HAL Id: tel-01449206
https://tel.archives-ouvertes.fr/tel-01449206
Submitted on 30 Jan 2017HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Rôle de la Thrombospondine-1 et du récepteur CD47
dans le développement de la fibrose rénale
Naïke Bige
To cite this version:
Naïke Bige. Rôle de la Thrombospondine-1 et du récepteur CD47 dans le développement de la fibrose rénale. Physiologie [q-bio.TO]. Université Pierre et Marie Curie - Paris VI, 2014. Français. �NNT : 2014PA066705�. �tel-01449206�
Université Pierre et Marie Curie
Ecole doctorale 394 « Physiologie, Physiopathologie et Thérapeutique »
Laboratoire INSERM-UMRS 1155
« Des maladies rénales rares aux maladies fréquentes, remodelage et réparation »
Rôle de la Thrombospondine-1 et du récepteur CD47
dans le développement de la fibrose rénale
Par Naïke Bigé
Thèse de doctorat de Physiologie et Physiopathologie
Dirigée par Monsieur le Professeur Jean-Jacques Boffa
Présentée et soutenue publiquement le 25 septembre 2014
Devant un jury composé de
Monsieur le Professeur Alexandre Hertig, PU-PH, président du jury Monsieur le Professeur Philippe Grimbert, PU-PH, rapporteur Monsieur le Professeur Hans-Kristian Lorenzo, MCU-PH, rapporteur
Madame le Professeur Marie-Noelle Peraldi-Gardin, PU-PH, membre du jury Monsieur le Professeur Ulrich Blank, DR-HDR, membre du jury
4
REMERCIEMENTS
Je remercie le Professeur Alexandre Hertig pour les connaissances néphrologiques et scientifiques qu’il m’a apportées et pour avoir accepté de présider cette thèse.
Je remercie le Professeur Philippe Grimbert pour ses commentaires pertinents sur ce travail à mi-parcours et pour avoir accepté d’en être rapporteur et membre du jury.
Je remercie le Professeur Hans Kristian Lorenzo pour avoir accepté d’être rapporteur de ce travail et membre du jury et pour ses suggestions pertinentes qui m’ont permis d’améliorer le manuscrit.
Je remercie le Professeur Marie-Noëlle Peraldi-Gardin pour l’enseignement qu’elle m’a prodigué tout au long de mon internat de néphrologie et au-delà, et pour avoir d’être accepté avec enthousiasme d’être membre du jury.
Je remercie le Professeur Ulrich Blank d’avoir accepté d’être membre du jury.
Je remercie le Professeur Jean-Jacques Boffa pour les connaissances néphrologiques médicales et scientifiques qu’il m’a transmises, pour m’avoir appris à prendre soin des patients avec humanité et humilité, pour m’avoir donné l’opportunité de réaliser cette deuxième thèse avec lui, pour la pertinence de sa réflexion scientifique et pour m’avoir guidée et encadrée dans ce travail de recherche avec tant de disponibilité et de gentillesse. J’espère que notre collaboration se poursuivra.
Je remercie le Professeur Pierre Ronco pour son enseignement de la Néphrologie tant médical que scientifique, pour la rigueur de sa réflexion scientifique et médicale et pour m’avoir permis de réaliser ce travail de recherche au sein de l’unité.
Je remercie le Professeur Christos Chatziantoniou pour son enseignement, pour m’avoir permis de réaliser ce travail de recherche au sein de l’unité et pour ses suggestions qui m’ont aidée dans la réalisation de ce travail.
Je remercie le Professeur Jean-Claude Dussaule pour les connaissances scientifiques qu’il m’a transmises et ses suggestions pertinentes qui m’ont aidée dans la réalisation de ce travail.
5 Je remercie Madame Chantal Jouanneau pour son apprentissage toujours passionné des techniques histologiques, son aide précieuse dans la réalisation de ce travail, sa disponibilité et ses encouragements pendant ces cinq années passées au laboratoire.
Je remercie Madame Sophie Vandermeersch pour son apprentissage des techniques de biologie moléculaire toujours dans la bonne humeur, sa disponibilité et son aide précieuse dans la réalisation de ce travail, sa bienveillance et sa gentillesse sans faille pendant ces cinq ans.
Je remercie Madame Sandrine Placier pour son aide tout au long de ce travail, qui fût indispensable dans la mise au point et la réalisation de la microscopie intravitale, pour son sa sympathie et son humour.
Je remercie le Docteur Fanny Lepeytre pour son implication et son aide précieuse dans ce travail, sa curiosité, son humour et sa spontanéité.
Je remercie le Docteur Jonathan Caron pour son aide précieuse dans ce travail, sa curiosité scientifique, médicale et culinaire et son humour parfois déroutant mais toujours au rendez-vous.
Je remercie Monsieur Emmanuel Douez pour sa motivation et son aide précieuse dans ce travail.
Je remercie le Docteur Nasim Shweke d’avoir ouvert la voie de la Thrombospondine 1. Je remercie Mesdames Caroline Martin, Jessy Renciot et Charlène Marmier et Monsieur Claude Kitou pour avoir pris soin des animaux et pour leur aide dans ce travail.
Je remercie Lara, Dom Guerrot, Pierre, Emmanuel, Laurent, Christos Junior, Alexeï, Mouna, Fala, Zeyneb, Tiffany, Aude, Ahmed, Timothée, Dom Cathelin, Guillaume, Yosu, Carlo, Boris, Cédric, Julie, Perrine, Clélia, Claire, Claudia, Christine, Marie-Christine, Catherine, Brigitte, Béatrice, Marie-Christine, Ychun, Edith et tous les membres de l’unité 702 puis 1155 pour les connaissances scientifiques que nous avons échangées, leur aide dans la réflexion scientifique, leur sympathie, leur bonne humeur et leur soutien.
6 Je remercie mes maîtres et collègues des services de Réanimation de Saint-Antoine, les Professeurs Bertrand Guidet, Eric Maury, Hafid Ait Oufella et Georges Offenstadt, et les docteurs Claire Pichereau, Mikael Alves, Arnaud Galbois, Ly Van Vong et Jean-Luc Baudel, et Saint-Louis, les Professeurs Elie Azoulay et Benoît Schlemmer et les docteurs Virginie Lemiale, Sandrine Valade, Marion Venot, Danielle Reuter, Emmanuel Canet et Guillaume Dumas de m’avoir permis de poursuivre ce travail de recherche pendant mon clinicat et pour leur soutien pendant ces quatre ans. Avec un merci tout particulier aux Docteurs Sandrine Valade, Marion Venot et Guillaume Dumas et Monsieur Igor Théodose pour leur soutien inconditionnel dans la dernière ligne droite.
Je remercie mes parents pour m’avoir donné la chance d’arriver jusqu’ici, pour avoir éveillé ma curiosité intellectuelle et mon esprit critique, pour m’avoir soutenue et encouragée dans les moments difficiles avec une grande bienveillance et toujours beaucoup d’humour.
Je remercie Yann et mes amis, Agnès, Claire, Vanessa, Emilie, la Kaout’ et la Mich’, Fannaille, la « Ritchie Family » qui s’est maintenant agrandie, Mayan, Ju, ma filleule Charlotte et sa petite sœur Elisa, Carole et son merveilleux Julien, Emilie, Gab et Cécile, Tibou, Jerem, Thuy et leur petit Paul pour leur soutien et pour tous les bons moments passés et à venir avec eux.
