repressors of gene
structure and processing of their precursors and the proteins they interact with (Ghildiyal and Zamore, 2009). Short interfering RNAs, or siRNAs, are ~22 nt and function to repress gene expression (Elbashir et al., 2001; Hamilton and Baulcombe, 1999). The microRNAs (miRNAs) are derived from genes with characteristic stem-loop structures and interact with members of the Argonaute (Ago) family of proteins (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). The piwi-interacting RNAs (piRNAs) are processed from single-stranded RNAs and associate with Piwi proteins (Aravin et al., 2007).
The discovery of siRNAs is closely intertwined with explorations of two related phenomena in animals and plants. Working with the nematode C. elegans, Fire and Mello demonstrated that exogenously introduced double-stranded RNA could be a potent and specific inhibitor of homologous genes in a process termed RNA interference (RNAi) (Fire et al., 1998). In plants, post-transcriptional gene silencing (PTGS) encompasses the phenomenon known as co-suppression, which describes the trans silencing of genes homologous to exogenous
transgenic or viral messages. Co-suppression was observed first in petunias, where attempts to overexpress genes by introducing extra copies resulted in the silencing of endogenous loci (Napoli et al., 1990; van der Krol et al., 1990). Hamilton and Baulcombe demonstrated the first connection between small RNAs and gene silencing (Hamilton and Baulcombe, 1999). Their key finding was that loci undergoing PTGS produces both sense and antisense small RNAs. These findings were generalized to animal RNAi, and the term siRNA was coined to describe the small, ~22 nt RNAs that repress gene expression (Elbashir et al., 2001). Thus siRNAs sensu
stricto are small RNAs that silence gene expression, regardless of their mechanism of biogenesis or action1. For this reason, siRNAs constitute the most diverse class of small non-coding RNAs.
Depending on the organism, siRNAs can silence gene expression transcriptionally or post-transcriptionally. In Schizosaccharomyces pombe, siRNAs act in the RNA-induced
initiation of transcriptional gene silencing (RITS) complex (Reinhart and Bartel, 2002; Verdel et al., 2004; Volpe et al., 2002). Loci recognized by the RITS complex undergo histone H3 lysine 9 methylation, resulting in heterochromatin formation and depressed transcription. In
Arabidopsis thaliana, cis-acting siRNAs (casiRNAs) are 24-nt RNAs that direct DNA
methylation and histone modification at homologous loci (Ghildiyal and Zamore, 2009; Mallory and Vaucheret, 2006). In contrast, natural antisense siRNAs and trans-acting siRNAs (nat-siRNAs and ta(nat-siRNAs, respectively) represent endogenous (nat-siRNAs that function
post-transcriptionally (Ghildiyal and Zamore, 2009; Mallory and Vaucheret, 2006). These examples highlight the evolutionary diversity of siRNAs, which makes them difficult to generalize about, but fascinating to study.
Characterization of heterochronic mutants in C. elegans provided key insights into the biology of small RNAs. C. elegans follow a well-characterized, highly reproducible
developmental program (Sulston and Horvitz, 1977). Heterochronic mutants, such as lin-4 and lin-14, show changes in the relative timing of developmental events (Chalfie et al., 1981). Loss of function (LOF) mutants in the lin-4 gene reiterate early cell fates at inappropriate times in development. In contrast, lin-14 LOF mutants precociously generate cell lineage patterns normally seen at later developmental stages. Lin-14 nulls are epistatic to lin-4 nulls, suggesting
that lin-4 is a negative regulator of lin-14 (Lee et al., 1993; Wightman et al., 1993). Intriguingly, gain of function (GOF) alleles of lin-14 were found that phenocopy the lin-4 null. Ambros and colleagues found that lin-4 encodes a 22 nt transcript, representing the first discovered miRNA (Lee et al., 1993). Concurrently with Ruvkun and colleagues, lin-4 was shown to have
homology to sequences in the 3´ untranslated region (UTR) of the lin-14 mRNA (Wightman et al., 1993). These sequences were deleted in the lin-14 GOF allele and, importantly, were
sufficient to recapitulate the temporal expression of lin-14 in a heterologous reporter (Wightman et al., 1993). These findings established the paradigm of gene regulation by miRNAs in animals: miRNAs basepair to complementary sequences in the 3´ UTRs of target mRNAs and repress expression2.
The recognition that lin-4 is part of a larger class of regulatory RNAs did not occur until it was discovered that another heterochronic gene, let-7, also gives rise to small RNAs (Reinhart et al., 2000). Due to their roles in developmental timing, these RNAs were called small temporal RNAs (stRNAs). Addressing the evolutionary importance of stRNAs, a broad survey of metazoa revealed the existence of let-7 small RNAs across Bilateria (Pasquinelli et al., 2000). This result, along with the contemporary discovery of siRNAs, prompted the survey of endogenously
expressed small RNAs in multiple model systems. These sequencing efforts identified numerous additional small RNA genes that necessitated a broader definition of the gene class, since many were not temporally regulated. These genes were thus classified as microRNAs, and were all found to originate from loci capable of forming characteristic stem-loop structures (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). Like lin-4, let-7 was found to
repress the expression of genes with homology in their 3´ UTRs (Reinhart et al., 2000), suggesting that these newly discovered miRNAs have similar functions in gene regulation.
Studies of transposon silencing Drosophila melanogaster identified the first piRNAs, which at the time were termed repeat associated siRNAs. In Drosophila, certain genetic loci protect the genome from homologous transposable elements in the germline (Aravin et al., 2007). Inspired by the role of PTGS in plant genome defense, Aravin et al. identified repeat associated siRNAs (rasiRNAs) derived from one such protective locus, Su(Ste) (Aravin et al., 2001). Su(Ste) was an appealing locus to search for siRNAs, as antisense transcription was observed there. The presence of an antisense transcript provided a mechanism for generating dsRNA, which was thought to be required for siRNA production. However, follow-up experiments demonstrated that dsRNA was not a requirement, suggesting a very different biogenesis mechanism (Vagin et al., 2006).
A similar class of RNAs was subsequently discovered in the mammalian testes. An abundant population of 28~30 nt RNAs were found in complex with rodent orthologs of Piwi (a subclass of the Argonaute family), resulting in the term piwi-interacting RNAs or piRNAs (Aravin et al., 2006; Girard et al., 2006; Lau et al., 2006). Piwi orthologs are required for male germline viability in mammals and flies3 (Aravin et al., 2007). The rasiRNAs from Drosophila were absent in Piwi null mutants and were subsequently found in Piwi complexes as well, leading to their reclassification as piRNAs (Aravin et al., 2003; Saito et al., 2006). The conservation of Piwi’s expression domain and germline protective functions in flies and mammals suggests deep conservation of the piRNA pathway in bilateria (Aravin et al., 2007; Cerutti and Casas-Mollano, 2006; Grimson et al., 2008). Consistently, piRNAs have been found
in highly divergent bilaterians, such as Nematostella vectensis (Grimson et al., 2008). The piRNAs represent an extremely diverse collection of RNA molecules. Individuals are each expressed at low levels, but combined, piRNAs represent the most abundant class of small regulatory RNAs in the male germline (Girard et al., 2006).
