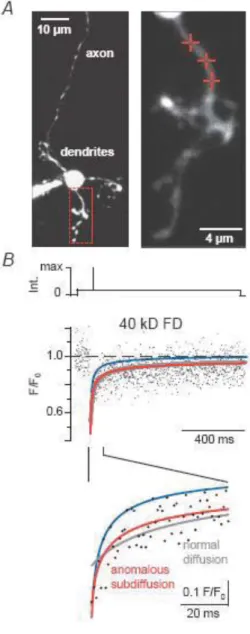
Restricted diffusion of calretinin in cerebellar granule cell dendrites implies Ca²⁺-dependent interactions via its EF-hand 5 domain
28
0
0
Texte intégral
Figure
Documents relatifs