HAL Id: hal-03015076
https://hal.archives-ouvertes.fr/hal-03015076
Submitted on 19 Nov 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
An oligomeric switch controls the Mrr-induced SOS
response in E. coli
Anaïs Bourges, Oscar Torres Montaguth, Wubishet Tadesse, Gilles Labesse,
Abram Aertsen, Catherine Royer, Nathalie Declerck
To cite this version:
Anaïs Bourges, Oscar Torres Montaguth, Wubishet Tadesse, Gilles Labesse, Abram Aertsen, et al..
An oligomeric switch controls the Mrr-induced SOS response in E. coli. DNA Repair, Elsevier, 2021,
97, pp.103009. �10.1016/j.dnarep.2020.103009�. �hal-03015076�
DNA Repair 97 (2021) 103009
Available online 6 November 2020
1568-7864/© 2020 The Authors. Published by Elsevier B.V. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
An oligomeric switch controls the Mrr-induced SOS response in E. coli
Anaïs C. Bourges
a,b, Oscar E. Torres Montaguth
c, Wubishet Tadesse
c, Gilles Labesse
b,
Abram Aertsen
c, Catherine A. Royer
a, Nathalie Declerck
b,d,*
aDepartment of Biological Sciences, Rensselaer Polytechnic Institute, Troy, NY, 12180, USAbCentre de Biochimie Structurale, CNRS, INSERM, Universit´e de Montpellier, 34090, Montpellier, France cDepartment of Microbial and Molecular Systems, KU Leuven, B-3001, Leuven, Belgium
dD´epartement MICA, INRA, 78350 Jouy-en-Josas, France
A R T I C L E I N F O
Keywords:
Pressure-induced SOS-response Methyl-directed endonuclease Quantitative fluorescence fluctuation microscopy and spectroscopy Live cell imaging
Protein modeling
A B S T R A C T
Mrr from Escherichia coli K12 is a type IV restriction endonuclease whose role is to recognize and cleave foreign methylated DNA. Beyond this protective role, Mrr can inflict chromosomal DNA damage that elicits the SOS response in the host cell upon heterologous expression of specific methyltransferases such as M.HhaII, or after exposure to high pressure (HP). Activation of Mrr in response to these perturbations involves an oligomeric switch that dissociates inactive homo-tetramers into active dimers. Here we used scanning number and brightness (sN&B) analysis to determine in vivo the stoichiometry of a constitutively active Mrr mutant predicted to be dimeric and examine other GFP-Mrr mutants compromised in their response to either M.HhaII activity or HP shock. We also observed in vitro the direct pressure-induced tetramer dissociation by HP fluorescence cor-relation spectroscopy of purified GFP-Mrr. To shed light on the linkages between subunit interactions and ac-tivity of Mrr and its variants, we built a structural model of the full-length tetramer bound to DNA. Similar to functionally related endonucleases, the conserved DNA cleavage domain would be sequestered by the DNA recognition domain in the Mrr inactive tetramer, dissociating into an enzymatically active dimer upon inter-action with multiple DNA sites.
1. Introduction
Restriction endonucleases (REs) constitute a primary defense against invading phages or other foreign DNA entering bacterial cells [1,2]. In
Escherichia coli K-12, Mrr (methylated adenine recognition and
restric-tion) has received special interest because of its involvement in the restricted cloning efficiency of methylated DNA isolated from plants and mammals [3]. More recently modification-dependent REs have emerged as useful tools for epigenetic studies [1,4,5]. Mrr, together with both
E. coli methylcytosine restriction systems McrA and McrBC, belongs to
the type IV family of REs that target modified, typically methylated, DNA with a loose sequence specificity [6]. Mrr cleaves N6-methyladenine (m6A)- or C5-methylcytosine (m5C)-containing DNA with no consensus nucleotide sequence context thus far identified [7,8]. To date, Mrr catalytic properties and specificity have not been investi-gated by in vitro experiments. Evidence that Mrr restricts methylated DNA comes from early observations of the mrr-dependent genotoxicity of various adenine or cytosine methyltransferases (MTases) such as M.
HhaII from Haemophilus haemolyticus [7]. In recombinant E. coli strains expressing conditionally such an MTase, the methylated chromosomal DNA becomes a substrate for Mrr endonuclease activity, leading to double-strand breaks and the SOS DNA repair response.
Mrr can also be activated in E. coli cells exposed to high hydrostatic pressure (HP), causing chromosomal DNA damage and induction of an SOS response similar to that observed in the presence of exogenous MTases [9]. HP represents an important environmental factor for un-derstanding bacterial adaptation to extreme conditions, as found in the deep biosphere where pressure can exceed 100 MPa [10,11]. Pressure is also an interesting thermodynamic parameter for monitoring the dena-turation and dissociation of biomolecules since its effects are more gradual and local than those inflicted by temperature or denaturants [12]. Moreover, HP has biotechnological applications in particular in the food-industry where it is used as a non-thermal pasteurization pro-cess called Pascalization or High-Pressure Propro-cessing (HPP). However, the efficacy of HPP can be compromised by resistant strains of foodborne pathogens [13]. Indeed E. coli can acquire extreme HP resistance in only
* Corresponding author at: Centre de Biochimie Structurale, CNRS, INSERM, Universit´e de Montpellier, 34090, Montpellier, France.
E-mail address: [email protected] (N. Declerck).
Contents lists available at ScienceDirect
DNA Repair
journal homepage: www.elsevier.com/locate/dnarepair
https://doi.org/10.1016/j.dnarep.2020.103009
a few selection steps [14,15]. Sub-lethal HP shocks (e.g. 100 MPa for 15 min) to E. coli K-12 strain MG1655 were shown to elicit typical SOS-mediated phenotypes, including nucleoid condensation and SulA-mediated filamentous growth upon return to atmospheric pressure (0.1 MPa) [16]. It was shown that this pressure induction of the SOS response is dependent on the RecA, LexA and RecBCD components of the bacterial DNA repair system triggered by the double-strand breaks generated by HP-activated Mrr [9,17]. Interestingly, Mrr mutant strains could be isolated that were no longer sensitive to either HP shock or M. HhaII MTase expression, suggesting mechanistic differences in the way both triggers activate Mrr [16].
Cellular localization and dynamics of Mrr fused to the green fluo-rescent protein (GFP) have previously been undertaken to investigate the activation mechanisms of Mrr upon exposure to HP or M.HhaII methylation of the host chromosome [16]. Wide-field microscopy im-ages of E. coli cells producing GFP-Mrr from a multi-copy plasmid showed that Mrr forms nucleoid-associated foci that coalesce after a HP shock or expression of the M.HhaII MTase [16]. Recently we charac-terized GFP-Mrr produced at low levels from a chromosomal insertion using scanning Number and Brightness (sN&B), a sensitive and quanti-tative fluorescence fluctuation microscopy method that allows direct determination of the intracellular concentration and oligomeric state of fluorescent proteins in single live cells [18]. Unexpectedly, whereas GFP-Mrr was tetrameric in untreated cells, we found that the intense foci observed after HP treatment or M.HhaII induction, that we thought corresponded to high-order oligomers of GFP-Mrr, were in fact made up of dimers. From these observations, we proposed a model for Mrr acti-vation by methylated DNA in which the equilibrium between inactive tetramer and active dimer is pulled toward active dimer that binds tightly to the large number of high affinity methylated DNA sites. In contrast, for the pressure-induced SOS response, we proposed that pressure pushes the tetramer-dimer equilibrium toward active dimer, which then binds to and cuts the DNA. In both cases, after chromosomal cleavage, the Mrr protein retains its dimeric conformation and remains irreversibly bound to DNA, localizing to foci associated with DNA damage and nucleoid condensation upon induction of the SOS response [19].