7
SOMMAIRE
ABBREVIATIONS ...11
INTRODUCTION ...12
I. Maladies rénales chroniques et fibrose rénale ...12
A. Epidémiologie des maladies rénales chroniques ...12
B. Mécanismes impliqués dans la fibrose rénale ...12
1. Rôle de l’inflammation ...13
2. Activation des myofibroblastes et production de matrice extra-cellulaire ...14
3. Altérations vasculaires...15
4. Rôle du TGF-β1 ...16
II. La Thrombospondine-1 ...18
A. Structure et partenaires ...18
B. Fonctions ...19
1. Adhésion et agrégation plaquettaire ...19
2. Activation du TGF-β1 ...20
3. Activité anti-angiogénique et modulation du tonus vasculaire ...21
4. Activité immunomodulatrice ...24
C. Rôle de la Thrombospondine-1 dans le développement des néphropathies expérimentales ...26
1. Par l’activation du TGF-β1 ...27
2. Par son activité anti-angiogénique...28
3. Par son activité anti-inflammatoire ...28
D. Thrombospondine-1 et pathologie rénale chez l’homme ...29
III. Le récepteur CD47 ...30
A. Structure et expression ...30
8 1. Thrombospondine-1 ...31 2. SIRPα et SIRPγ ...31 3. Intégrines ...32 C. Fonctions ...32 1. Phagocytose ...32 2. Mort cellulaire ...32 3. Effet immunomodulateur ...33 4. Effet anti-angiogénique ...34 D. CD47 et pathologie rénale ...35 OBJECTIFS ...37 RESULTATS ...38
I. Première partie : la Thrombospondine-1 joue un rôle pro-fibrosant et pro-inflammatoire au cours de l’obstruction urétérale ...39
A. Article “ Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction” ...40
1. Résumé ...40
2. Article...41
3. Figure supplémentaire ...55
4. Matériels et méthodes supplémentaires ...56
a. Animals and tissue samples ...56
b. Assessment of renal function ...56
c. Assessment of renal morphology ...56
d. Immunohistochemistry and Immunofluorescence ...57
e. In Situ End-Labeling for the Detection of Apoptotic Cells ...58
9 g. Determination of Tissue TGF-β1 and VEGF Concentrations by Enzyme-Linked
Immunosorbent Assay ...58
h. Western Blot Analysis ...59
i. Total RNA Extraction and Quantitative Real Time PCR ...59
j. Statistical Analyses ...60
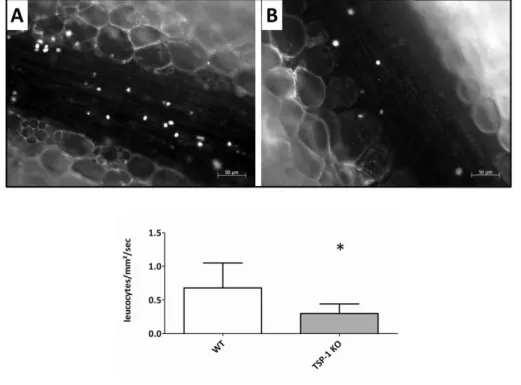
B. La Thrombospondine-1 favorise le rolling leucocytaire ...61
1. Matériels et méthodes ...61
2. Résultats ...62
II. Deuxième partie : Le récepteur CD47 participe aux altérations tubulaires et vasculaires au cours de l’obstruction urétérale ...63
A. Matériels et méthodes ...63
1. Animaux ...63
2. Extraction des ARN messagers, transcription inverse et PCR en temps réel ...63
3. Histologie et immunohistochimie ...64
4. ELISA TGF-β1 ...66
5. Analyses statistiques ...66
B. Résultats ...66
1. L’obstruction urétérale modifie l’expression de CD47 ...66
2. Le déficit génétique en CD47 limite la souffrance tubulaire après obstruction urétérale ...67
3. Le déficit génétique en CD47 limite les altérations vasculaires induites par l’obstruction urétérale ...68
4. Le déficit génétique en CD47 majore la fibrose induite par l’obstruction urétérale ...72
5. Le déficit génétique en CD47 majore l’expression de la Thrombospondine-1 et du TGF-β1 après obstruction urétérale ...72
10 6. Effet du déficit génétique en CD47 sur l’inflammation induite par l’obstruction
urétérale ...75
III. Troisième partie : Etude de l’expression de la Thrombospondine-1 au cours de la néphropathie hypertensive expérimentale ...77
A. Matériels et méthodes ...77
1. Animaux ...77
2. Histologie, immunohistochimie et immunofluorescence ...78
3. RT-qPCR ...79
4. Statistiques ...79
B. Résultats ...81
1. Lésions rénales induites par l’hypertension artérielle chez le rat ...81
2. Expression de la Thrombospondine-1 au cours de la néphropathie hypertensive chez le rat ...83
3. Effet du déficit génétique en Thrombospondine-1 sur le développement de la néphropathie vasculaire chez la souris ...88
DISCUSSION ...93
CONCLUSION ... 103
PERSPECTIVES... 105
REFERENCES ... 108
11
ABBREVIATIONS
αSMA: α Smooth Muscle Actin
ARNm: Acide ribonucléique messager AU: Unité arbitraire
Col3 : Collagène III CTRL: contrôle
ELISA: Enzyme Linked Immunosorbent Assay HTA: hypertension artérielle
KIM-1: Kidney Injury Molecule-1 KO: Knock-out
L-NAME: L-Nitro-Arginine-Methyl-Esther MCP-1: Monocyte Chemoattractant Protein-1 MEC: Matrice extra-cellulaire
MECA: Mouse Endothelial Cell Antigen NO: monoxyde d’azote
RT-qPCR : Reverse Transcription – quantitative Polymerase Chain Reaction TGF-β1: Transforming Growth Factor β
TSP-1: Thrombospondine-1
UUO: Obstruction urétérale unilatérale
VEGF-A: Vascular Endothelial Growth Factor A
12
INTRODUCTION
Malgré les traitements néphroprotecteurs, l’évolution des maladies rénales chroniques vers l’insuffisance rénale terminale reste inéluctable. Elle s’associe au développement d’une fibrose interstitielle qui se caractérise par une atrophie tubulaire, une raréfaction capillaire et l’accumulation de protéines de la matrice extra-cellulaire. Les traitements actuels permettent de ralentir l’extension de la fibrose mais ni de l’interrompre ni de la faire régresser. Afin d’identifier de nouvelles cibles thérapeutiques, il est indispensable de mieux comprendre les mécanismes impliqués dans la physiopathologie de la fibrose rénale. Nous nous proposons d’étudier le rôle de la Thrombospondine-1 (TSP-1) et de l’un de ces récepteurs, le CD47, dans ce processus.
I. Maladies rénales chroniques et fibrose rénale
A. Epidémiologie des maladies rénales chroniques
Les maladies rénales chroniques représentent un problème mondial de santé publique. En effet, l’incidence et la prévalence de l’insuffisance rénale chronique terminale sont en constante augmentation aux Etats-Unis comme en Europe 1–7. Quelle que soit leur étiologie, les maladies rénales chroniques partagent une évolution commune vers le développement d’une fibrose envahissant et détruisant progressivement le parenchyme rénal. Elle conduit ultimement au stade terminal de l’insuffisance rénale nécessitant un traitement de suppléance tel que la dialyse ou la transplantation rénale. En France, en 2012, l’incidence et la prévalence de l’insuffisance rénale chronique terminale s’élevaient respectivement à 154 et 1127 personnes par million d’habitants 7, contre 139 et 929 par million d’habitants en 2005 6.
B. Mécanismes impliqués dans la fibrose rénale
La fibrose est la conséquence d’un remodelage tissulaire inadapté après une agression tissulaire conduisant à des altérations morphologiques et fonctionnelles des cellules rénales 8,9. Les mécanismes impliqués dans ce processus sont multiples et complexes et font l’objet de nombreux travaux scientifiques. Ils sont résumés dans la Figure 1. Les
13 principaux événements impliqués sont les suivants : l’infiltration du parenchyme par des cellules inflammatoires, l’activation et l’expansion des myofibroblastes, la production de matrice extra-cellulaire (MEC), la raréfaction des capillaires péri-tubulaires, l’atrophie tubulaire et la gloméruloscérose 10,11. Nous détaillerons plus précisément les mécanismes mettant potentiellement en jeu la TSP-1 et le CD47.
1. Rôle de l’inflammation
L’inflammation joue un rôle majeur dans l’initiation et la pérennisation du processus de fibrose rénale. L’endocytose et la phagocytose des débris cellulaires générés par l’agression initiale induit le recrutement de cellules inflammatoires, incluant des polynucléaires neutrophiles, des monocytes/macrophages, des cellules dendritiques et des lymphocytes qui envahissent le parenchyme rénal. Une fois activées, ces cellules produisent des cytokines et des facteurs de croissance pro-fibrosants, tels que le TGF-β1 (Transforming Growth Factor- β1) 12,13. Ce dernier exerce également un effet chimiotactique et renforce le recrutement des monocytes/macrophages, de même que les facteurs MCP-1 (Monocyte Chemoattractant Protein-1) 14 et MIP-1 et -2 (Macrophage Inflammatory Protein-1 et -2)
10,11,15.