RNA interference and the biogenesis of siRNAs
During RNAi4, a dsRNA trigger post-transcriptionally silences homologous mRNAs. Long dsRNA is cleaved into ~21 nt siRNA duplexes (Zamore et al., 2000) in a process that required a dsRNA binding protein (Tabara et al., 2002) and the RNase III enzyme Dicer (Bernstein et al., 2001; Grishok et al., 2001; Knight and Bass, 2001). These duplexes are then loaded into Ago in a process that results in the incorporation of the guide strand (the siRNA) into RISC (Elbashir et al., 2001; Martinez et al., 2002; Nykanen et al., 2001). The other strand (the passenger strand) is then endonucleolytically cleaved by Ago, licensing RISC for RNAi (Matranga et al., 2005). During loading, the guide strand is 2´ O-methylated at its 3´ end5 (Forstemann et al., 2007; Tomari et al., 2007). In crystal structures of Ago, the 3´ end is bound to the PAZ domain (Lingel et al., 2003; Song et al., 2003; Wang et al., 2008c; Yan et al., 2003). The 5´ monophosphate end is anchored in the Mid domain of Ago, protecting it from 5´ to 3´ exonucleases (Wang et al., 2008c).
When the guide strand basepairs to near perfectly complementary target RNAs, the target is cleaved between the bases that pair to nucleotides 10 and 11 of the siRNA (Elbashir et al., 2001; Liu et al., 2004), generating fragments with 5´ monophosphates and 3´ hydroxyls (Elbashir
et al., 2001). Considerable molecular movement is required to accommodate the two turns of A-form helix A-formed upon target recognition (Bartel, 2009). The crystal structure of T.
thermophilus Ago complexed with a ssDNA guide strand demonstrate that the Watson-Crick surface of the 5´ end of the guide strand (corresponding to the seed region6) is solvent exposed (Wang et al., 2008c). This suggests a model in which the siRNA seed initially binds the target mRNA, followed by release of the siRNA 3´ end from RISC to form the remaining basepairs with the target (Yuan et al., 2005). The 3´ end of the guide strand then re-associates with the Ago PAZ domain (Wang et al., 2008b). The fully paired target is then position in the piwi domain active site, resulting in endonucleolytic cleavage through an RNase H-like mechanism (Liu et al., 2004; Song et al., 2004). Product release presumably also requires dissociation of the siRNA 3´ end from Ago. The 2´ O-methylation may exist to stabilize the siRNA during cycles of 3´ end release associated with catalysis.
In C. elegans, siRNAs derived from the trigger dsRNA are referred to as primary
siRNAs. In the presence of targets, the secondary siRNAs can be found derived from sequences 5´ of the regions targeted by primary siRNAs (Sijen et al., 2001). These secondary siRNAs require the action of RNA-dependent RNA polymerase (RDRP) but not Dicer, indicating a distinct biogenesis mechanism (Pak and Fire, 2007; Sijen et al., 2001; Sijen et al., 2007). Despite the relative location of secondary siRNAs, primary siRNAs do not act as primers for dsRNA synthesis7. In this form of RNAi, the precise sequence identity of discrete primary and secondary siRNAs are irrelevant so long as they are complementary to their targets. Processing of dsRNA triggers in C. elegans requires the dsRNA binding protein RDE-4, which binds
cooperatively to dsRNA (Parker et al., 2006) and positions the site of Dicer cleavage (Figure 1A). Thus the site of RDE-4 nucleation determines the register from which siRNAs are diced. If the nucleating RDE-4 binding event were stochastic, one would expect the register for each dsRNA to be established randomly.
Arabidopsis, with its ten Argonaute and four Dicer-like genes, utilizes multiple distinct siRNA pathways to regulate gene expression (Chen, 2009; Vaucheret, 2008). The tasiRNAs constitute an intriguing class of siRNAs in their biogenesis, function and evolution. Trans-acting siRNAs were discovered as a set of siRNAs that required components of both the co-suppression and miRNA pathways8 (Peragine et al., 2004; Vazquez et al., 2004). Intriguingly, these siRNAs post-transcriptionally targeted mRNAs with little homology to the genes from which they originate (Peragine et al., 2004; Vazquez et al., 2004). Loci encoding tasiRNAs, known as TAS genes, produce non-coding transcripts that are in turn converted into specific siRNAs. For example, the A. thaliana TAS3 gene produces a ~950 nt transcript that possesses two – and only two – discrete 21 nt regions of identity to Auxin Response Factor genes (Allen et al., 2005). In order for silencing to occur, specific tasiRNAs must reproducibly mature from a long precursor. Accordingly, tasiRNAs from a given locus are phased, i.e. their 5´ ends map with a distinctive 21 nt periodicity (Allen et al., 2005; Peragine et al., 2004; Vazquez et al., 2004; Yoshikawa et al., 2005). For TAS3, the register for this phase is set by miR390 cleavage of the precursor RNA, which is subsequently converted into dsRNA by the RDRP rdr6 (Allen et al., 2005). Dicer then recursively cleaves siRNAs from the terminus established by miR390, allowing the same siRNA species to be generated from discrete dsRNA precursors (Figure 1B). Likewise, all TAS genes
identified to date have miRNA complementary sites that are used to establish phasing and production of siRNAs with trans targets (Chen, 2009).
Biogenesis of metazoan miRNAs
Plant and animal genomes encode numerous miRNAs. Like the tasiRNAs, miRNA metabolism has evolved towards the singular goal of reproducibly generating discrete small RNA species. Unlike tasiRNAs, genes encoding miRNAs have characteristic stem-loop structures that are absolutely required for their processing (Bartel, 2004). In animals, nascent primary miRNA transcripts (pri-miRNAs) are recognized in the nucleus by the dsRNA binding protein DGCR-8 in complex with the RNase III enzyme Drosha (Denli et al., 2004; Gregory et al., 2004; Han et al., 2004; Landthaler et al., 2004; Lee et al., 2003). The pri-miRNA is
“cropped” by Drosha to liberate the pre-miRNA hairpin (Figure 2A). The Drosha:DGCR8 complex reproducibly cleaves each pri-miRNA at the same position by measuring ~10 base pairs from the single-strand/double-strand junction at the base of the stem (Han et al., 2006). Like all RNase III enzymes, Drosha cleavage leaves 2-nt 3´ overhanging ends. The nuclear pre-miRNA is recognized by Exportin-5 for transport to the cytoplasm (Lund et al., 2004). A second RNase III enzyme, Dicer, then cleaves the pre-miRNA to liberate the miRNA:miRNA* duplex (Lee et al., 2002). A ternary complex forms containing the pre-miRNA, Dicer and a dsRNA binding protein9 (Chendrimada et al., 2005; Forstemann et al., 2005). The Dicer PAZ domain anchors the 3´ overhang left by Drosha, using this reference to measure ~22 nt from the end of the hairpin to cleave the pre-miRNA (Macrae et al., 2006). The resulting miRNA:miRNA* duplex then associates with Argonaute in a process that favors loading of the miRNA strand (Schwarz et
al., 2003). The process of strand selection appears to sample the thermodynamic stabilities of the duplex termini. Disparity in the relative stabilities tends to favor the loading of the less stable 5´ end of the duplex.