In the present study we sought to further validate our model of Mrr activation and gain insight into the structural mechanism that couples Mrr oligomerization to catalysis. Using the same sN&B approach that we previously used to characterize the wild-type (WT) Mrr and an inactive catalytic mutant [19], we investigated the behavior of a constitutively active Mrr mutant that we predicted to be dimeric in unstressed cells, as well as the Mrr variants specifically compromised in their response to either HP shock or M.HhaII activity. Thanks to the high sensitivity afforded by sN&B, we were able to characterize these variants when produced at low, near physiological levels. We found that they behaved somewhat differently compared to when they were over-expressed for wide-field imaging. Determination of the in vivo stoichiometry of the GFP-Mrr protein fusions before and after a HP shock or MTase induction revealed that mutations affecting Mrr activity or activation perturb in one way or another the oligomeric switch that accompanies foci for-mation and the ensuing SOS response. Moreover, using a high-pressure microscopy system that we recently implemented for fluorescence cor-relation spectroscopy (FCS) measurements under pressure [20], we were also able to observe in vitro the HP-induced dissociation of purified WT GFP-Mrr oligomers, demonstrating that pressure alone can induce the oligomeric switch required for Mrr activation. Next, using homology modeling we sought to gain a structural understanding of the activation mechanism of Mrr. Based on structural and functional similarity with the MspJI methyl-directed restriction endonuclease [21,22], we con-structed a 3D model of the inactive full-length Mrr tetramer bound to DNA. This model provides a structural interpretation of the mutational effects observed in the present study, as well as of mutations reported in previous studies for which no satisfactory explanation could be pro-posed [23]. In light of this structure-function analysis, we propose that
the oligomeric switch that leads to Mrr activation is reminiscent of previously proposed mechanisms for other REs and involves sequestra-tion of the DNA cleavage domain by the DNA recognisequestra-tion domain in the inactive tetramer, and the formation of an enzymatically active dimer upon interaction with cognate DNA.
2. Material and methods
2.1. Bacterial strains and construction of mutants
Escherichia coli K-12 MG1655 [24] was used as parental strain for all strains used for microscopy experiments (Table S1). The genetic screen that led to the isolation of mutations compromising Mrr activation by either high pressure (V173A) or M.HhaII-dependent DNA methylation (H279Y) has been described in Gosh et al. [16]. Construction of the strains expressing the GFP-Mrr variants carrying these mutations from the PBAD arabinose inducible promoter at the mrr chromosomal locus
was performed as described earlier for the wild-type (WT) version and the catalytically compromised GFP-MrrD203A variant [19]. The GFP
variant used in these studies is a fast maturing, monomeric variant, GFPmut2 [25]. The mrr allele carrying the N111S/D124 G/V175 G tri-ple mutation rendering Mrr constitutively active was obtained by random mutagenesis using error prone PCR [26]. For the PCR reaction, 500 ng of plasmid pAA810 (a pACYC184 derivative containing the wild-type mrr gene [17]) was amplified using primers 5′-ATCGCTGCAGACGGTTCCTACCTATGAC-3′ and 5′-CGATAAGC
TTGCGTTTGCGGGGTTGAGG-3′ in the presence of unbalanced dNTP
concentrations (1 mM dCTP and dTTP, 0.2 mM dATP and dGTP) and 2 mM MnCl2. After the PCR reaction, the template plasmid was removed
by digestion with DpnI and the PCR products were digested with PstI and HindIII prior to ligation into the low copy number
pBAD33-gfp-mut2-T7tag plasmid [27]. Transformants of an E. coli SOS-lacZ reporter
strain [28] were plated on LB agar supplemented with 0.2 % glucose to prevent expression from the PBAD promoter. In order to identify
consti-tutive mutants, colonies were replica-plated on LB agar supplemented with 0.02 % arabinose and 0.04 % X-gal. Colonies displaying β-galac-tosidase activity were traced back in the original plate and sequenced.
2.2. Growth conditions and high-pressure shock
As previously described [19], E. coli MG1655 and derivative strains were grown at 37◦C in LB with appropriate antibiotics supplemented
with 0.002 % or 0.4 % arabinose for induction of GFP, Mrr, or GFP-Mrr proteins encoded from plasmid or chromosomal constructs, respectively. Expression of the M.HhaII MTase from the pTrc99A-hhaII plasmid [16] was induced in exponentially growing cells (OD600 ~0.15) by adding
1 mM IPTG (isopropyl β-D-thio-galactopyranoside) to the liquid cultures one hour before microscopy observations. For pressure treatment, cells from 500μL culture in late exponential phase (OD600 ~0.6) were
har-vested by 2 min centrifugation at 3500 rpm and re-suspended in 50μL
of LB to be pressurized in Micro-Tubes using a computer-controlled HUB440 high pressure generator (both from Pressure BioSciences, Inc., South Easton, MA). Pressure was typically maintained for 15 min at 100 Megapascal (100 MPa = 1000 bars, ~1000 atm, ~14,500 psi). After pressure release, the cell suspension was centrifuged and concen-trated to prepare microscopy sample. The strains carrying the pBAD-gfpmut2::mrr plasmid encoding WT Mrr or the MrrN111S/D124G/V175G
constitutively active variant were first grown in LB containing 0.4 % glucose to repress protein expression, then the cells were harvested and transferred to an agar pad supplemented with 0.4 % arabinose in order to induce expression of the fusion proteins during time-lapse microscopy experiments.
2.3. Two-photon scanning microscopy
previously described [19] using an Avalanche Photo Diobased de-tector (Perkin Elmer) and a femtosecond pulsed infrared laser (MaiTai, Newport/Spectra Physics, Mountain View, CA, USA) focused through a 60 × 1.2NA water immersion objective (Nikon APO VC). For GFP measurements, infrared light was filtered from detected light by using a 735 nm low-pass dichroic filter (Chroma Technology Corporation, Rockingham, VT) and emitted light was filtered with a 530/43 nm emission filter. Microscopy samples were prepared by depositing 3μL of
concentrated cell suspensions (OD600 of ~25) on an agar pads (2%
UltraPureTM LMP Agarose, Invitrogen) sandwiched between two cov-erslips No1 (VWR) coated with poly-L-Lysine (Sigma) and then mounted in an Attofluor coverslip holder. Two-photon scanning microscopy was performed by recording 50 raster scanned images of 256 × 256 pixels with a pixel dwell-time of 40 μs using an excitation laser power of
11 mW (at the microscope entrance) at 930 nm. Scanning Number and brightness (sN&B) analysis [18] of the fluorescence intensity average and fluctuations (variance) at each pixel over the scan series was used to calculate the intracellular concentration and molecular brightness of the diffusing fluorescent particles, as described previously in details [19,29,
30]. The stoichiometry of the GFP-fusion proteins was deduced from the molecular brightness value determined for free monomeric GFP in E. coli MG1655 strain expressing a chromosomal PBAD-gfpmut2 construct and
corrected for auto-fluorescence measured in the background strain.
2.4. Purification of GFP-Mrr
GFP-Mrr proteins were produced as N-terminal Strep-tag®II fusions from pRSET B (Invitrogen) expressed in E. coli BL21 (DE3) or T7 Express (New England Biolabs). Cells were grown to exponential phase in 700 mL of ampicillin-supplemented LB medium at 37◦C, and protein
expression was induced with 1 mM IPTG for 3 h at 30◦C. The cell pellet
was washed in 10 mM Tris pH 7.5, 100 mM NaCl and kept at − 80◦C.