Les macrophages jouent un rôle clef dans le développement de la fibrose rénale. Plusieurs stratégies d’inhibition des macrophages ont permis de réduire l’inflammation rénale et l’accumulation de MEC dans différents modèles expérimentaux de néphropathie
11,15
. Schématiquement, deux phénotypes de macrophages peuvent être distingués: les macrophages de type M1 activés par l’interféron γ et la voie Th1 et les macrophages de type M2 « désactivés » après exposition aux cytokines Th2. Les macrophages de type M1 favorisent le développement de la fibrose rénale. Ils sécrètent du TNFα (Tumor Necrosis Factor α) qui induit l’apoptose des cellules épithéliales tubulaires. En sécrétant des facteurs de croissance pro-fibrosant, tels que TGF-β1, le PDGF (Platelet Derived Growth Factor), le CTGF (Connective Tissue Growth Factor) et le FGF-2 (Fibroblast Growth Factor 2 ou basic Fibroblast Growth Factor), ils stimulent la transition épithélio- et endothélio-mésenchymateuse et la production de MEC par les myofibroblastes. A l’inverse, les macrophages de type M2 favoriseraient la résolution de l’inflammation et la réparation
14 rénale en limitant l’activation des macrophages M1 et des lymphocytes T effecteurs et en favorisant la différenciation des lymphocytes T régulateurs 16,17.
Les lymphocytes T sont également impliqués dans le processus de fibrose rénale mais leur rôle a été moins étudié. Dans le modèle d’obstruction urétérale unilatérale, la déplétion en lymphocytes T CD4+ limite les lésions de fibrose tandis que le transfert adoptif de lymphocytes T CD4+, mais pas T CD8+, chez des souris déficientes en lymphocytes restaure la fibrose rénale 18. De plus, la voie Th2 favoriserait le développement de la fibrose rénale à un degré plus important que la voie Th1 19. Des travaux récents ont également impliqué la voie Th17 dans la physiopathologie de glomérulonéphrites prolifératives 20–24 mais son rôle dans le processus de fibrose reste à déterminer.
2. Activation des myofibroblastes et production de matrice extra-cellulaire
Dans le parenchyme rénal normal, les fibroblastes siègent dans l’interstitium et maintiennent l’homéostasie de la MEC en produisant ses composants à un niveau basal. Ils expriment la protéine FSP-1 (Fibroblast specific Protein-1, ou S1004). Après une agression rénale, le TGF-β1 (Transforming Growth Factor β), le PDGF (Platelet Derived Growth Factor), le CTGF (Connective Tissue Growth Factor) et le FGF-2 (Fibroblast Growth Factor 2 ou basic Fibroblast Growth Factor) activent les fibroblastes qui deviennent alors des myofibroblastes, caractérisés par l’expression de l’actine du muscle lisse (αSMA : α smooth muscle actin). Ils produisent alors de grandes quantités de fibronectine et des collagènes I et III qui vont s’accumuler dans l’interstitium et les glomérules. L’origine des myofibroblastes dans le parenchyme rénal reste controversée. Initialement considérés comme issus de la différenciation de fibroblastes résidents, ils proviendraient également de la transformation d’autres cellules résidentes du parenchyme rénal et de cellules circulantes d’origine hématopoïétique. Sous l’influence de l’angiotensine II, de différents facteurs de croissance et cytokines (TGF-β1, FGF-2, CTGF et interleukine-1), et des métalloprotéases MMP-2 et MMP-9, les cellules épithéliales tubulaires peuvent acquérir le phénotype de myofibroblastes. Ce processus appelé transition épithélio-mésenchymateuse implique les voies de signalisation TGF-β1/Smad, intégrines/ILK et Wnt/β caténines 25,26. Les podocytes 27
15 et les cellules endothéliales 28 pourraient également évoluer vers un phénotype fibroblastique. Enfin, une proportion importante de myofibroblastes semble dérivée de cellules souches hématopoïétiques 29.
3. Altérations vasculaires
La fibrose rénale s’accompagne d’une raréfaction des capillaires péri-tubulaires mais aussi d’anomalies fonctionnelles des cellules endothéliales conduisant à une hypoxie chronique, participant au stress oxydatif, au recrutement des cellules inflammatoires et à la production de MEC 30 .
Au cours des maladies rénales chroniques, la diminution de la production de monoxyde d’azote (NO) favorise l’hypertension artérielle et le développement de lésions vasculaires et de la fibrose. Ce déficit est lié à une diminution de la biodisponibilité de l’arginine, le substrat de la NO synthase, et à une augmentation des inhibiteurs endogènes circulants de la NO synthase, en particulier la diméthylarginine asymétrique (ADMA). Le taux plasmatique d’ADMA est corrélé à la progression des maladies rénales chroniques et à la survenue d’événements cardio-vasculaires 31. L’inhibition de la NO synthase, par l’intermédiaire de l’endothéline, induit la sécrétion d’angiotensine II et la production de collagène I 32,33. Le découplage de la NO synthase endothéliale peut également conduire à la production de radicaux libres dérivés de l’oxygène. Le stress oxydatif ainsi généré induit des lésions cellulaires et le recrutement de cellules inflammatoires. Enfin, la diminution de la biodisponibilité du NO limite l’adaptation du tonus de la microcirculation rénale et favorise ainsi l’hypoxie tissulaire 30,31.
La raréfaction des capillaires péri-tubulaires, qui est un trait commun à toutes les maladies rénales chroniques, participe également à l’hypoxie cellulaire. Elle résulte d’un déséquilibre de l’angiogénèse induit par la diminution de facteurs pro-angiogéniques, tels que le vascular endothelial growth factor (VEGF), et l’augmentation de l’expression de facteurs anti-angiogéniques, tels que l’endostatine, l’angiostatine et la TSP-1 34. Les altérations du réseau capillaire péri-tubulaire sont corrélées étroitement aux lésions du compartiment tubulo-interstitiel 30 et au déclin de la fonction rénale 34.
16 La dysfonction endothéliale observée au cours des maladies rénales chroniques favorise l’inflammation tissulaire. Après une agression rénale, les cellules endothéliales activées, notamment par le TNFα, expriment la E- sélectine et la P-sélectine permettant le rolling des leucocytes, les molécules ICAM-1 et VCAM-1, induisant l’adhésion ferme des leucocytes à l’endothelium, et PECAM conduisant à leur transmigration 30.
Enfin, des travaux récents ont montré que les cellules endothéliales rénales pouvaient perdre leurs marqueurs spécifiques et exprimer des marqueurs mésenchymateux tels que FSP-1, αSMA et le collagène I suggérant l’existence d’une transition endothélio-mésenchymateuse 28.
4. Rôle du TGF-β1
Parmi les nombreux facteurs profibrosants impliqués dans le processus de fibrose rénale, le TGF-β1 joue un rôle clef. A l’état basal, le TGF-β1 est présent dans la MEC principalement sous forme latente. En réponse à une agression tissulaire, le TGF-β1 est libéré de ses peptides de latence pour se fixer sur ses récepteurs, activer sa voie de signalisation et induire la transcription de nombreux gènes régulant l’activation, la prolifération, l’apoptose, l’adhésion et la migration de nombreux types cellulaires 35. L’angiotensine II, qui joue un rôle majeur dans le développement des maladies rénales chroniques, induit l’activation et la sécrétion de TGF-β1 à différents niveaux 36.
Le TGF-β1 promeut la production de composants de la MEC par les cellules mésangiales, les fibroblastes et les cellules épithéliales tubulaires et empêche leur dégradation par les métalloprotéases 37. Il peut induire l’apoptose des cellules épithéliales tubulaires, des podocytes et des cellules endothéliales 35. En induisant la production de MCP-1, il participe au recrutement des monocytes/macrophages 38. De plus, l’existence d’une boucle d’amplification entre TGF-β1 et MCP-1 a été mise en évidence dans un modèle murin de glomérulonéphrite 39. Enfin, il module certaines voies de l’inflammation induites au cours de la fibrose rénale 35.
L’expression rénale du TGF-β1 et son taux urinaire sont corrélés à l’intensité de la fibrose tubulo-interstitielle chez des patients présentant une maladie rénale chronique 40. Plusieurs études ont démontré que l’inactivation du TGF-β1 41 et de la protéine Smad3 42, qui
17 transduit son signal, limite le développement de la fibrose au cours d’une néphropathie expérimentale. De plus, l’inactivation de la protéine Smad7 qui inhibe sa voie de signalisation contribue à l’accumulation de MEC dans un modèle d’obstruction urétérale unilatérale 43.
Figure 1. Mécanismes conduisant à la progression des maladies rénales chroniques vers la fibrose rénale (d’après Dussaule et al. Int. J. Exp. Pathol. 2011 9).
Les facteurs pro-fibrosants tels que le TGF-β1, l’endothéline et l’angiotensine II induisent des altérations morphologiques et fonctionnelles des cellules des différents compartiments rénaux orientent leur expression génique et protéique vers un phénotype fibrosant.