In mammals, the proteins that process miRNAs also function in siRNA pathways (Bartel, 2004). In other organisms, including plants, flies and nematodes, there are distinct pathways involved. In flies, Dicer-1 cleaves most pre-miRNA hairpins, while Dicer-2 typically cleaves long dsRNA (Ghildiyal and Zamore, 2009). Likewise, Ago-1 interacts with most miRNAs, and Ago-2 interacts with most siRNAs. The structural basis for this selectivity appears to be the degree of pairing between passenger and guide strands of the small RNA duplex. Whereas extensively paired duplexes are recognized by Dicer-2/R2D2 and loaded into Ago-2, duplexes with central mismatches preferentially interact with Dicer-1/Loqs and load their guide strands into Ago-1(Forstemann et al., 2007; Tomari et al., 2007). Some miRNAs, such as miR-277, are cleaved by Dicer-1/Loqs, but are subsequently loaded into Ago-2 in a Dicer-2 dependent
manner.
Pri-miRNA hairpins are often found in genomic clusters with other pri-miRNA hairpins, or within introns of spliced RNAs (Kim et al., 2009). Most are transcribed by RNA polymerase II10. Consistent with Pol II transcription, capped and polyadenylated pri-miRNA transcripts can be found (Lee et al., 2004). Clustered miRNAs are generally correlated in their expression, suggesting transcription of the entire cluster from a single promoter (Bartel, 2004). Consistently, single transcripts containing multiple pri-miRNA hairpins have been found for several clustered miRNAs, making these miRNAs polycistronic (Kim et al., 2009).
In humans, approximately 40% of miRNAs are embedded in the sense strand of introns of pre-mRNAs (Kim et al., 2009; Rodriguez et al., 2004). RT-PCR analyses of processing intermediates suggest that Drosha can crop intronic pri-miRNAs prior to splicing (Kim and Kim, 2007; Morlando et al., 2008). Despite breaking the covalent bond between splice sites, Drosha processing does not appear to be refractory to splicing. Therefore, either spliceosomal
complexes form concurrently with Drosha cleavage (Figure 2A, right panel), or the ~10 base pairs remaining from the pri-miRNA hairpin are sufficient to hold the intron together until the spliceosome can assemble. By emerging within introns, newly evolving miRNAs genes can piggyback on existing sequences required for transcription, including those required to achieve the same spatial and temporal regulation as their host genes.
Gene regulation by miRNAs: mechanisms and models
The interactions between lin-4/lin-14 and let-7/lin-41 ingrained the idea that miRNAs basepair to multiple extensively complementary sites in the 3´ UTRs of their target mRNAs, resulting in translational repression of these mRNAs. With such a short sequence, putative interacting regions were identified that maximized basepairing to the small RNA. Building on this idea, the first breakthrough in understanding miRNA targeting at a genome-scale came from a search for mRNAs with varying degrees of complementarity to recently discovered miRNAs in Arabidopsis thaliana (Reinhart et al., 2002; Rhoades et al., 2002). Near perfect pairing allows these miRNAs to cleave their target mRNAs, strongly downregulating their expression
(Hutvagner and Zamore, 2002; Llave et al., 2002; Tang et al., 2003). Intriguingly, a similar search in animal genomes failed to detect any significant targeting of these types (Lewis et al., 2003; Rhoades et al., 2002). Inspection of targets genetically identified in C. elegans is
consistent with these results, as none of the classic miRNA/mRNA interactions were paired around the scissile phosphate. In nematodes, this may be to avoid runaway RNAi-like amplification of silencing that might result from slicing by RISC.
Successful characterization of animal miRNA targeting required a comparative genomic approach. In mammals, single-genome searches for targets on the basis of pairing or
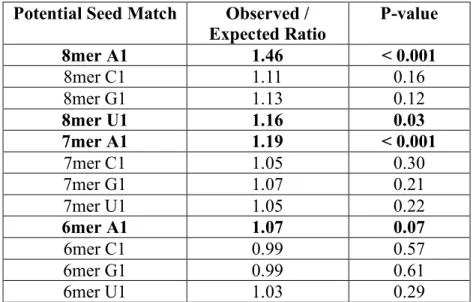
thermodynamic stability fail to identify any more targets than expected by chance (Bartel, 2009). Instead, targets were identified on the basis of preferential conservation of sequences that pair with miRNAs (Enright et al., 2003; Lewis et al., 2003; Stark et al., 2003). Systematic searches for regions of perfect Watson-Crick base pairing between different portions of miRNAs and putative targets identified nucleotides 2-7 of the miRNA as the most critical, a region referred to as the miRNA seed (Lewis et al., 2003). Seed sites are often supplemented by either an
additional basepair to position 8 of the miRNA, the presence of an adenosine moiety across from position 1 of the miRNA, or both (Lewis et al., 2005) . These seed sites, the 7merM8, 7merA1 and 8mer, respectively, represent the vast majority of target specificity, as sites with
supplemental pairing to the 3´ end of the miRNA are rare (Bartel, 2009).
Both plant and animal miRNAs repress targets primarily by mRNA destabilization. In plants, canonical targets pair extensively to miRNAs, resulting in target cleavage in a process that resembles canonical RNAi (Jones-Rhoades et al., 2006). In mammals, HOXB8 regulation by miR-196 is the only known example of plant-like targeting (Yekta et al., 2004). Instead, most mammalian miRNA targets are repressed more subtly, with a typical seed site contributing ~25% reduction in expression (Baek et al., 2008; Lim et al., 2005; Selbach et al., 2008). Most sites appear to act independently; i.e. the fold-change attributed to multiple sites equals the product of the fold change attributed to individual sites. Notably, sites that occur within close proximity to
one another appear to function cooperatively, with total repression exceeding the product of the repression measured from individual sites (Grimson et al., 2007).
Just as HOXB8 is an exception to the general rules of animal miRNA targeting,
APETALA2 is an exception to plant targeting. APETALA2 protein is reduced in the presence of miR172, its mRNA is sliced in its 3´ UTR by miR172 as assayed by RACE11, and yet its mRNA levels remain unchanged (Aukerman and Sakai, 2003; Chen, 2004). This suggests that miR172 translationally represses APETALA2. Other miR172 targets TOE1 and TOE2 also appear to be translationally repressed (Aukerman and Sakai, 2003), suggesting that translational repression may be a common theme, though the majority of investigated targets clearly show reduction in their mRNA levels in the presence of cognate miRNA (Jones-Rhoades et al., 2006). Thus like animal miRNA targets, the mRNA destabilization of plant miRNA targets explains the bulk of miRNA regulation with a few notable exceptions.