Cells were resuspended in 20 mL of lysis buffer (100 mM sodium phosphate buffer pH 7, 125 mM Na2SO4, 50 mM NaCl, 2.5 mM MgCl2,
0.5 mM DTT,) supplemented with 1.5 mM benzamidine, 1 mg/mL lysozyme and 1μg/mL DNase I. After sonication and centrifugation, the
supernatant was injected on a high capacity Strep-Tactin resin (IBA) in purification buffer (20 mM Tris pH8, 150 mM NaCl and 1 mM EDTA) supplemented with 2.5 mM desthiobiotin for protein elution. Analytical size exclusion chromatography (SEC) was performed on a Superdex 200 HR10/300 column (GE healthcare) equilibrated with 100 mM Tris pH8, 150 mM NaCl and 1 mM EDTA at a flow rate of 0.5 mL/min. Elution fractions of 400μL were collected and the fluorescence intensity of
100μL aliquots were measured in 96-well microplate (Greiner) using an
infinite M1000 PROplate reader (TECAN, Switzerland) using exciting and emission wavelengths at 488 (+/- 15) nm and 528 (+/- 20) nm band-path, resp. Purified GFPmut2, either His-tagged or Strep-tagged, was used for calibration of the fluorescence intensities and it was found to remain monomeric under the conditions used for in vitro ex-periments. All protein aliquots were kept at − 80◦C in purification
buffer.
2.5. High pressure FCS
In order to make direct observation of fluorescent proteins at high pressure, we constructed a microscopy set-up similar to that described previously by Gratton and Mueller [20,31]. These authors developed a pressure cell made of a glass capillary, sealed on one end and connected to a high-pressure pump on the other end, which could resist several thousand bars. Here we used a capillary of internal diameter 50μm and
total diameter 400μm, with pressure plug connections drilled to the
appropriate diameter and fixed with epoxy glue on both ends to facili-tate sample loading. The capillary was mounted in a stainless-steel holder the size of an Attofluor coverslip holder with slits for the capil-lary, and the plug/seal ensemble on either end of the capillary was connected to a high-pressure line. A peristaltic pump was used to load
the sample in a thin fused silica capillary. For high pressure experiments the HP line was blocked by closing the valve on one end of the capillary and connecting the other end to an automated high-pressure pump from Pressure BioSciences (South Easton, MA) on the other. FCS experiments under high pressure were performed using the same set up as for two-photon scanning microscopy, except that the water objective was replaced by a 60 × 1.4NA oil immersion objective (Nikon APO VC), and glycerol, rather than oil, was used as the coupling medium. This matched the capillary refraction index better than oil and minimized the distortion due to the capillary curvature. For each sample the fluores-cence signal was recorded for 100 s at a 500 kHz frequency. The SimFCS software was used for FCS data acquisition and analysis.
2.6. Structure modeling
Due to important sequence divergence, comparative modeling of Mrr relied on fold-recognition to identified optimal templates for the N-ter-minal and C-terN-ter-minal domains independently. Various templates sharing with Mrr sequence identity in the range of 15–25 % were detected for both domains using the server @TOME-2 [32]. Sequence-structure alignments were refined using the dedicated editor Vito [33] to maximize coverage and optimally place insertions/dele-tions. MODELLER [34] was used to build full models of each domain in the desired oligomeric state and the quality of the models was evaluated using Qmean [35]. The catalytic CAT domain (residues 131–304) was first modeled as a tetramer using the tetrameric structures of the AspBHI (PDB:4OC8 [36]) and MspJI (PDB:4FOQ [21]) Type IIM modification-dependent restriction enzymes in combination. We next modeled the protomer of the Mrr N-terminal domain (residues 1–90) bound to a 30-bp DNA fragment based on PDB:1F5T which provided the best sequence-structure alignment in the region of the predicted helix-turn-helix DNA recognition motif. Modeling of the full-length Mrr tetrameric assembly on DNA was then performed using as template the structure of the DNA-bound MspJI tetramer (PDB:4R28) in which the divergent N-terminal DNA recognition domain was manually replaced by the modeled Mrr wH domain, preserving as well, the fold and ori-entations of the linker region (residues 91–130).
3. Results
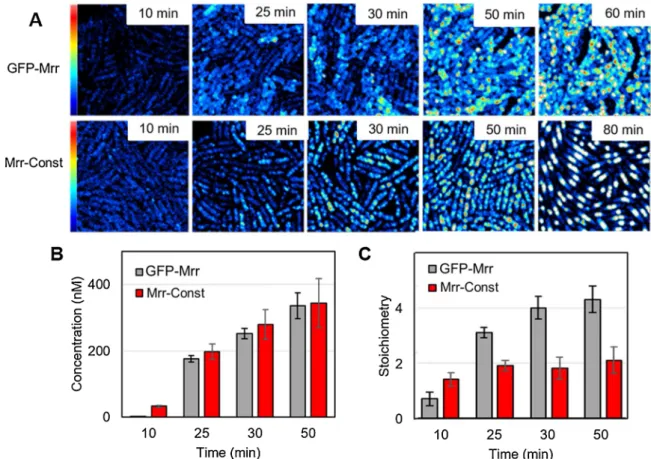
3.1. A constitutively active variant of Mrr is dimeric
We have proposed previously [19] that the active form of Mrr is a dimer, and that the activation mechanism upon HP shock that triggers the SOS-response is based on the pressure-induced dissociation of the inactive tetrameric form into the active dimer. Here, we investigate the structure-function relationships between oligomerization of Mrr and its activity. We first sought to ascertain whether constitutive activity could be linked to a permanently dimeric form of the protein. To this end, we determined by quantitative fluorescence microscopy the oligomeric state of a GFP-Mrr protein fusion carrying a triple mutation (N111S/D124 G/V175G) we isolated by screening for mutant
gfpmut2-mrr alleles rendering Mrr constitutively active. Using scanning
N&B in live E. coli cells, we compared the number and brightness of fluorescent particles detected in strains expressing wild-type GFP-Mrr (WT Mrr) or the GFP-MrrN111S/D124G/V175G constitutive mutant (Mrr-Const), both of which were under the control of the arabinose-inducible PBAD promoter in a plasmid construct (Table S1).
Expression of the constitutive mutant Mrr led to strong cell fila-mentation, indicating it inflicts DNA damage and triggers the SOS response. Because of this very toxic effect, observations of the strains carrying the wild-type or mrrN111S/D124G/V175G mutant gene fusion were
performed after growth under repressing condition in liquid culture then shifting the cells onto agarose pads supplemented with the inducing sugar.
immobile foci in the cells, without HP treatment or induction of the M. HhaII methyltransferase, whereas WT Mrr formed small, very mobile foci when observed under the same conditions (Fig. 1A and Supple-mentary materials S2A and S2B). Scanning N&B analysis of the scan series revealed that the wild-type and mutant GFP-Mrr fusions were produced at a similar rate and concentration in both strains (Fig. 1B). However, by the end of the time course their molecular brightness was 2-fold higher in the strain expressing WT Mrr, compared to that measured in the strain expressing the constitutive mutant. The stoichi-ometry values deduced by comparison with a strain expressing the GFP alone indicated the formation of tetramers in case of WT Mrr whereas Mrr-Const remains dimeric even at the highest concentration allowing accurate intensity fluctuation measurements (Fig. 1C). Note that the brightness of the freely diffusing GFP reporter did not change overtime during the induction process on the agar pad and that over-expression of this monomeric GFP variant up to 1.2 μM did not lead to aggregation as
deduced from its constant and low molecular brightness (Supplementary Fig. S3). The observation that a constitutively active variant of Mrr is also constitutively dimeric strongly supports our model for Mrr activa-tion based on the dissociaactiva-tion of inactive tetramers into active dimers.