18
II. La Thrombospondine-1 A. Structure et partenaires
La famille des Thrombospondines (TSP) comporte cinq membres répartis en deux groupes. Le groupe A réunit la Thrombospondine-1 (TSP-1) et la thrombospondine 2 (TSP-2) qui sont des glycoprotéines homotrimériques de structure homologues comportant un domaine de séquences répétées de type 1 (TSR1) et une séquence homologue à celle du procollagène. Les TSP 3 à 5, qui constituent le groupe B, se distinguent par leur structure homopentamérique comportant une séquence répétée de type 2 (TSR2) supplémentaire et par l’absence de TSR1 et du domaine procollagénique.
La TSP-1, première décrite en 1971, a été identifiée par analyse du contenu des granules α de plaquettes stimulées par la thrombine 44. Il s’agit d’une glycoprotéine homotrimérique de 450 kDa qui peut être exprimée et/ou sécrétée dans la MEC par de nombreux types cellulaires. Ses différents domaines lui permettent d’interagir avec de nombreux partenaires et lui confèrent ainsi des activités biologiques variées (Figure 2) 45–47.
Son domaine N-terminal constitué d’environ 200 acides aminés, se lie à l’héparine, aux protéoglycanes et aux intégrines α3β1, α4β1, α5β1 et α6β1. Ce domaine assure également la clairance de la TSP-1 par le récepteur à lipoprotéine de faible densité (LRP).
Il existe ensuite un domaine d’oligomérisation suivi d’un domaine procollagénique, également nommé domaine de type C de Von Willebrand (VWC), qui participe à l’activité anti-angiogénique de la TSP-1.
Le domaine suivant TSR1 (domaine de répétition de type 1) est constitué du motif TSPc répété trois fois et contient les deux séquences indispensables à l’activation du TGF-β1. Il participe aussi à l’activité anti-angiogénique de la TSP-1 en interagissant avec les intégrines β1 et le CD36.
Enfin, la partie carboxy-terminale de la TSP-1, appelée domaine de signature, hautement conservée au sein de la famille des TSP, comporte des domaines EGF (Epidermal Growth Factor)-like répétés trois (TSR2), suivis de treize séquences de fixation au calcium et enfin d’un domaine globulaire terminal lectine like permettant la fixation au récepteur CD47 ou IAP (Integrin Associated Protein).
19
Figure 2. Structure, partenaires et activités de la Thrombospondine-1 (adapté de Lawler. Curr Op Cell Biol, 2003 47; Sid et al. Crit Rev Oncol Hematol, 2004 45; Carlson et al. Cell Mol Life Sci, 2008 46)
La Thrombospondine-1 est une glycoprotéine homotrimérique matricielle de 450 kDa. Ses différents domaines lui permettent d’interagir avec de nombreux partenaires et lui confèrent ainsi des activités biologiques variées grâce auxquelles elle participe à la régulation de l’adhésion cellulaire, l’apoptose, l’angiogénèse et l’inflammation au cours du remodelage tissulaire.
B. Fonctions
1. Adhésion et agrégation plaquettaire
La TSP1 induit l’adhésion des plaquettes à l’endothelium et leur agrégation. L’interaction de la TSP-1 exprimée à la surface endothéliale avec les récepteurs CD36 et CD47 présents à la surface des plaquettes induit l’expression des intégrines β1 et β3 et l’adhésion ou l’agrégation plaquettaire 48–50. En inhibant l’effet anti-thrombotique du NO, la TSP-1 favorise également l’adhésion et l’agrégation plaquettaire 51.
20
2. Activation du TGF-β1
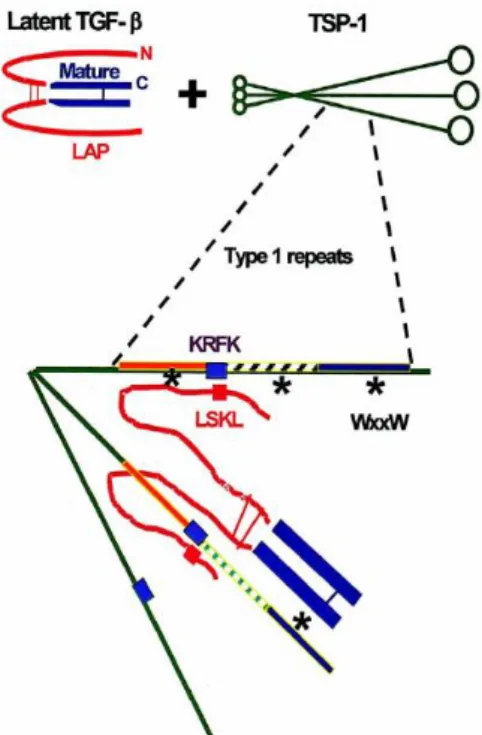
La TSP-1 est un activateur endogène du TGF-β1 52 qui est un facteur pro-fibrosant jouant un rôle majeur dans le développement de la fibrose rénale 12. La TSP-1 forme un complexe trimoléculaire avec le TGF-β1 mature et son peptide de latence, le Latency Associated Peptide (LAP), grâce à deux motifs situés dans le domaine TSR1. Chacun des motifs WxxW situés dans le TSR1 se fixe au TGF-β1 latent créant un complexe stable. La séquence KRFK se lie ensuite à la séquence LSKL du LAP permettant la libération du TGF-β1 mature qui pourra ensuite se fixer sur ses récepteurs 53,54 (Figure 3).
Figure 3. Activation du TGF-β latent par la Thrombospondine-1 (d’après Murphy-Ullrich et Poczatek. Cytokine Growth Factor Rev, 2000 54).
Les domaines WxxW et KRFK de la Thrombospondine-1 se lient respectivement au TGF-β latent et au peptide de latence du TGF-β (LAP) permettant la libération du TGF-β actif dans la matrice extra-celllulaire.
21
3. Activité anti-angiogénique et modulation du tonus vasculaire
Les effets anti-angiogéniques de la TSP-1 résultent de mécanismes multiples incluant une diminution de la disponibilité de facteurs de croissance pro-angiogéniques et de l’activation de leur voie de signalisation, l’inhibition de la migration et de la croissance des cellules endothéliales et l’induction de leur apoptose, et l’inhibition de la voie de signalisation du monoxyde d’azote (NO) (Figure 4).
En 1990, Good et al. décrivent pour la première fois le rôle anti-angiogénique de la TSP-1. Elle inhibe la migration de cellules endothéliales mais aussi la néovascularisation cornéenne stimulée par le basic fibroblast growth factor (bFGF) et le VEGF 55. Le phénotype des souris knock-out pour le gène de la TSP-1 est caractérisé par une augmentation de la densité vasculaire rétinienne 56, des capillaires cardiaques et musculaires, de la concentration musculaire en VEGF et de la capacité musculaire à l’effort 57.
L’activation du TGF-β1 participe à l’effet anti-angiogénique de la TSP-1. En effet, chez la souris, l’angiogénèse et la croissance tumorale sont ralenties lorsque les cellules tumorales sont transfectées par les séquences de la TSP-1 activant le TGF-β1 (TSR1 et 2, séquence KRFK). La croissance de ces tumeurs est restaurée par l’injection du peptide compétitif LSKL inhibant l’activation du TGF-β1 latent par la TSP-1 58.
Via CD36, la TSP-1 favorise l’apoptose des cellules endothéliales en activant la voie des caspases 59,60, en stimulant l’expression de Bax et en réduisant celle de Bcl-2 60. La triple interaction TSP-1/intégrines β1/CD36 inhibe la migration des cellules endothéliales 61. Le traitement par les peptides compétitifs, ABT-510 et ABT-526, mimant la séquence du TSR2 se fixant au CD36, inhibe la croissance tumorale dans des modèles animaux 62–64 et permet une stabilisation tumorale chez certains patients présentant des sarcomes des tissus mous à des stades avancés 65.