The precise mechanism of miRNA-mediated repression of gene expression is still unclear despite12 intense investigation. Proposed models include inhibition of translation initiation, inhibition of elongation, increased ribosome drop-off, nascent polypeptide degradation,
endonucleolytic cleavage, mRNA deadenylation and repressive compartmentalization (Eulalio et al., 2008). It seems unlikely that there is a single mechanism capable of unifying the underlying data. Instead, one interpretation of these reports is that RISC is a promiscuous complex that interfaces with multiple effectors, the dominance of which depends on the particular system used. Regardless of the exact mechanism, models of miRNA action can be grouped into two general classes: those that affect translation, and those that affect mRNA stability.
11
The idea that miRNAs affect translation stems from studies of lin-14 regulation by lin-4. During nematode development, LIN-14 protein is high during the L1 stage, but drops
significantly in L2, concomitant with expression of the lin-4 small RNA (Lee et al., 1993; Wightman et al., 1993). Despite significant reduction in protein concentration, lin-14 mRNA levels are comparable between L1 and L2 (Olsen and Ambros, 1999). Moreover, the distribution of lin-14 mRNA in polysome gradients were indistinguishable between stages L1 and L2,
suggesting either that translation was inhibited at a step after initiation or that LIN-14 protein was rapidly degraded (Olsen and Ambros, 1999).
Translation repression was the best-supported model for miRNA-mediated repression until it was discovered that the miRNAs could globally downregulate target mRNA levels. Lim and colleagues performed microarray analyses of mammalian cells transfected with miRNAs and found that genes that were significantly downregulated at the mRNA level were enriched in seed-matches to the transfected miRNA (Lim et al., 2005). Importantly, luciferase reporters bearing the 3´ UTRs of down-regulated genes recapitulated the effect at the protein level with a similar magnitude of repression. Subsequent studies using quantitative mass-spectrometry found that, for most seed-match targets, repression at the protein level recapitulated repression
measured at the RNA level, implying little translational repression for most mammalian miRNA targets (Baek et al., 2008; Selbach et al., 2008).
In mammals, miRNAs can associate with any of four members of the Argonaute family. Only one of these, Ago2, can cleave complementary targets (Liu et al., 2004). Whether or not RISCs vary in other ways is still unclear. Different Agos may interact with different effectors, perhaps contributing to the breadth of post-transcriptional mechanisms observed in the literature. In addition, miRNAs may have different target specificities depending upon the makeup of their
RISCs. In Drosophila and Arabidopsis, Ago family members have clearly specialized in the small RNAs with which they interact and the mechanism by which they function (Hutvagner and Simard, 2008). Differences in mammalian Ago function and specificity remain largely
unexplored.
Gene regulation by miRNAs: functions and themes
The groundbreaking studies of lin-4, let-7 and their targets shaped early thinking about miRNA regulation. Both lin-4 and let-7 act as switches: their activity turns off the expression of their targets. By reducing LIN-14 protein levels to near inconsequential levels, lin-4 expression allows cells to adopt a new gene expression program and developmental fate. Furthermore, specific developmental defects were attributable to deregulation of discrete pairs of miRNA and target13. As additional miRNAs were discovered, their diverse expression patterns indicated that this paradigm of developmental switching could not be universally true. Elucidating the
functions of these miRNAs would require accurate identification and characterization of their targets.
The magnitude of repression and relative confidence in target predictions made
Arabidopsis a choice model system for early studies of miRNA function14. Predicted targets of
13
These and other similar observations have lead some to believe that miRNAs evolved to regulate a few critical targets (Xiao et al., 2007). However, both experimental and computation evaluation of miRNA targeting have demonstrated that animal miRNAs can influence the expression levels of hundreds of mRNAs (Lim et al., 2005) and have hundreds of targets conserved above background (Friedman et al., 2009). As conservation is a direct measure of evolutionary pressure, one must reject the hypothesis that most conserved miRNAs have evolved to act on a few targets.
14
Arabidopsis miRNAs were strikingly enriched in transcription factors (Rhoades et al., 2002). Moreover, many of these transcription factors are involved in meristem specification. Analyses of transgenic plants where specific miR165/miR166 complementary sites were mutated in PHABULOSA/PHAVOLUTA found expanded expression domains (Mallory et al., 2004), and bulk loss of miRNAs delayed developmental timing and overproliferation of meristem similar to reiteration of early cell fates in heterochronic mutants (Park et al., 2002). These results all support a switch-like model for miRNA function, whereby miRNA expression during development ensures forward progression through cell fate and differentiation programs.
In addition to functioning as switches, animal miRNAs can play more subtle roles in regulating gene expression. Broadly speaking, miRNA:target interactions can result in switching, tuning or neutral changes in target function by affecting target expression levels (Bartel and Chen, 2004; Shkumatava et al., 2009; Stark et al., 2005). Targeting by miRNAs in animals typically results in a less than 2-fold decrease in expression, with higher degrees of repression achieved by increasing site number. Intuitively, smaller changes are more likely associated with tuning interactions whereas larger fold changes may represent switching interactions. However, distinctions in the mode of regulation depend on both the magnitude of miRNA-mediated repression and on the thresholds at which changes in gene product
concentrations have functional consequences (Bartel, 2009). Like other targeting events, neutral interactions would reduce expression levels, but with no functional consequence upon which selection might act. Thus sites mediating beneficial tuning and switch-like interactions are expected to be under positive selection, whereas sites mediating neutral interactions are not
expected to be conserved15. Lastly, mRNAs may be anti-targets: genes for which
downregulation by miRNAs are injurious and would thus be under selection to avoid targeting.
3´ untranslated regions: regulatory functions
From birth through death, all cellular RNAs exist as ribonucleoproteins (RNPs). A typical eukaryotic mRNA, at the very least, forms a RNP with a cap-binding complex, and poly(A) binding proteins; most are associated with additional proteins and RNPs (Cech, 2006; Moore, 2005). Thus when considering the metabolism of an mRNA, one must consider the metabolism of the mRNP. Messenger RNAs are composed of 5´ untranslated regions (UTRs), open reading frames (ORFs) and 3´ UTRs. In metazoans, most known cis-acting elements that regulate mature mRNAs are located in 3´ untranslated regions (Martin and Ephrussi, 2009; Sonenberg and Hinnebusch, 2009). This topology likely evolved for two reasons: 1) placement of cis-regulatory sequences outside of the ORF allows these elements to evolve independently from coding sequences; and 2) cap-scanning during translation initiation is capable of
dissociating bound complexes in the 5´ UTRs16.
Myriad RNA binding proteins provide a toolbox for post-transcriptional regulation through the 3´ UTR. These proteins can control the localization, translation, and stability of the mRNAs they interact with. These are broadly used mechanisms, as suggested by a recent study of mRNA localization in Drosophila embryos by high throughput in situ hybridization. This survey included 3000 transcripts and found that 71% of these RNAs had distinct spatial patterns in the early embryo (Lecuyer et al., 2007). How these patterns emerge in Drosophila is still
15
under investigation, but similar studies in the C. elegans germline have pointed to the primacy of 3´ UTRs in specifying spatial expression patterns. When 3´ UTRs are fused to reporter genes transcribed throughout the germline, these reporters recapitulated the expression patterns of the endogenous genes, suggesting that these 3´ UTRs carry all of the necessary information to specify expression domains (Merritt et al., 2008).