3.2. Mrr mutations uncouple oligomerization and activation
A previous genetic screen has led to the identification of mutations that seem to uncouple Mrr activation by high pressure and by M.HhaII- dependent DNA methylation, i.e. mutants activated by either HP or MTase induction but not both [16]. Two of these Mrr variants have been
previously characterized by conventional epifluorescence microscopy using GFP fusions over-expressed from inducible gene constructs carried by a multi-copy plasmid [16]. In these prior experiments, after induction of M.HhaII, the strain producing the GFP-MrrV173A variant was found to
elicit the SOS response similar to the strain expressing wild-type GFP-Mrr, exhibiting large mid-cell foci and a significant decrease in viability, whereas this strain was only moderately affected after a HP shock. Inversely, the viability of a strain over-expressing GFP-MrrH279Y was decreased after a pressure shock but was only modestly affected by expression of the MTase and no large convergent foci were observed ([16] and Supplementary Fig. S4A).
Here we used 2-photon scanning microscopy to examine the behavior of HP resistant GFP-MrrV173A and MTase resistant GFP-
MrrH279Y (called hereafter Mrr-HPR and Mrr-MTR, respect.) when these variants were expressed at very low levels from a single copy of a chromosomal construct inserted at the natural mrr locus and fused to gfp (Fig. 2A). Very few foci were apparent in unstressed cells expressing the wild-type or mutant gfp-mrr chromosomal inserts, in contrast to the multiple distinct foci previously observed in strains over-producing these GFP fusions (supplemental Fig. S4A). However, a pressure shock of 15 min at 100 MPa led to the formation of large foci in most cells producing WT Mrr or Mrr-MTR, but in only a few cells producing Mrr- HPR or the catalytically inactive mutant GFP-MrrD203A (Mrr-Inact)
previously characterized [19]. Foci appeared in all strains after induc-tion of the M.HhaII MTase, indicating that the Mrr variants still responded to exogenous DNA methylation, although the SOS response was not elicited as strongly as in the wild-type strain (Fig. 2B).
Fig. 1. Effect of constitutive activity on Mrr stoichiometry. (A) Average fluorescence intensity maps obtained by 2-photon scanning microscopy of Escherichia K12
MG1655 single live cells expressing GFPmut2 protein fusion with wild-type Mrr (GFP-Mrr) or a constitutive mutant (Mrr-Const carrying N111S/D124 G/V175G triple mutation) from a plasmid-based PBAD promotor as a function of time after transfer on an agar pad supplemented with 0.4 % arabinose. Image size is 20 × 20μm and
maximum intensity is 2.5 counts per 40μs pixel dwell-time. (B) Concentration and (C) stoichiometry values of GFP-Mrr wild-type and constitutive mutant retrieved
from scanning number and brightness (sN&B) analysis of the scan series shown in panel A, deduced from the background corrected molecular brightness values measured for WT or mutant GFP-Mrr relative to that measured in an isogenic strain producing the GFPmut2 monomer (see supplementary Figure S3). Above 60 min, the concentration of the fluorescent proteins is too high for accurate determination of their molecular brightness. Error bars represent the standard deviation of the means for 6 fields of view (FOVs) per time points.
N&B analysis of the raster scanned images was then performed to determine the oligomeric state of the different GFP-Mrr proteins before and after perturbation by either HP or induction of the MTase. The Mrr- HPR variant showed slightly lower stoichiometry (3.5 +/- 0.5) than tetrameric (3.9 +/- 0.4) WT Mrr before perturbation, but not as low as the Mrr-Inact (2.8 +/- 0.6) previously reported [19] (Fig. 2B). This in-dicates that the Mrr tetramer stability is slightly diminished by the V173A mutation. Interestingly, in contrast to WT Mrr, the average stoichiometry of the Mrr-HPR variant remained nearly unchanged after HP shock, indicating that this mutant protein was either more resistant than the wild-type to pressure-induced dissociation and/or re-associated more readily than the wild-type upon pressure release. In contrast, in-duction of the M.HhaII MTase led to dissociation of the Mrr-HPR to dimer, as observed for WT Mrr, indicating that the V173A mutation has no or little impact on the protein response to the presence of hyper-methylated DNA. These observations confirm that this mutation alters the Mrr activation process by HP but not MTase expression, and that uncoupling of these two processes is accompanied by modification in the oligomeric properties of the Mrr protein.
N&B analysis of the strain producing the Mrr-MTR variant revealed a tetrameric stoichiometry prior to any perturbation. As observed for the Mrr-HPR variant, after HP stress the stoichiometry of Mrr-MTR was only slightly lower, 3.5 +/- 0.5, again suggesting that the tetramer was less
prone to HP-induced dissociation and/or re-associated more readily than WT Mrr. As observed for the wild-type and other mutant proteins, induction of the M.HhaII MTase led to dissociation of Mrr-MTR tetra-mers into ditetra-mers, (Fig. 2B). This behavior was unexpected since the H279Y mutation carried by this mutant was previously found to diminish the M.HhaII-dependent activation of Mrr when highly expressed from a plasmid construct in the strain used for the genetic screen [16]. In this strain, Mrr-MTR formed multiple small foci and nucleoide hyper-condensation did not occur following MTase expres-sion, whereas in the strain used in the present study which produces low Mrr-MTR levels, the chromosome-encoded variant associated into large foci similar to those observed for the wild-type (Supplementary Fig. S4). This concentration-dependent behavior suggests that the mutation may stabilize the Mrr-MTR tetramer. When this variant is expressed from the plasmid construct at concentrations much higher than its target sites, the tetramer-dimer equilibrium could not be so easily pulled to dimer. In
vitro characterization of the purified Mrr-MTR protein fusion by size
exclusion chromatography (SEC) indeed showed that this variant remained largely oligomeric after dilution, indicating higher intrinsic tetramer stability. In contrast, the Mrr HPR prepared and analyzed under the same conditions was readily dissociated into dimers at lower concentration (Supplementary Fig. S5), similar to what was previously observed for WT Mrr [19].
Fig. 2. Effect of pressure or M.HhaII MTase on
the cellular localization and stoichiometry of GFP-Mrr and variants. (A) Fluorescence in-tensity of E. coli single live cells with a chro-mosomal PBAD-gfp::mrr insertion encoding wild-
type Mrr (GFP-Mrr) or variants carrying single mutations abolishing Mrr catalytic activity (D203A carried by Mrr-Inact) or affecting Mrr activation by either pressure (V173A carried by Mrr-HPR) or induction of the M.HhaII MTase (H279Y carried by Mrr-MTR). Average fluores-cent intensity maps retrieved from 2-photon sN&B analysis are shown for unstressed cells and for cells after a pressure stress (15 min at 100 MPa) or IPTG induction of the plasmid- encoded M.HhaII MTase. Image size is 20 × 20μm and maximum intensity is 2.5
counts per 40μs pixel dwell-time. B)
Stoichi-ometry values of GFP-Mrr WT and variants as observed in A, deduced from the background corrected molecular brightness values relative to that of monomeric GFP. Solid and open red crosses indicate the observation of, resectively, strong or mild cell filamentation indicative of the SOS response. Error bars represent the standard deviation of the stoichiometry values calculated in at least 8 independent experi-ments, each averaging the brightness values measured inside individual bacterial cells within 6 to 8 FOVs (~500 cells).