L’inhibition de la voie du VEGF et du NO à différents niveaux joue également un rôle important dans l’effet anti-angiogénique de la TSP-1. En limitant l’activité de métalloprotéases et en augmentant leur élimination, la TSP-1 s’oppose à la libération du VEGF à partir de la MEC 66. Elle peut aussi séquestrer le VEGF, mais aussi le FGF et l’HGF, dans la MEC et empêcher leur fixation sur leurs récepteurs cellulaires 67. Grâce à son domaine N-terminal qui possède des sites de fixation aux protéoglycanes des cellules
22 endothéliales, la TSP-1 se comporte comme un inhibiteur compétitif du VEGF 66,68. Enfin, grâce aux TSR et à son interaction avec les intégrines β1 et les récepteurs CD36 69,70 et CD47
71
, elle inhibe la phosphorylation du récepteur VEGF-R2 bloquant ainsi la voie de signalisation du VEGF et notamment la production de NO par la NO synthase endothéliale 72. En aval, la TSP-1 inhibe à plusieurs niveaux la voie signalisation du NO dans les plaquettes, les cellules endothéliales et les cellules musculaires lisses vasculaires 27. In vitro, elle bloque l’activation de la guanylate cyclase soluble par le NO, donc la production de GMP cyclique (GMPc) et par conséquent l’activation de la protéine kinase dépendante du GMPc 73.
En inhibant la vasodilatation induite par le NO 74, la TSP-1 module le tonus vasculaire. Chez les souris knock-out pour la TSP-1, l’administration de TSP-1 ou de CD47 empêche la vasodilatation induite par l’acétycholine et la perfusion de TSP-1 ou d’anticorps agonistes de CD47 induit une hypertension artérielle transitoire 72. De plus, les souris knock-out pour la TSP-1 présentent une meilleure survie et récupération tissulaire après ischémie-reperfusion de différents organes, tels que la peau 74,75, le foie 76, et le muscle 75,77,78. Ces résultats soulignent que la TSP-1 endogène limite l’adaptation de la perfusion tissulaire au cours de l’ischémie et contribue au développement des lésions tissulaires.
Le récepteur C47 joue un rôle critique dans l’inhibition de la voie du NO par la TSP-1. En effet, in vitro, l’expression de CD47, mais pas celle de CD36, est nécessaire pour inhiber la production de GMPc, l’adhésion et la prolifération des cellules endothéliales induites par le NO à des concentrations physiologiques de TSP-1. De plus, l’inhibition du CD47, mais pas celle de CD36, par des anticorps bloquants ou des oligonucléotides antisens permet de restaurer la réponse des cellules vasculaires au NO 79. In vivo, lésions d’ischémie-reperfusion sont réduites de façon similaire chez les souris knock-out pour CD47 et knock-out pour TSP-1
76,78,80
, tandis que les lésions observées chez souris knock-out pour CD36 sont similaires à celles des souris sauvages 75.
La TSP-1 serait également capable d’inhiber la prolifération des cellules endothéliales progénitrices. Dans un modèle d’hypertension artérielle, le nombre de cellules endothéliales progénitrices circulantes est inversement proportionnel au taux de TSP-1 81. In vitro, les cellules endothéliales progénitrices de souris diabétiques ont une adhésion et un pouvoir d’endothélialisation diminués lorsqu’elles surexpriment la TSP-1 82. De plus, l’exposition de
23 cellules progénitrices endothéliales humaines à la TSP-1 limite leur prolifération et la formation de néovaisseaux 83.
Le rôle anti-angiogénique de la TSP-1 dans les pathologies humaines non tumorales a été suggéré par des travaux concernant l’artérite oblitérante des membres inférieurs. Le taux plasmatique de TSP-1 est plus élevé chez les patients souffrant d’artérite que chez les témoins 83 et son expression est augmentée dans les tissus ischémiques et corrélée à la diminution de la densité capillaire 84.
Figure 4. Activité anti-angiogénique de la Thrombospondine-1 (adapté de Isenberg et al. Nat Rev Cancer. 2009 85).
Les effets anti-angiogéniques de la TSP-1 résultent de mécanismes multiples incluant une diminution de la disponibilité du facteur de croissance pro-angiogénique VEGF-A et de l’activation de sa voie de signalisation, l’inhibition de la migration et de la croissance des cellules endothéliales et l’induction de leur apoptose, et l’inhibition de la voie de signalisation du monoxyde d’azote (NO). Ces mécanismes impliquent les récepteurs CD36 et CD47. Ce dernier joue un rôle critique pour l’inhibition de la voie du NO.
24
4. Activité immunomodulatrice
Lors de la description initiale du phénotype des souris knock-out pour TSP-1, Lawler
et al. rapportaient une augmentation du nombre de leucocytes circulants et une
inflammation pulmonaire mimant à un moindre degré l’inflammation présente chez les souris knock-out pour TGF-β1 suggérant un effet anti-inflammatoire de la TSP-1 via l’activation du TGF-β1 52,86. Les résultats variables des études ultérieures menées in vitro soulignent la complexité des effets immunologiques de la TSP-1 (Figure 5).
En activant le TGF-β1, la TSP-1 confère aux cellules présentatrices d’antigènes un phénotype tolérogène 87,88 et favoriserait la différenciation des lymphocytes T régulateurs 89. La liaison de la TSP-1 aux récepteurs CD47 90,91 et CD36 87,91,92 des cellules présentatrices d’antigènes, leur confère également un phénotype tolérogène, en diminuant leur sécrétion de TNFα 90,91, d’interleukine 12 87,90,91 et d’interleukine 10 91. Après une stimulation prolongée, les cellules dendritiques exposées à la TSP-1 deviennent non stimulables et le blocage de l’interaction TSP-1-CD47 restaure leur production de cytokines 91, suggérant le rôle critique du CD47. Enfin, la fixation de la TSP-1 au récepteur CD47 exprimé par les lymphocytes T CD4 naïfs ou mémoires inhiberait leur activation dépendante du TCR 93, les orienterait vers un phénotype régulateur 94, et induirait leur apoptose par l’intermédiaire de la protéine BNIP3 (BCL2/adenovirus E1B 19 kDa protein-interacting protein 3) 95,96.
D’autres études retrouvent des résultats contradictoires. L’interaction de la TSP-1 avec les intégrines α4β1 limiterait l’adhésion des lymphocytes T activés à VCAM-1 mais stimulerait leur production de MMP2 et 9 favorisant leur pouvoir d’invasion tissulaire 97. L’interaction triple entre le CD36 présent sur les cellules présentatrices de l’antigène, le CD47 exprimé par les lymphocytes T et la TSP-1 matricielle favoriserait l‘activation et l’expansion clonale des lymphocytes T de patients souffrant de polyarthrite rhumatoïde 98. La liaison de la TSP-1 au CD36 exprimé par les macrophages favoriserait la sécrétion de l’interleukine 6, une cytokine pro-inflammatoire, tandis qu’elle inhiberait celle de l’interleukine 10, une cytokine anti-inflammatoire. Néanmoins, la sécrétion d’interleukine 10 serait restaurée en présence de TGF-β1 par les macrophages leur conférant un rôle de tolérance 99.
25 Tandis que les études réalisées in vitro semblent complexes et parfois contradictoires, la plupart des études menées in vivo retrouvent un effet pro-inflammatoire net de la TSP-1. Au cours de l’ischémie-reperfusion, le déficit ou le blocage de la TSP-1 s’accompagne d’une protection tissulaire et d’une diminution de l’infiltrat inflammatoire quel que soit l’organe lésé : la peau 74, le foie 76 ou le muscle 77. Après ischémie de la patte, la sécrétion de cytokines pro-inflammatoires et l’activité phagocytaire des macrophages sont diminuées chez les souris TSP-1 KO comparées aux souris sauvages 77. Chez les souris TSP-1 KO, on note également une réduction de l’expression du facteur chimiotactique MCP-1 et de l’infiltration macrophagique au cours de la cicatrisation cutanée 100. Enfin, la TSP-1 via l’activation du TGF-β1 induirait l’engagement des lymphocytes T dans la voie Th17. Dans un modèle d’encéphalite auto-immune liée à une infiltration cérébrale par les lymphocytes Th17, l’administration du peptide LSKL, qui bloque l’activation du TGF-β1 par la TSP-1, retarde la survenue des symptômes neurologiques 101. Dans ce même modèle, les souris TSP-1 KO présentent des formes atténuées de cette encéphalite grâce à une réduction de la sécrétion d’interleukine 17 et d’interféron γ. Ce bénéfice est annulé par la restauration de l’activation du TGF-β1 102.
Outre son effet sur l’immunité adaptative, la TSP-1 intervient également dans la régulation de l’immunité innée. Elle favoriserait l’adhésion des monocytes à l’endothelium en induisant l’augmentation de l’expression des molécules d’adhésion VCAM-1 et ICAM-1 103 et leur transmigration grâce au CD47 104. Elle faciliterait la phagocytose des cellules endommagées grâce au CD36 105. Au cours du sepsis bactérien ou fungique, la mortalité des souris TSP-1 KO est réduite et s’associe à une diminution de la libération des cytokines pro-inflammatoires, du recrutement des leucocytes 106, et à une augmentation de l’activité phagocytaire des macrophages et de la clairance microbienne 106,107.