Examples of subcellular mRNA localization are prevalent throughout eukaryotes: Bicoid (bcd) localizes to the anterior pole of Drosophila oocytes, Vg1 localizes to the vegetal pole in Xenopus oocytes, MBP localizes to the myelin lamellae of mammalian oligodendrocytes, and ASH1 localizes to the bud in S. cerevisiae (Martin and Ephrussi, 2009). All of these genes require cis-acting elements in their 3´ UTRs to achieve their specific localization and expression domains. Though all localized to 3´ UTRs, there are a variety of mechanisms by which these cis-acting elements direct localization (St Johnston, 2005). Some mRNAs localize by active transport via myosin transport over actin fibers (ASH1) or dynein/kinesin transport over microtubules (Drosophila grk and osk). Other mRNAs can be localized via passive diffusion and selective anchoring at specific sites (Drosophila germ cell-less), or by local protection from degradation (D. rerio nanos).
Translational control is particularly important for mRNAs that must be stored or localized before their protein products are required. For example, posterior localization of the Oskar protein is critical for abdominal and germline development in Drosophila (Lehmann and
Nusslein-Volhard, 1986). Nurse cells supply oskar mRNA to developing oocytes, where it must accumulate in the posterior pole. Premature translation of oskar mRNA is detrimental to
development, and thus it must be translationally repressed in anterior regions, but active in the posterior (Kim-Ha et al., 1995). This is accomplished by sequence-specific binding by Bruno to
the oskar 3´ UTR. Bruno recruits Cup, an eIF-4E binding protein that interacts with the 5´ end of oskar mRNPs to inhibit translation initiation (Nakamura et al., 2004).
During early embryogenesis, the developing embryo relies entirely on stored maternal mRNAs before the onset of zygotic transcription. Moreover, it is critical that these mRNAs are translationally quiescent until fertilization occurs. Regulation of mRNAs encoding oscillatory proteins, such as those required for cell cycle progression, is further complicated by the fact that each cycle must rely on the same pool of mRNA, indicating that the cyclical regulation of protein levels need be entirely post-transcriptional (Groisman et al., 2002). This has been characterized extensively in Xenopus, where key maternal mRNAs, such as cyclin B1, are translationally arrested and held in a deadenylated state (Sheets et al., 1994; Stebbins-Boaz et al., 1996). Upon fertilization, cycles of mRNA polyadenylation and deadenylation correlate with oscillations in protein levels, consistent with the function of poly(A) binding proteins as
stimulators of cap-dependent translation. These cycles are controlled by coordinated action of a cyoplasmic poly(A) polymerase and a poly(A) specific ribonuclease, which act on mRNAs as specified by cytoplasmic polyadenylation elements found in their 3´ UTRs (Kim and Richter, 2006).
Continuing with the theme of embryonic development, changes in expression patterns at the maternal to zygotic transcription reveals yet another essential role for post-transcriptional regulation. Once embryonic transcription can drive development, the maternal program is cleared. In zebrafish, zygotic transcription triggers accelerated decay of maternal mRNAs. Embryonically expressed miR-430 promotes this decay, as maternal mRNAs are enriched for miR-430 seed sites in their 3´ UTRs (Giraldez et al., 2006). Likewise, genes with narrow
(Houseley and Tollervey, 2009). The best characterized are the AU-rich elements (AREs), which are found in multiple copies in the 3´ UTRs of immediate early genes, such as c-fos (Chen et al., 1995). When bound by proteins such as TTP, these elements often reduce mRNA half-life by accelerating deadenylation and decay (Carballo et al., 1998).
More generally, cytoplasmic localization of mRNPs is often interconnected with regulation of translation and stability. P-bodies and stress granules represent two ends of a spectrum of foci associated with non-translating mRNAs; P-bodies are sites rich in RNA decay factors17 and devoid of translation factors, whereas stress granules contain many translation initiation components (Buchan and Parker, 2009). Genetic and chemical interference with translation and mRNA decay, along with time-lapse microscopy, suggest that these are dynamic granules that can dock with one another, and transition in their identity depending upon the proteins present (Buchan and Parker, 2009). Together, these foci act as sites of mRNA storage and decay, which allows for cells to dynamically and efficiently regulate protein output without new transcription. Cationic amino acid transporter-1 (CAT-1) regulation is an interesting example that highlights the importance of combinatorial post-transcriptional control and localization. In liver cells, CAT-1 mRNA is repressed by miR-122 targeting, leading to its localization in P-bodies. When these cells are stressed, CAT-1 mRNA relocalized from P-bodies to polysomes. This process requires additional cis-acting elements in the CAT-1 3´ UTR that are bound by HuR, a protein known to stabilize mRNAs containing AREs. Thus the 3´ UTR of CAT-1 integrates multiple regulatory signals that control CAT-1 protein levels.
The impact of alternative pre-mRNA processing on mRNA regulation
Alternative pre-mRNA processing can significantly alter the cis-regulatory elements present in 3´ UTRs. Alternative cleavage and polyadenylation can alter the identity of 3´ UTRs without impinging upon ORFs, or it can synergize with alternative splicing to redefine ORF and 3´ UTR identities (Licatalosi and Darnell, 2010). In humans, nearly all genes undergo
alternative pre-mRNA processing. Analyses of RNA-seq data indicate that at least 98% of multi-exon genes are alternatively spliced (Wang et al., 2008a). In addition to alternative splicing, an estimated 50% of human genes undergo alternative polyadenylation (Tian et al., 2005). Genes with alternative poly(A) sites are classified as having tandem UTRs or alternative last exons (ALEs) depending on the relative locations of alternative poly(A) signals (Figure 3A). Genes with tandem UTRs possess multiple poly(A) sites 3´ of the stop codon. Genes with
alternative last exons have at least two different terminal exons, the selection of which couples splicing with cleavage and polyadenylation.
Although there are numerous mechanisms of RNA 3´ end formation (Wilusz and Spector), mRNAs mature almost exclusively through the process of cleavage and
polyadenylation. Despite relatively simple catalytic requirements, the process of cleavage and polyadenylation requires at least 14 protein factors in mammals (Figure 3B). The poly(A) signal, AAUAAA and variants thereof, is recognized by CPSF160. However, AAUAAA is insufficient to specify 3´ end formation18. Efficient cleavage and polyadenylation requires additional
auxiliary sites upstream and downstream of the poly(A) signal (Figure 3B). These are
degenerate, U-rich sequences that bind complexes stabilizing CPSF recruitment, including the
endonuclease CPSF-73, to the poly(A) site. A solution structure of the CstF-64 RRM indicates a preference for binding UU dinucleotides (Perez Canadillas and Varani, 2003), consistent with the enrichment for U in the downstream sequence element. The dependencies on auxiliary
sequences create opportunities to regulate poly(A) site selection by improving or hindering recognition of these elements.