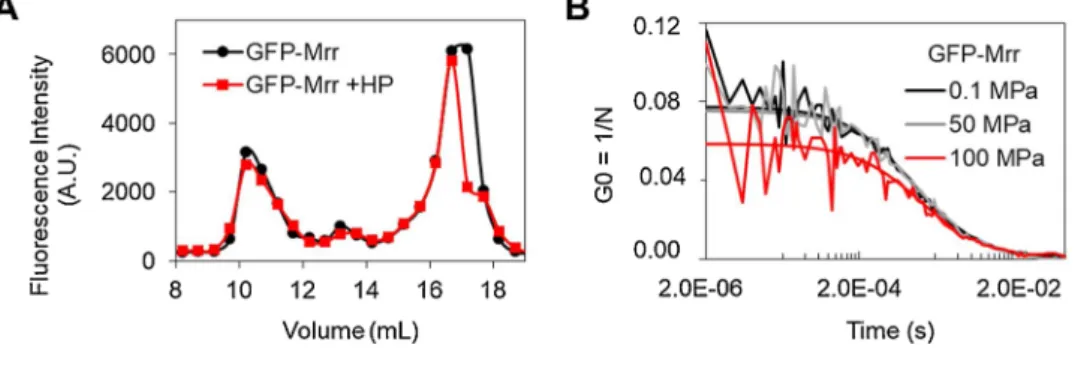
3.3. Pressure leads to reversible dissociation of GFP-Mrr oligomers in vitro
The change in GFP-Mrr stoichiometry observed by sN&B in vivo was monitored after exposure of the cells to a HP shock of 100 MPa for 15 min and return to atmospheric pressure. However, these observations did not conclusively demonstrate that the Mrr protein dissociates because of HP. Indeed, pressure could conceivably change the confor-mation of the nucleoid, increasing Mrr affinity for chromosomal DNA and pulling the protein toward dimer, much as occurs upon hyper- methylation of DNA after MTase expression. We thus sought to examine in vitro whether pressure acts directly on the Mrr protein and leads to the dissociation of Mrr oligomers. We first compared the SEC profile of the purified Strep-tagged GFP-Mrr before and after a HP shock (Fig. 3A). No difference was observed, indicating that if pressure had any effect on the oligomeric properties of the purified protein in vitro, this effect was fully reversible after return to atmospheric pressure. In order to observe the protein behavior directly under pressure, we implemented a high-pressure sample chamber with a 2-photon scanning microscope using a silica capillary system derived from Muller and Gratton [27] and described in Bourges et al. [20]. Here we used this set-up to measure FCS profiles of a pressurized Strep-tagged GFP-Mrr solution in 2-photon excitation mode. As a control, we carried out the same experiment for the purified Strep-tagged GFP protein. The G(0) value of the FCS curves (corresponding to the plateau value of G(τ) at
short times) is inversely proportional to the number of particles diffusing in the effective volume, Veff, and directly proportional to their
bright-ness. Dissociation of a higher order oligomer would lead to both an in-crease in the number of particles and a dein-crease in their brightness, and hence a lower value for G(0). As expected, pressure had no effect on the G(0) value obtained for free monomeric GFP (Supplementary Fig. S6A). In contrast, we observed a large decrease in the G(0) value for the pu-rified Strep-tagged GFP-Mrr at 100 MPa, the pressure limit of our capillary system (Figs. 3B and S6B). Unfortunately, the presence of free GFP, due to proteolytic cleavage in the protein extracts (see Supple-mentary Fig. S5) precluded accurate stoichiometric calculations of the brightness values. Nonetheless, these results demonstrate that HP directly leads to dissociation of Mrr oligomers.
3.4. Structural model of tetrameric Mrr catalytic domain
We next turned to homology modeling to obtain structural insight into the coupling between oligomerization and catalysis in Mrr and its
variants. The Mrr protein has been predicted to contain an N-terminal winged helix (wH) DNA binding domain comprising a helix-turn-helix (HTH) DNA recognition motif, and a C-terminal catalytic (CAT) domain comprising the canonical PD-(D/E)XK phosphodiesterase/metal binding motif found in a wide variety of DNA processing proteins including many restriction and modification enzymes [[39]. Recogni-tion of methylated DNA is thought to be carried out by the wH domain, whereas cleavage by the CAT domain occurs at some distance from the recognition sequence, as is typical for type IV REs [6]. We first con-structed a homology model of the Mrr-CAT domain (residues 131–304) based on sequence alignment to the cleavage domain of two distantly related methyl-directed restriction enzymes MspJI and AspBHI [21,36] (Supplementary Fig. S7). These type IIM REs present an “Mrr-like” ID-(Q/E)XK variation of the catalytic motif in the C-terminal domain [37], and as predicted for Mrr, they recognize methylated DNA through the N-terminal domain. Moreover, both MspJI and AspBHI are homo-tetramers, therefore we could use the MspJI crystal structure as a template to model a tetrameric Mrr-CAT structure (Fig. 4A). Despite the low sequence identity (~18 % over 170 residues), good quality models were automatically generated, in which the overall three-dimensional fold and the tetrameric organization of the catalytic core are pre-served with rather well-conpre-served monomer-monomer and dimer-dimer interfaces. The architecture of the putative active site seems also well conserved, in particular residues predicted to be directly implicated in metal ion-mediated catalysis (E172, D203, Q219, K221 and E231) [37]. Interestingly, in the different protomers of the modeled tetramer, the glycine-rich loop 193–203 containing the catalytic residue D203 ap-pears with different conformations. These could correspond to distinct activation states in the Mrr enzyme. This catalytic loop extends toward the dimer-dimer interface where the DNA helix to be cleaved is expected to bind (Fig. 4B). It is plausible that substitution of the aspartyl side- chain of residue 203, besides abolishing catalysis, may also compro-mise the interactions between dimers and the stability of the Mrr tetramer, as suggested by the reduced stoichiometry measured in vivo for the Mrr-Inact variant carrying the D203A mutation. Similarly, residues V173 and V175 are located in a conserved helix of the scaffold forming the ID-(Q/E)XK phosphodiesterase active site and against which the catalytic loop can pack through a network of hydrophobic and electro-static interactions. Replacement of these residues by a smaller residue such as alanine would create local cavities that could modify the active site shape. This could modify the alternative packing of the catalytic loop at the dimer interface, leading to Mrr variants with reduced activity
Fig. 3. Reversible pressure-induced
dissocia-tion of purified wild-type GFP-Mrr in vitro. (A) Size exclusion chromatography of purified Strep-tagged GFP-Mrr before (black curve) and after (red curve) high pressure (HP) treatment (15 min at 100 MPa). The GFP-Mrr protein fusion was mostly present as oligomers detected in the fluorescent peak eluting at 10.5 mL, as well as in the minor peak at 13 mL corre-sponding to an apparent molecular weight of 130 kDa compatible with a dimer. No change is observed following the HP treatment after re-turn to atmospheric pressure, indicating that HP has no or reversible effect on GFP-Mrr oligo-meric properties in vitro. (B) Fluorescence cor-relation spectroscopy (FCS) curves of purified Strep-tagged GFP-Mrr under pressure. The decrease in the G(0) value observed at 100 MPa is due to an increase in the number (N) of diffusing particles detected in the 2-photon excitation volume, indicating the HP-induced dissociation of GFP-Mrr oligomers into more numerous entities of lower oligomeric state (see related Supplementary Figure S6).
and/or altered oligomeric properties affecting the Mrr activation process.