26
Figure 5. Activité immunomodulatrice de la Thrombospondine-1 (études in vitro)
Abréviations utilisées : LT : lymphocytes T, CPA : cellules présentatrices d’antigène, IL : interleukine, T reg : lymphocytes T régulateurs, Th17 : lymphocytes Th17.
Les résultats des études menées in vitro soulignent la complexité des effets immunologiques de la TSP-1 qui intervient dans la régulation de l’immunité innée et acquise en modulant le recrutement des cellules inflammatoires et l’orientation de leur phénotype.
C. Rôle de la Thrombospondine-1 dans le développement des néphropathies expérimentales
Chez l’adulte sain, l’expression de la TSP-1 au sein du parenchyme rénal est très faible et se limite à la capsule de Bowman et à l’interstitium. Au cours des néphropathies, elle peut être exprimée et sécrétée par différents types cellulaires (podocytes, cellules mésangiales, interstitielles, endothéliales, musculaires lisses et tubulaires) selon la nature de l’agression rénale et le stade évolutif de la maladie.
27
1. Par l’activation du TGF-β1
En 1998, Hugo et al. décrivent pour la première fois in vivo l’expression de la TSP-1 dans trois modèles de glomérulonéphrite expérimentale chez le rat (néphrite de Heymann, néphrite induite par les anticorps anti-Thy1 et la puromycine). Ils montrent une expression corticale de novo de l’ARNm et de la protéine de TSP-1 par les tubules, les macrophages et surtout les myofibroblastes. Cette expression est superposable à celle du TGF-β1 actif, précède la survenue de la fibrose tubulo-interstitielle et est corrélée à son extension 108.
La même équipe montre que l’injection intra-rénale d’oligonucléotides sens anti-TSP-1 conduit à ders résultats concordants 109. Les oligonucléotides anti-sens réduisent l’activation glomérulaire du TGF-β1, l’accumulation glomérulaire de protéines de la MEC et la protéinurie dans un modèle de glomérulonéphrite membrano-proliférative par les anticorps anti-Thy1 chez le rat. L’inhibition spécifique de l’activation du TGF-β1 par la TSP-1 par le peptide compétitif LSKL, qui mime la séquence du LAP se fixant à la TSP-1, reproduit les mêmes résultats 110. L’injection intra-péritonéale de LSKL diminue également les lésions tubulaires, l’infiltration interstitielle par les myofibroblastes et l’accumulation de collagène provoquées par une obstruction urétérale unilatérale 111. Dans ce modèle, l’inhibition de l’activation du TGF-β1 par la TSP-1 s’accompagne d’une réduction de la transcription et de l’expression de TSP-1, suggérant qu’il existe un rétrocontrôle positif du TGF-β1 actif sur la TSP-1 111. L’axe TSP-1-TGF-β1 est également impliqué dans la néphropathie diabétique. L’administration de LSKL réduit l’accumulation interstitielle de fibronectine, les altérations podocytaires et l’abuminurie chez des souris diabétiques soumises à une néphrectomie unilatérale 112. De plus, vingt semaines après l’induction d’un diabète par la streptozocine, l’expression glomérulaire de TGF-β1, des protéines Smad2 et 3 phosphorylées et de PAI-1 est diminuée chez les souris knock-out pour TPS-1 comparées à des contrôles sauvages. Cette diminution de l’activation de la voie de signalisation du TGF-β1 s’accompagne d’une réduction des lésions glomérulaires, de l’infiltrat inflammatoire interstitiel et de la protéinurie 113.
28
2. Par son activité anti-angiogénique
Trois études se sont intéressées au rôle anti-angiogénique de la TSP-1 dans des modèles expérimentaux de néphropathie chez le rongeur. En 2001, Kang et al. montrent que les altérations vasculaires (raréfaction des capillaires glomérulaires et tubulaires, diminution de l’expression du VEGF et de la prolifération des cellules endothéliales) associées au vieillissement rénal 114 et à la réduction néphronique 115 chez le rat sont proportionnelles à l’expression glomérulaire et tubulo-interstitielle de TSP-1. De plus, il existe une relation linéaire entre l’expression, tubulaire et glomérulaire, de la TSP-1, et le nombre de macrophages infiltrant l’interstitium et les glomérules, respectivement 115.
En 2005, Thakar et al. identifient par PCR micro-array les gènes surexprimés dans des reins de rat après ischémie-reperfusion induite par clampage du pédicule vasculaire rénal pendant 30 minutes. Le gène codant pour la TSP-1 est l’un des deux les plus exprimés. L’expression de la TSP-1 est maximale trois heures après reperfusion puis décroit au cours du temps pour se normaliser à quarante-huit heures. La TSP-1 est essentiellement exprimée par les cellules tubulaires proximales lésées et colocalise avec la caspase 3 suggérant un rôle de la TSP-1 dans l’apoptose cellulaire induite par l’ischémie. Ceci est confirmé par l’induction de l’expression de la caspase 3 par les cellules tubulaires proximales cultivées en présence de TSP-1. Enfin, les souris TSP-1 KO sont significativement protégées contre les lésions tubulaires et l’insuffisance rénale aiguë induites par l’ischémie-reperfusion 116.
3. Par son activité anti-inflammatoire
L’activité pro-inflammatoire de la TSP-1 dans le développement des néphropathies a été suggérée pour la première fois en 2001 par Kang et al. En effet, ils montrent dans un modèle de réduction néphronique par néphrectomie des cinq sixième chez le rat, une relation linéaire entre l’expression tubulaire et glomérulaire de TSP-1 et l’infiltration macrophagique 115.
Après induction d’un diabète par la streptozocine, les souris TSP-1 KO présentent une glomérulosclérose et une fibrose interstitielle moins marquées que les animaux sauvages. Cette protection s’accompagne d’une réduction de l’accumulation interstitielle de macrophages et lymphocytes 113.
29 Trois semaines après l’injection d’anticorps anti-membrane basale, les souris
knock-out pour TSP-1 présentent des lésions de glomérulonéphrite extra-capillaire et une
dysfonction rénale moins sévères que les animaux sauvages. En effet, le nombre de croissants extra-capillaires et de dépôts de fibrine est réduit, ainsi que l’infiltration interstitielle par les lymphocytes CD4 et CD8 et les macrophages 117.
D. Thrombospondine-1 et pathologie rénale chez l’homme
En 1994, Mc Gregor et al. étudient pour la première fois chez l’homme l’expression des Thrombospondines sur des biopsies de rein normal et au cours de différentes glomérulopathies. Dans le rein humain normal, les auteurs retrouvent une expression très faible limitée à l’interstitium. En revanche, l’expression des TSP est intense dans les synéchies floculo-capsulaires et l’interstitium au cours de la hyalinose segmentaire et focale et au sein des croissants cellulaires des glomérulonéphrites extra-capillaires tandis que les croissants fibreux sont peu marqués. Deux équipes ont étudié l’expression de la TSP-1 au cours de la néphropathie diabétique chez l’homme 118,119. Toutes deux montrent une expression mésangiale, notamment dans les nodules de Kimmelstiel-Wilson, mais aussi podocytaire, tubulaire et interstitielle, dont l’intensité et l’extension croissent avec la sévérité des lésions histologiques. Elle s’accompagne également d’une augmentation de l’expression du CTGF, du TGF-β1 et des protéines Smad2 et 3 phosphorylées intervenant dans la voie de signalisation du TGF-β1 activé. Ces trois études confirment les résultats obtenus au cours des néphropathies expérimentales. L’expression de la TSP-1 est variable selon le type de pathologie rénale. Elle concerne le compartiment et/ou les cellules impliquées dans le processus inflammatoire et/ou fibrosant, et son importance est corrélée à la sévérité des lésions histologiques.
30
III. Le récepteur CD47
A. Structure et expression
Le récepteur CD47 est un récepteur membranaire de 50kDa appartenant à la superfamille des immunoglobulines. Identifié pour la première fois en 1990 comme une protéine de la membrane plasmique des monocytes/macrophages associée à l’intégrine αVβ3, il a été initialement appelé IAP (Integrin Associated Protein) 120. Il comporte un domaine N-terminal extra-cellulaire immunoglobuline-like puis 5 domaines transmembranaires et enfin une courte extrémité C-terminale cytoplasmique 120,121 (Figure 6). Il possède quatre isoformes issues d’un épissage alternatif, hautement conservées entre la souris et l’homme et ayant une distribution tissulaire spécifique 122. Initialement identifié à partir de leucocytes et de cellules placentaires 120,121, des études ultérieures ont montré que son expression est ubiquitaire. Au sein des tissus, il est exprimé par diverses cellules, notamment les hématies, les plaquettes, les cellules musculaires lisses vasculaires, les cellules endothéliales et de nombreuses cellules cancéreuses 122.