Alternative polyadenylation can be a highly dynamic process. An important example of regulated alternative polyadenylation comes from studies of immunoglobulin regulation in B-cells. Immature B-cells express a membrane bound form of IgM, whereas differentiated plasma B-cells express a secreted form of IgM. This variation is due to alternative polyadenylation of the IgM heavy chain mRNA (Alt et al., 1980; Takagaki et al., 1996). Use of a proximal alternative last exon results in the secreted form, whereas a distal poly(A) site generates the membrane bound form. B-cell activation was found to correlate with increased expression of CstF-64, which is critical for recognition of downstream U-rich elements (Takagaki et al., 1996). In addition, overexpression of this protein was sufficient to drive secretion of IgM. The
proximal site had apparently lower affinity for CstF-64, demonstrating how control over the concentration of a general factor can affect specific sites.
Recent studies have identified a number of conditions in which cells tend to express many transcripts with shorter UTR isoforms19. For example, depolarization of neurons leads to changes in UTR composition primarily through differential ALE usage (Flavell et al., 2008).
19 This is presumably due to changes in poly(A) site selection; however, because measurements
Activation of T-cells results in a global shift towards shorter UTRs, but this shift occurs primarily through changes in tandem UTRs (Sandberg et al., 2008). Expanding upon the observations made in T-cells, this study found a general correlation between proliferation and tandem UTR shortening. More recently, it was found that oncogenic transformation lead to more significant usage of short UTRs than cell lines with comparably proliferative cells (Mayr and Bartel, 2009). Importantly, it was shown that oncogene transcripts bearing shorter UTRs were more efficiently expressed, leading to more robust transformation.
Alternative splicing independent of polyadenylation can also affect the composition of 3´ UTRs. Alternative splicing events that change the reading frame typically generate
pre-termination codons (PTCs). The presence of a PTC often elicits a surveillance phenomenon known as nonsense-mediated decay (NMD)20. If the poly(A) site remains unchanged, PTCs effectively lengthen the 3´ UTR, sometimes referred to as a “faux UTR” (Amrani et al., 2004). Some models of NMD postulate a mechanism for sensing the distance from stop codon to poly(A) tail, whereby increasing distance elicits NMD. Other models propose that the presence of an exon junction complex21 3´ of the stop codon can allow discrimination between PTCs and “normal” stop codons (Le Hir et al., 2000; Moore, 2005). This is an attractive model since most 3´ UTRs lack introns, thus the presence of an EJC in a UTR context is unusual and provides specificity (Maquat, 2004). Like miRNA-mediated repression, NMD is likely an opportunistic
20
NMD was originally described as a quality control mechanism for clearing transcripts with nonsense mutations resulting from transcription errors or splicing errors, but is also used as a regulatory mechanism for down-regulating expression levels.
21
process that utilizes multiple regulatory pathways22 to downregulate targets. Intriguingly, there are a number of auto- and cross-regulatory loops controlling the levels of RNA binding proteins that utilize NMD as a regulatory mechanism (Lareau et al., 2007).
Advances in our understanding of the processing and regulation of genes all point to one conclusion: genes are complicated. Efforts to understand this complexity will undoubtedly reveal new and exciting facets of biology.
Figure Legends
Figure 1. A) RNAi in C. elegans. Long dsRNA from a variety of origins serves as a primary
trigger for generating siRNAs. The dsRNA is bound cooperatively by RDE-4. Binding can occur in any number of registers (two are shown). The relative position of RDE-4 binding determines where Dicing occurs and thus which discrete siRNAs are made. Diced siRNA duplexes are then loaded into RDE-1 (requiring the DExD helicase DRH-1, not shown). Targets are mRNAs with homology to the trigger dsRNA. Amplification of RNAi is associated with the formation of secondary siRNAs; these siRNAs are found exclusively 5´ of the region
homologous to the trigger. B) Trans-acting siRNA biogenesis in Arabidopsis. Shown is the pathway for TAS3. A non-coding transcript is targeted for cleavage by miR390. The RNA-dependent RNA polymerase RDR6 acts on the 5´ fragment to generate dsRNA. This dsRNA is then recursively cleaved by a DCL, measuring from the miR390-defined end. Resulting siRNAs are loaded into AGO. Two distinct siRNAs from TAS3 (colored yellow and orange) have significant complementarity to members of the Auxin Response Family of transcription factors, ARF3 and ARF4, and target their mRNAs for cleavage.
Figure 2. A) Biogenesis of metazoan miRNAs. Shown are the names of human orthologs of
processing factors. Pri-miRNA hairpins can be found in a variety of gene architectures.
Monocistronic miRNAs are often found in autonomously transcribed non-coding loci that can be spliced or unspliced. Mono- and polycistronic miRNAs occur in clusters within introns or exons of non-coding transcripts or within introns of coding transcripts. Pri-miRNA hairpins are
recognized by the Drosha:DGCR8 complex (termed the “microprocessor”) cotranscriptionally. DGCR8 recognizes pri-miRNA stems with its dsRNA binding domains, positioning Drosha for cleavage relative to the base of the stem in a process known as “cropping”. Exportin-5:Ran-GTP then recognizes the minihelix of the release pre-miRNA for export to the cytoplasm. Dicer in complex with a dsRNA binding partner recognize the pre-miRNA hairpin, measuring ~22 bp from the 3´ end of the stem for cleavage (dicing). The resulting duplex is then loaded into Ago and the passanger strand is either cleaved by Ago or dissociates. B) Critical regions for miRNA targeting. Shown are the seed site types and a schematic of the miRNA associated with Ago. The domains of Ago relative to the miRNA are labeled in grey. Fading 3´ nucleotides indicate these positions are buried in crystal structures of Argonaute in complex with a guide strand.
Figure 3. A) Classification of UTR types and their relationship to mRNA processing events.
Cleavage and polyadenylation sites are indicated by arrowheads. Thick boxes represent coding exons, thin boxes represent UTRs. B) Model of poly(A) site recognition in metazoans. Proteins known to associate into discrete complexes are boxed. Arrangement approximately reflects known binding locations. CFII(m), CPSF and CstF all have components known to associate with Pol II CTD.
References
Allen, E., Xie, Z., Gustafson, A.M., and Carrington, J.C. (2005). microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121, 207-221.
Alt, F.W., Bothwell, A.L., Knapp, M., Siden, E., Mather, E., Koshland, M., and Baltimore, D. (1980). Synthesis of secreted and membrane-bound immunoglobulin mu heavy chains is directed by mRNAs that differ at their 3' ends. Cell 20, 293-301.
Amrani, N., Ganesan, R., Kervestin, S., Mangus, D.A., Ghosh, S., and Jacobson, A. (2004). A faux 3'-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432, 112-118.
Aravin, A., Gaidatzis, D., Pfeffer, S., Lagos-Quintana, M., Landgraf, P., Iovino, N., Morris, P., Brownstein, M.J., Kuramochi-Miyagawa, S., Nakano, T., et al. (2006). A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203-207.
Aravin, A.A., Hannon, G.J., and Brennecke, J. (2007). The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science (New York, NY 318, 761-764. Aravin, A.A., Lagos-Quintana, M., Yalcin, A., Zavolan, M., Marks, D., Snyder, B., Gaasterland,
T., Meyer, J., and Tuschl, T. (2003). The small RNA profile during Drosophila melanogaster development. Developmental cell 5, 337-350.