3.5. Model of the full-length Mrr tetramer in complex with DNA
In order to gain further insight into the mechanism of Mrr activation and the structural consequences of the mutations affecting this mecha-nism, we built a chimeric model of the full-length Mrr tetramer bound to DNA. Particularly relevant for this purpose was the available crystal structure of the MspJI tetramer in complex with DNA (PDB:4R28, [22]). The DNA recognition domain at the N-terminus of this type II RE adopts a totally different fold from that predicted for Mrr and could thus not serve as a template. We chose the wH domain of the DtxR transcription factor bound to a 50-mer DNA fragment (PDB:1F5T, [38]) to model a protein-DNA complex for the N-terminal domain of Mrr (residues 1–93). For the long linker region that connects the wH and CAT domains (residues 94–130), structural modeling predicts two putative alpha he-lices at equivalent positions to those seen in the MspJI linker region (Supplementary Fig. S7). In the MspJI tetrameric structure, this linker region adopts two distinct conformations, leading to different orienta-tions of the DNA recognition domain relative to the CAT domain in the full-length monomers and dimers [21,22]. We retained the same two conformational types of the linker region to model the full-length Mrr tetramer bound to DNA (Fig. 4C and D). In this model, an “open” dimer can interact with DNA simultaneously through the wH recognition domain of one protomer and the catalytic CAT domain of the other protomer, while the “closed” dimer has its wH domains tightly packed against the CAT domains and the linker helices (Supplementary Fig. S8). In the closed configuration, the HTH DNA recognition motifs of the wH domains remain exposed, but a DNA fragment bound at these sites would make no further stabilizing contacts with the protein. Instead, the multiple intra- and inter-subunit interactions made by the wH domains in the closed configuration with the catalytic core as well as the long linker region are predicted to greatly contribute to the overall stability of the tetramer. As in the MspJI template structure, this organization precludes engagement of the DNA double helix for cleavage at the cat-alytic interface, keeping the catcat-alytic residues far from the DNA, thereby maintaining the enzyme in an inactive state. Disruption of such a tetrameric assembly is thus required to permit productive binding of the DNA substrate to the Mrr enzyme.
In light of the 3D model of the full-length Mrr tetramer, we further examined the structural effect of Mrr mutations that could not be inferred from the modeling of the conserved catalytic domain alone (Supplementary Fig. S8). Based on this model, the substitution of a histidine residue with a more hydrophobic tyrosine at position 279 could improve the interaction between the catalytic core and the protein N-terminus in the closed configuration (Supplementary Fig. S8). This would stabilize the compact conformation of the polypeptide chain (in agreement with the position of the mutation site in the closed configu-ration shown in Fig. 4C and Supplementary Fig. S8) and thereby stabi-lize the tetramer. Alternatively, it is possible that such a mutation could affect dynamic transitions that allow formation of the active dimer without directly impacting tetramer stability. Conversely, mutations to smaller residues located in the linker region, as found in the Mrr constitutive variant (N111S and D124G), could increase the linker flexibility and/or alter domain packing in the tetramer, facilitating the switch to the active dimer. The third mutation carried by this constitu-tive mutant, V175G located in the vicinity of the catalytic loop (Fig. 5B), if not fortuitous, could reduce the enzymatic activity and may thereby avoid lethality of the mutant strain producing Mrr dimers, allowing for its isolation. The V173A mutant, also positioned near the catalytic loop, could likewise lower the enzymatic activity, such that the effect of pressure might be diminished, particularly at high concentrations (when expressed from a plasmid) that favor the inactive tetramer.
4. Discussion
We sought here to identify the molecular mechanism by which the type IV Mrr restriction enzyme is activated and triggers the SOS response in E. coli after exposure to elevated pressure or DNA methylation by an exogenous MTase. We found that a constitutively active mutant Mrr is dimeric in live E. coli cells in absence of any perturbation and that other variants compromised in their activity or activation process display altered oligomeric properties, as well. We also demonstrated by high pressure FCS experiments that pressure dissociates purified Mrr tetra-mers directly. These observations provide strong support for our previ-ously published model for Mrr activation based on an oligomeric switch triggered by HP shock or MTase expression [19]. In this model we proposed that Mrr is maintained in an inactive tetrameric form in un-stressed cells and that it can be converted into a catalytic dimer under perturbative conditions by two distinct mechanisms. High pressure, which is known to dissociate oligomers, would push the Mrr tetramer-dimer equilibrium toward the active dimer, which recognizes and cleaves at putative cryptic sites present in the bacterial chromo-some. In contrast, expression of a specific MTase such as M.HhaII creates a large number of high affinity methylated DNA sites and thereby drastically decreases the molar ratio between Mrr subunits and target sites. Mrr binding to these sites eventually pulls the tetramer-dimer equilibrium toward the active dimer in which each protein subunit in-teracts with a DNA target site. The fact that it has been possible to isolate mutations that render Mrr less sensitive to either HP or MTase expres-sion supports the existence of mechanistic differences in the manner by which these two perturbations activate Mrr. The Mrr dimers released after HP shock or favored by MTase induction would then remain associated with the DNA, forming foci upon nucleoide condensation following the recruitment of the SOS response machinery at the dsDNA cleavage sites.
The three-dimensional model of the full-length Mrr tetramer pro-posed here provides insights into the structural features that may govern the coupling between Mrr activity and oligomerization. Given the functional and structural similarities between Mrr and the methyl- specific MspJI RE used as template, we are confident that this model represents a reasonable depiction of the Mrr tetramer, most notably for the C-terminal CAT domain forming the conserved catalytic core. This CAT tetramer presents two types of dimeric interfaces (Fig. 4A). One involves an extended interaction surface between loops of the central β-sheet structure of each protomer. It is this interface that associates the CAT domains back-to-back to form the “open” or “closed” dimer (Sup-plementary Fig. S8) as seen in the full-length structure of the MspJI tetramer [21]. However, neither one of these dimeric configurations likely corresponds to the active dimer. Given the structural features of the MspJI phosphodiesterase superfamily [37,39], it is most likely that the catalytic dimer of Mrr is formed by helical contacts between conserved elements of the PD-(D/E)XK motif, comprising the catalytic loop that folds at this dimeric interface (associating the green and yel-low, or cyan and pink protomers shown in Fig. 4). In this dimeric configuration, the CAT domains are associated head-to-head via a less extensive interface compared to the back-to-back dimeric form. There-fore the intrinsic stability of this catalytic dimer is expected to be low unless stabilized by additional protein-DNA or inter-subunits interac-tion, as seen in other DNA modifying enzymes of this structural family [39–41]. In MspJI, mutations introduced in the conserved helices forming this interface resulted in much reduced ds cleavage activity, demonstrating that the stability of the head-to-head CAT dimer is crucial for catalysis [21]. Yet, the back-to-back dimer being much more favor-able, we propose that it assembles readily upon protein folding, then further dimerizes head-to-head to form an inactive tetramer in which the catalytic sites at the dimer-dimer interface are not accessible or properly orientated for dsDNA cleavage. Hence, under normal conditions, the active head-to-head dimer would never be populated enough to have deleterious cryptic endonuclease activity on the bacterial genome. In the
presence of specific target DNA (following the entrance of foreign methylated DNA or expression of an exogenous MTase), the strong in-teractions made by the N-terminal recognition domains with the large number of high affinity sites could destabilize the inactive tetramer by disrupting the back-to-back dimeric interface, thereby favoring the active head-to-head dimer on the DNA substrate.