Figure 6. Structure du récepteur CD47 (Lindberg et al. J Cell Biol. 1993 121).
Le récepteur CD47 est un récepteur membranaire de 50kDa appartenant à la superfamille des immunoglobulines. Il comporte un domaine N-terminal extra-cellulaire immunoglobuline-like puis 5 domaines transmembranaires et enfin une courte extrémité C-terminale cytoplasmique.
31
B. Partenaires
Grâce à son interaction avec de multiples partenaires, CD47 intervient dans de nombreux processus cellulaires tels que l’activation, la prolifération, l’adhésion, la migration et l’apoptose. Il joue également un rôle critique pour l’inhibition de la voie du NO. Ces principaux partenaires et effets cellulaires sont résumés sur la Figure 7.
1. Thrombospondine-1
CD47 est l’un des récepteurs de la TSP-1. En jouant un rôle critique dans l’inhibition de la voie du NO par la TSP-1, il lui confère des propriétés anti-angiogéniques (Figures 4 et 7). La liaison du CD47 à la TSP-1 jour un rôle modulateur dans l’immunité innée et adaptative (Figures 5 et 7). Les propriétés du couple TSP-1-CD47 ont déjà été décrites plus haut et ne seront donc pas développées ici.
2. SIRPα et SIRPγ
Le récepteur CD47 peut se lier à deux protéines de la famille SIRP (Signal Regulatory Protein) 123, SIRPα et SIRPγ 124 (Figure 7). SIRPα a été le second ligand identifié du récepteur CD47 125. Il est aussi appelé SHPS1 126, p84 et CD172a. SIRPα est exprimé par les monocytes, la plupart des macrophages tissulaires, les granuleux, certaines cellules dendritiques dans les tissus lymphoïdes, certaines cellules progénitrices médullaires et par les neurones 79. Il s’agit d’une protéine transmembranaire comportant trois domaines extra-cellulaires immunoglobuline-like, un domaine transmembranaire et un domaine cytoplasmique contenant quatre motifs ITIM 126. SIRPα peut moduler les réponses cellulaires à des facteurs de croissance ou d’autres molécules grâce à ses motifs ITIM qui, une fois phosphorylés par les kinases de la famille Src, recrutent et activent les phosphatases SHP-1 et -2 (Src homology domain 2-containing phosphatases-1 et -2) 123. Le rôle de l’interaction CD47-SIRPγ est très peu connu. In vitro, elle induit la migration transendothéliale des lymphocytes T 127.
32
3. Intégrines
Le CD47 a été initialement caractérisé grâce à son association au sein d’une même membrane plasmique avec les intégrines αvβ3 et nommé Integrin Associated Protein (IAP)
120
. Depuis, des interactions entre CD47 et d’autres intégrines ont été décrites. Le couplage du complexe CD47-intégrines β3 aux protéines Gi hétérotrimériques inhibe la production d’AMP cyclique 128 et donc certaines voies de signalisation intra-cellulaires. In vitro, la TSP-1 module l’interaction de CD47 avec certaines intégrines 48,49,128–130.
C. Fonctions 1. Phagocytose
L’inhibition de la phagocytose est la fonction la mieux caractérisée du complexe CD47-SIRPα 124 (Figure 7). L’interaction du CD47 présent à la surface les hématies avec la protéine SIRPα exprimée par les macrophages spléniques prévient la phagocytose des hématies saines 131 mais aussi des hématies opsonisées par le complément ou par des anticorps 132, suggérant le rôle du complexe CD47-SIRPα dans la physiopathologie des anémies hémolytiques auto-immunes. Les souris déficientes en CD47 sont plus sensibles à une anémie hémolytique auto-immune expérimentale que les souris sauvages. De plus, les souris NOD (non obeses diabetic) déficientes en CD47 développent spontanément une anémie hémolytique auto-immune létale en 180 à 280 jours 133.
2. Mort cellulaire
In vitro, le complexe TSP-1-CD47-intégrines β3 induit l’apoptose de cellules tumorales
par un mécanisme indépendant des caspases 130,134 impliquant l’inhibition de la production d’AMP cyclique par les protéines Gi hétérotrimériques 128. L’interaction TSP-1-CD47 induit également l’apoptose des lymphocytes T par l’intermédiaire de la protéine BNIP3 95,96 (Figure 7).
33
3. Effet immunomodulateur
Par ses interactions avec la TSP-1 (Figure 5), les intégrines et SIRPα, CD47 joue un rôle immunomodulateur en participant à l’homéostasie des cellules dendritiques et des lymphocytes, et en participant à l’adhésion et à la transmigration des leucocytes.
Le nombre de cellules dendritiques et de lymphocytes T CD4 et CD8 spléniques est réduit chez les souris knock-out pour SIRPα, comme pour CD47 135. La transfection de cellules hématopoïétiques par le récepteur CD47 améliore la reconstitution médullaire et la fonctionnalité des lymphocytes T, B et NK après greffe de moelle 136. La liaison du récepteur CD47 à la surface des lymphocytes T avec la protéine SIRPα à la surface des cellules dendritiques empêche la maturation des cellules dendritiques et leur sécrétion d’interleukines, et limite l’activation et la prolifération lymphocytaire T CD4 137. L’interaction des deux molécules est notamment impliquée dans le développement des réponses Th17 et Th1 au cours des maladies auto-immunes, les souris mutantes pour SIRPα et knock-out pour CD47 développant des formes moins sévères d’encéphalite ou colite auto-immune expérimentale 138.
In vitro, l’interaction de la TSP-1 à la surface des lymphocytes T avec le récepteur
CD47 exprimé par les cellules dendritiques module également leur activation et leur différenciation. Elle limite la sécrétion de cytokines pro-inflammatoires, telles que le TNFα et l’interleukine 12 90,91 et l’interleukine 10 91par les cellules dendritiques. De plus, elle inhibe l’activation des lymphocytes T 93, les oriente vers un phénotype régulateur 94, et induit leur apoptose par l’intermédiaire de la protéine BNIP3 95,96. A l’inverse, l’interaction triple entre le CD36 présent sur les cellules présentatrices de l’antigène, le CD47 exprimé par les lymphocytes T et la TSP-1 matricielle favoriserait l‘activation et l’expansion clonale des lymphocytes T de patients souffrant de polyarthrite rhumatoïde 98.
La liaison du récepteur CD47 exprimé par les cellules endothéliales et épithéliales à la protéine SIRPα présente à la surface des neutrophiles 139 et des monocytes 140 favorise également leur migration transendothéliale et transépithéliale. L’augmentation de l’expression des molécules d’adhésion VCAM-1 et ICAM-1 induite par liaison de la TSP-1 au CD47 favorise également l’adhésion des monocytes à l’endothelium 103. Enfin, CD47 participe à l’adhésion et au recrutement des lymphocytes T en modulant la conformation
34 des intégrines LFA-1 et VLA-4 141, et à leur migration transendothéliale grâce à son interaction avec SIRPγ 127.
In vivo, le déficit en CD47 76,80,142,143, l’inhibition de son expression par des oligonucléotides antisens 144 ou le blocage de l’interaction TSP-1-CD47 par des anticorps 145 s’associe à une réduction de l’infiltrat inflammatoire dans les modèles d’ischémie-reperfusion cérébrale 80, médullaire 142, hépatique 76 et rénale 143. Le rôle de CD47 dans l’immunité innée au cours des infections bactériennes est plus équivoque. Dans un modèle de péritonite bactérienne, la mortalité des souris knock-out pour CD47 est largement supérieure à celle des souris hétérozygotes. Cette surmortalité s’associe à un défaut de migration des neutrophiles, de stress oxydatif et de phagocytose 146. A l’inverse, l’administration de si-RNA dirigés contre CD47 ou contre la TSP-1 empêche le développement d’une méningite chez des souris nouvelles-nées infectées par E.coli K1. Cette protection serait liée à un défaut de maturation des cellules dendritiques 103.