Aravin, A.A., Naumova, N.M., Tulin, A.V., Vagin, V.V., Rozovsky, Y.M., and Gvozdev, V.A. (2001). Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr Biol 11, 1017-1027.
Aukerman, M.J., and Sakai, H. (2003). Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. The Plant cell 15, 2730-2741.
Baek, D., Villen, J., Shin, C., Camargo, F.D., Gygi, S.P., and Bartel, D.P. (2008). The impact of microRNAs on protein output. Nature 455, 64-71.
Bartel, D.P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281-297.
Bartel, D.P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215-233. Bartel, D.P., and Chen, C.Z. (2004). Micromanagers of gene expression: the potentially
widespread influence of metazoan microRNAs. Nature reviews 5, 396-400.
Bernstein, E., Caudy, A.A., Hammond, S.M., and Hannon, G.J. (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363-366.
Buchan, J.R., and Parker, R. (2009). Eukaryotic stress granules: the ins and outs of translation. Molecular cell 36, 932-941.
Carballo, E., Lai, W.S., and Blackshear, P.J. (1998). Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science (New York, NY 281, 1001-1005.
Carroll, S.B. (2000). Endless forms: the evolution of gene regulation and morphological diversity. Cell 101, 577-580.
Cech, T.R., Moras, D., Nagai, K., Williamson, J.R. (2006). The RNP World. In The RNA World, Third Edition, T. Gaasterland, Cech, T.R., Atkins, J.F., ed. (Cold Spring Harbor, Cold Spring Harbor Laboratory Press).
Cerutti, H., and Casas-Mollano, J.A. (2006). On the origin and functions of RNA-mediated silencing: from protists to man. Current genetics 50, 81-99.
Chen, C.Y., Xu, N., and Shyu, A.B. (1995). mRNA decay mediated by two distinct AU-rich elements from c-fos and granulocyte-macrophage colony-stimulating factor transcripts: different deadenylation kinetics and uncoupling from translation. Molecular and cellular biology 15, 5777-5788.
Chen, X. (2004). A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science (New York, NY 303, 2022-2025.
Chen, X. (2009). Small RNAs and their roles in plant development. Annual review of cell and developmental biology 25, 21-44.
Chendrimada, T.P., Gregory, R.I., Kumaraswamy, E., Norman, J., Cooch, N., Nishikura, K., and Shiekhattar, R. (2005). TRBP recruits the Dicer complex to Ago2 for microRNA
processing and gene silencing. Nature 436, 740-744.
Denli, A.M., Tops, B.B., Plasterk, R.H., Ketting, R.F., and Hannon, G.J. (2004). Processing of primary microRNAs by the Microprocessor complex. Nature 432, 231-235.
Elbashir, S.M., Lendeckel, W., and Tuschl, T. (2001). RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes & development 15, 188-200.
Enright, A.J., John, B., Gaul, U., Tuschl, T., Sander, C., and Marks, D.S. (2003). MicroRNA targets in Drosophila. Genome biology 5, R1.
Eulalio, A., Huntzinger, E., and Izaurralde, E. (2008). Getting to the root of miRNA-mediated gene silencing. Cell 132, 9-14.
Fire, A., Xu, S., Montgomery, M.K., Kostas, S.A., Driver, S.E., and Mello, C.C. (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806-811.
Flavell, S.W., Kim, T.K., Gray, J.M., Harmin, D.A., Hemberg, M., Hong, E.J., Markenscoff-Papadimitriou, E., Bear, D.M., and Greenberg, M.E. (2008). Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60, 1022-1038.
Forstemann, K., Horwich, M.D., Wee, L., Tomari, Y., and Zamore, P.D. (2007). Drosophila microRNAs are sorted into functionally distinct argonaute complexes after production by dicer-1. Cell 130, 287-297.
Forstemann, K., Tomari, Y., Du, T., Vagin, V.V., Denli, A.M., Bratu, D.P., Klattenhoff, C., Theurkauf, W.E., and Zamore, P.D. (2005). Normal microRNA maturation and germ-line stem cell maintenance requires Loquacious, a double-stranded RNA-binding domain protein. PLoS biology 3, e236.
Ghildiyal, M., and Zamore, P.D. (2009). Small silencing RNAs: an expanding universe. Nature reviews 10, 94-108.
Giraldez, A.J., Mishima, Y., Rihel, J., Grocock, R.J., Van Dongen, S., Inoue, K., Enright, A.J., and Schier, A.F. (2006). Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science (New York, NY 312, 75-79.
Girard, A., Sachidanandam, R., Hannon, G.J., and Carmell, M.A. (2006). A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442, 199-202.
Gregory, R.I., Yan, K.P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., and Shiekhattar, R. (2004). The Microprocessor complex mediates the genesis of microRNAs. Nature 432, 235-240.
Grimson, A., Farh, K.K., Johnston, W.K., Garrett-Engele, P., Lim, L.P., and Bartel, D.P. (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing.
Grimson, A., Srivastava, M., Fahey, B., Woodcroft, B.J., Chiang, H.R., King, N., Degnan, B.M., Rokhsar, D.S., and Bartel, D.P. (2008). Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455, 1193-1197.
Grishok, A., Pasquinelli, A.E., Conte, D., Li, N., Parrish, S., Ha, I., Baillie, D.L., Fire, A.,
Ruvkun, G., and Mello, C.C. (2001). Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106, 23-34.
Groisman, I., Jung, M.Y., Sarkissian, M., Cao, Q., and Richter, J.D. (2002). Translational control of the embryonic cell cycle. Cell 109, 473-483.
Hamilton, A.J., and Baulcombe, D.C. (1999). A species of small antisense RNA in
posttranscriptional gene silencing in plants. Science (New York, NY 286, 950-952. Han, J., Lee, Y., Yeom, K.H., Kim, Y.K., Jin, H., and Kim, V.N. (2004). The Drosha-DGCR8
complex in primary microRNA processing. Genes & development 18, 3016-3027. Han, J., Lee, Y., Yeom, K.H., Nam, J.W., Heo, I., Rhee, J.K., Sohn, S.Y., Cho, Y., Zhang, B.T.,
and Kim, V.N. (2006). Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125, 887-901.
Houseley, J., and Tollervey, D. (2009). The many pathways of RNA degradation. Cell 136, 763-776.
Hutvagner, G., and Simard, M.J. (2008). Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol 9, 22-32.
Hutvagner, G., and Zamore, P.D. (2002). A microRNA in a multiple-turnover RNAi enzyme complex. Science (New York, NY 297, 2056-2060.
Jacob, F., and Monod, J. (1961). Genetic regulatory mechanisms in the synthesis of proteins. Journal of molecular biology 3, 318-356.
Jones-Rhoades, M.W., Bartel, D.P., and Bartel, B. (2006). MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol 57, 19-53.
Kim, J.H., and Richter, J.D. (2006). Opposing polymerase-deadenylase activities regulate cytoplasmic polyadenylation. Molecular cell 24, 173-183.