As previously, we propose a distinct mechanism for pressure-induced Mrr activation. Mrr affinity for chromosomal DNA methylated by E.coli endogenous dam and dcm MTases is insufficient to trigger the
conformational switch leading to dissociation to the active dimer. Hence, the active dimer must be formed under pressure, and subse-quently bind to DNA. Internal cavity volume has been shown to be the primary determinant of pressure unfolding of proteins [42], and likely is the major contributing factor to pressure-induced oligomer dissociation. Although calculation of cavity volume is highly speculative in case of modeled structure, the lower packing efficiency, and hence more solvent excluded void volume, at the back-to-back interface relative to the head-to-head interface could favor pressure-induced release of the
Fig. 4. Homology modeling of Mrr. (A)
Tetra-meric model of Mrr catalytic domain (CAT, residues 131-304) based on the crystal structure of the MspJI type IIM restriction enzyme (PDB:4FOQ, [21]). The amino acid side-chain of mutated residues are shown as spheres colored in red for the mutation abolishing Mrr catalytic activity (D203A carried by Mrr-Inact), in orange and dark blue for the mutations uncoupling Mrr activation by pressure (V173A carried by Mrr-HPR) or co-expression of M. HhaII MTase (H279Y carried by Mrr-MTR), respectively, and in dark grey for the V175G mutation found in the Mrr constitutive mutant (Mrr-Const) together with N111S and D124G outside the CAT domain. The C-termini of the Mrr polypeptides chains are indicated as grey spheres (B) Zoom on the dimer-dimer interface showing two superimposed conformations of the catalytic loop (residues 193-203 colored in dark blue or dark grey) viewed in the modeled structure of the Mrr-CAT tetramer. The pre-dicted position of a DNA helix bound at the catalytic interface is shown as a dashed orange circle, as proposed for the active DNA/MspJI complex (21). The side-chains of residues V173, V175 and D203 are in stick representation. (C) Model of the full-length Mrr monomer showing the winged helix (wH) DNA binding domain (residue 1-93) in the “open” (cyan) and “closed” (magenta) conformation and the superimposed C-terminal catalytic domain. The N-terminal wH domain and the linker (residues 94-130 colored in red) are shown at positions equiva-lent to those seen in the inactive MspJI/DNA complex structure used as template (PDB:4R28, [22]). Colored spheres represent the side chains atoms of mutated residues in the catalytic domain colored as in panel A, or in the linker region colored in green (N111 and D124). Grey spheres correspond to the Mrr N-terminus where the GFP-tag is fused. (D) Model of the Mrr tetramer in complex with DNA in which two monomers (colored in cyan and yellow) are in the “open” conformation and interact with DNA with either the wH recognition domain (in yellow) or the catalytic CAT domain (in cyan), while the other two monomers (colored in green and magenta) are in the “closed” conformation with the wH domain packed against the CAT domain (see also Supplemen-tary Figure S8). The side chain of the catalytic residue D203 of each monomer is shown as red sphere. The DNA fragment shown in cartoon representation was extracted from the template structure (PDB:1F5T [38]) used for modeling the Mrr-wH/DNA complex. The predicted po-sition of a DNA fragment bound at one of the cleavage site as shown in panel B is indicated as a dashed orange cylinder.
active dimer. High pressure could also disrupt other intra- and inter-subunit interactions involving other protein regions, in particular the linker region, that greatly contribute to the overall stability of the tetrameric structure.
In Mrr as in the MspJI restriction enzyme used for homology modeling, replacement of the catalytic residue D203 or D334, resp., (the first “D” of the PD-(D/E)XK conserved motif) abolishes the endonuclease activity [21,23], yet this essential residue is not seen in direct contact with DNA in the modeled or X-ray determined structure of the complex (Fig. 4D). In the MspJI co-crystals with a specific oligonucleotide, one of the recognition domains of the “open” dimer interacts with the meth-ylated MspJI recognition sequence, but none of the active centers formed at the catalytic dimer-dimer interfaces is in position for hydro-lyzing the bound DNA molecule and cleavage does not occur. The proper positioning of the DNA molecule at one of the two paired catalytic sites requires reorientation of the recognition domain, but in the tetrameric configuration, this latter is held in place via the extensive interaction network it makes with the protein subunits forming the “closed” dimer. Since crystallization was carried out using a ratio of specific DNA site-to-tetramer of 2:1, it is likely that this non-productive tetrameric complex corresponds to an inactive or resting state in which the enzyme is maintained in the absence of sufficient specific targets to bind. Indeed, MspJI activity is strongly dependent on the concentration of DNA tar-gets, with maximal activity observed for a site-to-tetramer ratio of 4:1 [21]. This suggests that efficient cleavage by this restriction enzyme occurs when all four recognition domains are engaged in specific con-tacts with DNA rather than in protein concon-tacts, thereby allowing the domain rearrangement required for the cleavage process. In the case of Mrr, the concentration-dependent response to MTase expression
observed in vivo for the wild-type as well as mutant GFP-Mrr fusions (Fig. 2 and Supplementary Fig. S4) indicates that the molar ratio be-tween the Mrr enzyme and its DNA target sequences is also crucial for the activation mechanism. Dissociation of the Mrr tetramer upon interaction with hyper-methylated DNA or exposure to high pressure would similarly permit an alternative conformation of the recognition domain compatible with the productive binding of the DNA duplex at the active site of the catalytic domain. After reconfiguration of the dimer (and eventually of the catalytic loop) each protomer of the catalytic dimer can cleave one strand of the DNA, generating double-strand breaks.
Based on the present experimental and modeling studies, we inferred a structural mechanism for the allosteric regulation of Mrr that trans-forms the inactive tetramer into the catalytic dimer ready to cleave dsDNA (Fig. 5). In this model we consider Mrr in equilibrium between states resulting from alternative conformations and interactions be-tween protein domains and subunits that compete with productive protein-DNA interactions. In Fig. 5 we indicate how these equilibria are most influenced by intrinsic (mutations and protein concentration) or external perturbations (HP or exogenously methylated DNA). In Sup-plementary Fig. S10 we represent the conformational state of wild-type and mutant Mrr which, according to this model and in agreement with our quantitative fluorescence microscopy data, is predominant in un-stressed cells and after exposure to HP shock or expression of M.HhaII MTase.
Besides the effect of HP and DNA hyper-methylation already dis-cussed, this model explains some intriguing observations regarding the effect of mutations that alter Mrr activity or activation. In particular, the existence of two types of dimers provides a structural explanation for
Fig. 5. Schematics of the proposed conformational switch leading to the Mrr catalytic dimer ready for dsDNA cleavage. Grey squares and triangles represent Mrr CAT
catalytic domains and wH DNA recognition domains, respectively, with the linker region in between indicated as a thin black line. The double strand DNA helix is shown as thin blue lines. a) After synthesis, Mrr protomers readily associate back-to-back to form an inactive dimer where the catalytic sites are inoperative (red crosses). b) The back-to-back dimers further assemble head-to-head and form an inactive compact tetramer that binds nonspecifically to cryptic sites (white rect-angles) on the chromosomal DNA. c) Exposure to high pressure disrupts the back-to-back dimeric interface and favors the active head-to-head dimer where both catalytic centers are in position for dsDNA cleavage (green circles). DNA methylation (by an exogenous MTase or carried by foreign DNA) provides high affinity sites (blue rectangles) for the binding of the wH domains, disrupting their interactions with the CAT domains and pulling the tetramer-dimer equilibrium towards the active dimer. Binding of the DNA helix at the catalytic interface further stabilizes the active dimer and allows ensuing double strand breaks. Green and red arrows indicate, resp., the positive or negative effect of external perturbations (high pressure, DNA methylation) and intrinsic factors (mutations, protein concentration) on the conformational equilibria involved in Mrr activation.