4. Effet anti-angiogénique
Le récepteur CD47 joue un rôle critique pour l’inhibition de la voie du NO par la TSP-1 et inhibe ainsi la croissance et la migration des cellules endothéliales, la relaxation des cellules musculaires lisses et l’agrégation plaquettaire 85 (Figure 4). Ainsi, les souris knock-out pour CD47, tout comme celles knock-out pour TSP-1, sont partiellement protégées des lésions provoquées par l’ischémie-reperfusion de différents organes : la peau 75,78,147, le foie
76, le cerveau 80 et le rein 143,148. De même, le blocage de l’interaction entre CD47 et TSP-1
par des anticorps 75,78,145,147 ou l’utilisation d’oligonucléotides anti-CD47 75,78,147 améliore la récupération tissulaire après une ischémie cutanée ou musculaire. On observe également une diminution des lésions cutanées induites par l’irradiation chez les souris knock-out pour TSP-1 ou CD47 84. Le couple CD47-TSP-1 intervient également dans la régulation mitochondriale. Le nombre et la taille des mitochondries sont augmentés dans les muscles des souris knock-out pour TSP-1 ou CD47 qui présentent une capacité à l’effort accrue 80.
35
Figure 7. Principaux partenaires et effets cellulaires de CD47 (d’après Soto-Pantoja et al. Expert Opin Ther Targets 2013 149).
Grâce à son interaction avec ses principaux partenaires, la Thrombospondine-1, les intégrines et SIRPα, CD47 intervient dans de nombreux processus cellulaires tels que la phagocytose, l’apoptose, la prolifération et la survie cellulaire.
D. CD47 et pathologie rénale
Peu de données sur la fonction du récepteur CD47 au cours des pathologies rénales ont été publiées.
In vitro, des anticorps bloquants le récepteur CD47 inhibent la migration des
leucocytes à travers une couche de cellules épithéliales tubulaires proximales humaines (HK-2) 150.
In vivo, l’expression de TSP-1 et CD47 augmente 24 heures après
ischémie-reperfusion induite par clampage transitoire de l’artère rénale chez la souris. Les lésions tubulaires et l’insuffisance rénale sont réduites chez les souris knock-out pour CD47. Cette protection s’accompagne d’une réduction de la production de cytokines et chémokines
pro-36 inflammatoires, de l’infiltration par les polynucléaires neutrophiles et les macrophages, d’une diminution de l’apoptose des cellules rénales et de la production de radicaux libres dérivés de l’oxygène. Enfin, la greffe de cellules hématopoïétiques de souris sauvages n’altère par cette protection suggérant le rôle du récepteur CD47 exprimé par les cellules rénales, et non pas de celui présent sur les cellules circulantes, dans le développement des lésions 143. Dans un modèle de greffe rénale chez le rat, la perfusion des greffons par des anticorps dirigés contre le CD47 avant la greffe améliore la perfusion des greffons, et limite les lésions tubulaires et la dysfonction rénale secondaires à l’ischémie-reperfusion. De façon spectaculaire, seuls les rats greffés avec un rein perfusé par des anticorps anti-CD47 survivent 148.
Au niveau glomérulaire, on note l’expression de CD47 par les cellules mésangiales et de SIRPα par les podocytes chez le rat. Dans un modèle de glomérulonéphrite membrano-proliférative induite par les anticorps anti-Thy1, la mésangiolyse s’accompagne d’une perte de l’expression de CD47 par les cellules mésangiales et de la diminution de la phosphorylation de SIRPα exprimé par les podocytes suggérant un rôle de l’interaction CD47-SIRPα dans la communication cellulaire au sein du glomérule. Cependant, aucune donnée ne permet d’affirmer le rôle joué par ces deux molécules dans cette étude descriptive 151. Chez l’homme, une équipe a étudié l’expression glomérulaire de CD47 et des intégrines. Dans le glomérule normal, les cellules épithéliales et mésangiales expriment CD47 et αvβ3. La glomérulonéphrite aiguë post-infectieuse, la glomérulonéphrite membrano-proliférative et la néphropathie diabétique s’accompagnent d’une diminution de leur expression 152.
37
OBJECTIFS
Le premier objectif de ce travail était de définir le rôle de la Thrombospondine-1 (TSP-1) dans le développement de la fibrose rénale. Nous nous sommes particulièrement intéressés à l’activation du TGF-β1 et aux propriétés anti-angiogéniques et immunomodulatrices de cette glycoprotéine matricielle. Pour cela, nous avons tout d’abord réalisé une étude descriptive de l’expression de la TSP-1 au cours de l’obstruction urétérale unilatérale chez le rat et de la réparation rénale accompagnant la désobstruction. Nous avons ensuite démontré le rôle de la TSP-1 dans le développement de la fibrose rénale induite par l’obstruction urétérale unilatérale et déterminé les mécanismes physiopathologiques impliqués grâce à l’utilisation de souris génétiquement déficientes en TSP-1.
Dans une seconde partie de ce travail, nous avons analysé le rôle joué par CD47, l’un des récepteurs majeurs de la TSP-1, dans le développement des lésions induites par l’obstruction urétérale unilatérale. Dans ce but, nous avons analysé les lésions tubulaires et vasculaires, la fibrose interstitielle et l’inflammation dix jours après obstruction urétérale unilatérale chez des souris sauvages et génétiquement déficientes en CD47.
Enfin, dans une troisième partie, nous avons tenté de déterminer le rôle de la TSP-1 dans le développement de la néphroangiosclérose, première cause d’insuffisance rénale chronique terminale. Pour cela, nous avons d’abord étudié l’expression de la TSP-1 au cours d’une néphropathie hypertensive expérimentale induite par un inhibiteur pharmacologique de la NO-synthase, le L-NAME, chez le rat. Nous avons ensuite comparé les lésions rénales développées par des souris sauvages et génétiquement déficientes en TSP-1 rendues hypertendues par administration d’angiotensine II et de L-NAME.
38
39
I. Première partie : la Thrombospondine-1 joue un rôle fibrosant et pro-inflammatoire au cours de l’obstruction urétérale
40
A. Article “ Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction”
1. Résumé
La Thrombospondine-1 (TSP-1) est un activateur endogène du TGF-β1 et présentent des propriétés anti-angiogéniques qui pourraient compromettre la réparation rénale. Nous avons testé si la TSP-1 est impliquée dans le développement de la fibrose rénale après obstruction urétérale unilatérale (UUO), un modèle classique de fibrose rénale.
Chez le rat, l’obstruction urétérale maintenue pendant dix jours induit une inflammation, une atrophie et une dilatation tubulaire, une apoptose et une prolifération des cellules tubulaires ainsi que la raréfaction des capillaires péri-tubulaires conduisant au développement d’une fibrose interstitielle. Parallèlement à l’accumulation de collagène III et de TGF-β1 associée à ces lésions, l’expression de la TSP-1 augmente dans l’interstitium et apparaît de novo dans les cellules tubulaires lésées. La levée de l’obstruction urétérale au dixième jour permet une réparation progressive du parenchyme rénal et une récupération fonctionnelle. Sur une période s’étendant du septième au quinzième jour après désobstruction, nous avons observé la réapparition des capillaires péri-tubulaires et un retour à l’état basal du contenu rénal en VEGF. De plus, l’expression de la TSP-1 décroit parallèlement à celle du collagène III et du TGF-β1.
Grâce à l’utilisation de souris knock-out pour la TSP-1, nous avons pu démontrer l’effet délétère de la TSP-1 sur le parenchyme rénal dans le modèle d’UUO. En effet, après dix jours d’UUO, les tubules rénaux et le réseau capillaire péri-tubulaire sont mieux préservés chez les souris génétiquement déficientes en TSP-1 que chez les animaux sauvages. Des études supplémentaires ont montré que la TSP-1 joue, dans ce modèle, un rôle pro-inflammatoire médié, au moins en partie, par le facteur chimiotactique MCP-1 et par l’activation de la voie Th17.
L’ensemble des expériences menées montrent que la TSP-1 représente un médiateur anti-angiogénique, pro-fibrosant, et pro-inflammatoire important dans le développement des lésions rénales. Le blocage de son action représente une thérapeutique potentielle dans la prise en charge des maladies rénales chroniques.
41
55
3. Figure supplémentaire
Figure 1S: Representative example of cell proliferation revealed by PCNA antibody (A) and cellular apoptosis revealed by TUNEL assay (B). The number of proliferative cells in SHAM group was negligible. Ten days after UUO, there was a dramatic increase in proliferative and apoptotic cells. After 7 days of RUUO, cellular proliferation and apoptosis further increased compared with UUO. Fifteen days after RUUO, the number of proliferating and apoptotic cells significantly decreased compared with UUO rats. Values are mean ± SEM, * P<0.05, ** P<0.01, *** P<0.005, **** P<0.0001