Kim, V.N., Han, J., and Siomi, M.C. (2009). Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10, 126-139.
Kim, Y.K., and Kim, V.N. (2007). Processing of intronic microRNAs. The EMBO journal 26, 775-783.
Kim-Ha, J., Kerr, K., and Macdonald, P.M. (1995). Translational regulation of oskar mRNA by bruno, an ovarian RNA-binding protein, is essential. Cell 81, 403-412.
Knight, S.W., and Bass, B.L. (2001). A role for the RNase III enzyme DCR-1 in RNA
interference and germ line development in Caenorhabditis elegans. Science (New York, NY 293, 2269-2271.
Lagos-Quintana, M., Rauhut, R., Lendeckel, W., and Tuschl, T. (2001). Identification of novel genes coding for small expressed RNAs. Science (New York, NY 294, 853-858. Landthaler, M., Yalcin, A., and Tuschl, T. (2004). The human DiGeorge syndrome critical
region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol 14, 2162-2167.
Lareau, L.F., Inada, M., Green, R.E., Wengrod, J.C., and Brenner, S.E. (2007). Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446, 926-929.
Lau, N.C., Lim, L.P., Weinstein, E.G., and Bartel, D.P. (2001). An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science (New York, NY 294, 858-862.
Lau, N.C., Seto, A.G., Kim, J., Kuramochi-Miyagawa, S., Nakano, T., Bartel, D.P., and
Kingston, R.E. (2006). Characterization of the piRNA complex from rat testes. Science (New York, NY 313, 363-367.
Le Hir, H., Izaurralde, E., Maquat, L.E., and Moore, M.J. (2000). The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. The EMBO journal 19, 6860-6869.
Lecuyer, E., Yoshida, H., Parthasarathy, N., Alm, C., Babak, T., Cerovina, T., Hughes, T.R., Tomancak, P., and Krause, H.M. (2007). Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131, 174-187. Lee, R.C., and Ambros, V. (2001). An extensive class of small RNAs in Caenorhabditis elegans.
Science (New York, NY 294, 862-864.
Lee, R.C., Feinbaum, R.L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843-854. Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., Lee, J., Provost, P., Radmark, O., Kim, S.,
et al. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415-419.
Lee, Y., Jeon, K., Lee, J.T., Kim, S., and Kim, V.N. (2002). MicroRNA maturation: stepwise processing and subcellular localization. The EMBO journal 21, 4663-4670.
Lee, Y., Kim, M., Han, J., Yeom, K.H., Lee, S., Baek, S.H., and Kim, V.N. (2004). MicroRNA genes are transcribed by RNA polymerase II. The EMBO journal 23, 4051-4060. Lehmann, R., and Nusslein-Volhard, C. (1986). Abdominal segmentation, pole cell formation,
and embryonic polarity require the localized activity of oskar, a maternal gene in Drosophila. Cell 47, 141-152.
Lewis, B.P., Burge, C.B., and Bartel, D.P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15-20.
Lewis, B.P., Shih, I.H., Jones-Rhoades, M.W., Bartel, D.P., and Burge, C.B. (2003). Prediction of mammalian microRNA targets. Cell 115, 787-798.
Licatalosi, D.D., and Darnell, R.B. (2010). RNA processing and its regulation: global insights into biological networks. Nature reviews 11, 75-87.
Lim, L.P., Lau, N.C., Garrett-Engele, P., Grimson, A., Schelter, J.M., Castle, J., Bartel, D.P., Linsley, P.S., and Johnson, J.M. (2005). Microarray analysis shows that some
microRNAs downregulate large numbers of target mRNAs. Nature 433, 769-773. Lingel, A., Simon, B., Izaurralde, E., and Sattler, M. (2003). Structure and nucleic-acid binding
of the Drosophila Argonaute 2 PAZ domain. Nature 426, 465-469.
Liu, J., Carmell, M.A., Rivas, F.V., Marsden, C.G., Thomson, J.M., Song, J.J., Hammond, S.M., Joshua-Tor, L., and Hannon, G.J. (2004). Argonaute2 is the catalytic engine of
mammalian RNAi. Science (New York, NY 305, 1437-1441.
Llave, C., Xie, Z., Kasschau, K.D., and Carrington, J.C. (2002). Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science (New York, NY 297, 2053-2056.
Macrae, I.J., Zhou, K., Li, F., Repic, A., Brooks, A.N., Cande, W.Z., Adams, P.D., and Doudna, J.A. (2006). Structural basis for double-stranded RNA processing by Dicer. Science (New York, NY 311, 195-198.
Mallory, A.C., Reinhart, B.J., Jones-Rhoades, M.W., Tang, G., Zamore, P.D., Barton, M.K., and Bartel, D.P. (2004). MicroRNA control of PHABULOSA in leaf development:
importance of pairing to the microRNA 5' region. The EMBO journal 23, 3356-3364. Mallory, A.C., and Vaucheret, H. (2006). Functions of microRNAs and related small RNAs in
plants. Nature genetics 38 Suppl, S31-36.
Maquat, L.E. (2004). Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5, 89-99.
Martin, K.C., and Ephrussi, A. (2009). mRNA localization: gene expression in the spatial dimension. Cell 136, 719-730.
Martinez, J., Patkaniowska, A., Urlaub, H., Luhrmann, R., and Tuschl, T. (2002). Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell 110, 563-574. Matranga, C., Tomari, Y., Shin, C., Bartel, D.P., and Zamore, P.D. (2005). Passenger-strand
cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 123, 607-620.
Mayr, C., and Bartel, D.P. (2009). Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673-684.
Merritt, C., Rasoloson, D., Ko, D., and Seydoux, G. (2008). 3' UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol 18, 1476-1482.
Moore, M.J. (2005). From birth to death: the complex lives of eukaryotic mRNAs. Science (New York, NY 309, 1514-1518.
Morlando, M., Ballarino, M., Gromak, N., Pagano, F., Bozzoni, I., and Proudfoot, N.J. (2008). Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol 15, 902-909.
Nakamura, A., Sato, K., and Hanyu-Nakamura, K. (2004). Drosophila cup is an eIF4E binding protein that associates with Bruno and regulates oskar mRNA translation in oogenesis. Developmental cell 6, 69-78.
Napoli, C., Lemieux, C., and Jorgensen, R. (1990). Introduction of a Chimeric Chalcone
Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. The Plant cell 2, 279-289.
Nykanen, A., Haley, B., and Zamore, P.D. (2001). ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell 107, 309-321.
Olsen, P.H., and Ambros, V. (1999). The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Developmental biology 216, 671-680.
Pak, J., and Fire, A. (2007). Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science (New York, NY 315, 241-244.
Park, W., Li, J., Song, R., Messing, J., and Chen, X. (2002). CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol 12, 1484-1495.
Parker, G.S., Eckert, D.M., and Bass, B.L. (2006). RDE-4 preferentially binds long dsRNA and its dimerization is necessary for cleavage of dsRNA to siRNA. RNA (New York, NY 12, 807-818.