mutations favoring a dimeric form of Mrr which can range from totally inactive or constitutively active. Our structural predictions suggest that residues D203, V173 and V175 are within or in the vicinity of the cat-alytic loop at the head-to-head dimer interface where the DNA double helix can bind and be cleaved (Fig. 4B). Thus, replacement of these residues is likely to destabilize this catalytic interface and lead to Mrr variants displaying both a lower degree of oligomerization (but in the inactive back-to-back dimeric form) and altered catalytic efficiency. Since HP-induced dissociation of WT Mrr favors the active head-to-head dimer, these mutants appear resistant to HP treatment. However, interaction with numerous high affinity, specific DNA targets produced by induction of the MTase can lead to formation of active dimers despite destabilization of this interface. Stabilized by interactions with the DNA substrate, the MrrV173A catalytic dimer can cause damage to the bacte-rial chromosome sufficient to trigger an SOS response, whereas MrrD203A
missing the catalytic aspartyl side-chain remains inactive (Fig. 2). In contrast, the N111S/D124G mutations located in the linker region would disrupt key interactions in the inactive tetramer and release the catalytic dimer, thereby rendering the enzyme constitutively active (and potentially lethal in the absence of the V175G mutation).
Another puzzling observation is the distinct responses to external perturbations exhibited by the Mrr-MTR variant when expressed at high concentration from a plasmid or at low concentration from a chromo-somal construct. Our in vivo and in vitro characterization of this variant suggests that the H279Y mutation carried by this variant may increase the stability of the Mrr tetramer (Fig. 2B and Supplementary Fig. S5). According to our structural model, this stabilizing effect could enhance interactions between the catalytic domain and the protein N-terminus, maintaining the DNA-recognition domain in the “closed” configuration that stabilizes the tetrameric assembly and prevents competing in-teractions with cognate DNA (Fig. 4C and Supplementary Fig. S8). The stability of the Mrr-MTR tetramer is expected to be further increased at high protein concentration (increasing also the enzyme-to-site ratio), which could explain why foci formation was not observed upon M.HhaII induction when over-produced in the strain used previously for epi-fluorescence microscopy (Supplementary Fig. S4). However, despite overall stabilization of the Mrr-MTR tetramer, the H279Y mutation would diminish, but not prevent, dissociation of the tetramer at high pressure (and may increase the population of the inactive dimer, relative to the active one), limiting the amount of DNA cleavage by the released active dimer which could more readily re-associate into the stable compact tetramers after returning to atmospheric pressure.
Our 3D-model of the full-length Mrr tetramer also provides further interpretation of previously reported mutational effects that have remained so far unexplained (Supplementary Fig. S9). In an earlier structural model of monomeric Mrr bound to DNA, two conserved arginine residues (R68 and R77) whose substitution inactivates the Mrr enzymes were not seen in direct contact with DNA [23], whereas in the present model of the Mrr wH domain based on the DtxR/DNA complex structure, these residues are located in the recognition helix and in the β-sheet “wing” of the HTH motif, respectively, and both make crucial non-specific contacts to the phosphate backbone of the DNA, as seen in the template structure. The S92P mutation isolated by screening for spontaneous HP-resistant mrr mutant alleles [17] involves a non-conserved residue which is not expected to be crucial for either DNA recognition or catalysis but which, according to our model, is flanking the linker region connecting the wH and CAT domain. Introduction of a proline residue in this region is likely to affect the structure and/or dynamics of the helical linker where the conformational switch between the open and closed dimer takes place. Using another HP-based assay for isolating point mutations that abolish Mrr activation, Aertsen and col-leagues identified four independent Mrr variants with amino acid sub-stitutions (R181L, Y184D, G185S, Q192P) all situated in the same region of the CAT domain, again outside the conserved elements of the catalytic core [23]. This unexpected finding suggested to the authors that HP-induced Mrr activation may depend on yet uncharacterized
protein-protein interactions in addition to protein-DNA interactions. This prediction is indeed supported by our model which shows that these mutations occur at the subunit interface of the Mrr tetramer where the HP-induced dissociation would be initiated. Interestingly, a close ho-molog of Mrr from Salmonella Typhimurium LT2, although it is not activated by high pressure, can be readily activated by different spon-taneous mutations, indicating that LT2 Mrr can easily adopt an active conformation that elicits the cellular SOS response [43]. The most effi-cient single amino substitutions that render LT2 Mrr constitutively active involve Gly186 and Ile202 which according to our homology model are also implicated in intra- and inter-subunits contacts that are disrupted upon the oligomeric switch leading to activation.
Overall these studies validate our previously published push/pull model for pressure and MTase effects on Mrr activity [19] and provide an example of how proteins use alternative oligomeric interactions for allosteric modulation of endonuclease activity. Although structural models based on distant homologs should not be over-interpreted and details in the interaction surfaces remain highly speculative, we believe that the general activation mechanism inferred for Mrr is valid and is in fact reminiscent to that proposed earlier for other restriction enzymes. Indeed, the coupling between catalysis and oligomerization seems to be a common trait of multi-modular endonucleases that require more than one DNA recognition site for efficient cleavage. In case of MspJI, it re-mains to be established whether activation of this RE in the presence of its target DNA involves a tetramer-dimer switch, as we propose for Mrr, or a reconfiguration of the tetramer allowing productive binding of the DNA substrate by the catalytic dimer. Besides MspJI, the endonuclease FokI, the best characterized member of Type IIs restriction enzymes, presents striking functional and structural similarities with Mrr. This unusual RE is also composed of a N-terminal DNA recognition domain made of wH motifs and a C-terminal PD-(D/E)XK catalytic domain that cleaves non-specifically some distance away from its DNA target sequence. Although FokI can interact specifically with DNA as a monomer, full activation of its nuclease activity requires formation of a dimeric complex upon binding of two recognition sequences [41,44]. In the free FokI dimer as in the monomeric FokI/DNA complex, the pro-tomer adopts a compact structure comparable to that seen in the “closed” form of the MspJI subunits where the cleavage domain is packed alongside the recognition domain in a position that does not permit DNA cleavage [22,45]. Similar to what we propose for Mrr based on the structure of the MspJI/DNA complex, the sequestration of the cleavage domain in an inactive oligomeric state (monomeric for FokI, tetrameric for Mrr) involves an extensive set of proteprotein teractions, in particular in the linker region. These intramolecular in-teractions must be disrupted and replaced by intermolecular interactions with DNA for the enzyme to reach the active state where the head-to-head dimer interface is competent for binding and double strand cleavage of the DNA. The fact that mutations that relax FokI cleavage specificity mapped not in the recognition domain but at inter-domain interfaces further validated the activation mechanism postulated for FokI [45]. We believe that this mechanism applies as well to Mrr and other non-conventional REs whose activity is stimulated in the presence of multiple target sequences [46]. As previously proposed [47], the simultaneous recognition of multiple DNA sites by these restriction en-zymes would ensure that single binding events do not lead to chromo-somal DNA damage at cryptic sites in the host cell.
Authors statement
Bourges developed and performed all the fluorescence microscopy and spectroscopy experiments, as well as purification and in vitro char-acterization of the proteins ; O.E. Torres Montaguth and W. Tadesse obtained the Mrr mutants ; G. Labesse performed the 3D modeling and contributed to manuscript editing ; A. Aertsen contributed conceptual-ization, ideas and the reviewing of the manuscript; C. Royer contributed to the conceptualization, methodological development, supervision, and