HAL Id: hal-02787501
https://hal.inrae.fr/hal-02787501
Submitted on 5 Jun 2020
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Développement d’outils sous Galaxy pour la
reconstruction phylogénétique et pour l’analyse
statistique liée à la métagénomique.
Thi Ngan Ta
To cite this version:
Thi Ngan Ta. Développement d’outils sous Galaxy pour la reconstruction phylogénétique et pour l’analyse statistique liée à la métagénomique.. Bio-informatique [q-bio.QM]. 2017. �hal-02787501�
2
eannée de Master Bio-Informatique et Génomique
Développement d’outils sous Galaxy pour la reconstruction
phylogénétique et pour l’analyse statistique liée à la
métagénomique
TA Thi Ngan - Année universitaire 2016/2017
Sous la direction de :
Maria Bernard de l’unité GABI – INRA Jouy en Josas
Mahendra Mariadassou de l’unité MaiAGE – INRA de Jouy en Josas
Géraldine Pascal de l’unité GenPhySE – INRA de Toulouse
R
EMERCIEMENTS
Je tiens particulièrement à remercier Géraldine PASCAL, Maria BERNARD et Mahendra MARIADASSOU, mes maitres de stage, pour leur patience, pour toute leur aide qu’ils m’ont apporté dans la recherche de stage, pour leurs précieux conseils tout au long de mon stage ainsi que pour avoir pris le temps de relire et corriger plusieurs fois mon rapport. Ils m’ont donné une grande occasion pour travailler dans une super condition. J'ai beaucoup appris de connaissances et de l’expérience qui me suivra dans tous mes futurs travaux.
Je veux donner un grand merci à Maria BERNARD qui m’a accueilli dans son équipe, pour son attention et sa bonne humeur, pour ses innombrables idées et ses remarques pertinentes pour l’avancée du projet et pour m’avoir accompagné et conseillé.
Je tiens à remercier toute l'équipe d’accueil de l’unité GABI et de l’unité MaIAGE, en particulier tous les membres ainsi que la responsable Mrs. Denis Laloë de l’équipe PSGen, pour son accueil chaleureux et la bonne ambiance. Merci aux secrétaires de GABI pour leurs aides. Je tiens à remercier tous les personnes de l’INRA Jouy en Josas.
Je tiens à remercier aussi les autres stagiaires présents, Frédéric, Anna, Raphaëlle, Julie, Sam, Lien, Hoang, ... pour leur bonne humeur tous les jours, aussi pour l’aide apportée et surtout pour ces moments de rire lors des pauses déjeuner. Merci à Marco, Mathieu, Kenza, Andrea, Tatiana,... pour leurs conseils et leurs aides.
Je tiens à remercier également toute l’équipe pédagogique du Master BIG (Bioinformatique et Génomique) et les enseignants responsables de la formation, pour nous avoir assuré la partie théorique de celle-ci.
T
ABLE DES MATIERES
Remerciements ... 1 INTRODUCTION ... 1 1.1 Métagénomique Amplicon ... 1 1.1.1 Les enjeux ... 1 1.1.2 Le séquençage d’amplicons ... 11.1.3 Les bases de données et les outils d’analyse ... 1
1.2 Développement d’outil sous Galaxy liée à la métagénomique amplicon ... 3
1.2.1 La plateforme Galaxy ... 3
1.2.2 FROGS: Find, Rapidly, OTUs with Galaxy Solution... 3
1.2.3 Les besoins en analyses statistiques ... 4
2 MATERIELS ET METHODES ... 5
2.1 Démarche méthodologique pour développement d’outil sous Galaxy ... 5
2.1.1 Galaxy ... 5
2.1.2 Les langages de programmation ... 5
2.2 Démarche méthodologique pour la reconstruction d’arbre phylogénétique ... 6
2.2.1 PyNAST ... 6
2.2.2 MAFFT ... 6
2.2.3 FastTree2... 7
2.3 Démarche méthodologique pour l’analyse statistique liée à la métagénomique ... 7
2.3.1 Phyloseq ... 7
2.3.2 DESeq2 ... 9
3 RESULTATS... 9
3.1 FROGS Tree : l’outil pour la reconstruction d’arbres phylogénétiques ... 10
3.2 FROGS Phyloseq : les outils sous Galaxy pour l’analyse statistique des données d’écologie microbienne ... 11
3.2.1 Import data ... 11
3.2.2 Visualisation des compositions ... 13
3.2.3 L’alpha diversité ... 14
3.2.4 Diversité bêta ... 15
3.2.5 Visualisation de structure ... 17
3.2.6 Regroupement hiérarchique des échantillons ... 18
3.2.7 L'analyse de variance multivariée (MANOVA) ... 19
3.2.8 FROGS DESeq2: deux outils pour l’analyse différentielle des abondances ... 20
4 DISCUSSION ... 23 5 CONCLUSION ET PERSPECTIVES ... 25 6 Références ... 7 Annexe 1 ... 8 Annexe 2 ... 9 Annexe 3 ...
1
1 INTRODUCTION
1.1 M
ETAGENOMIQUEA
MPLICON 1.1.1 Les enjeuxLes micro-organismes jouent un rôle essentiel pour le bon fonctionnement des écosystèmes1,2. Les communautés microbiennes varient dynamiquement en réponse aux gradients tant spatiaux que temporels et la plupart des micro-organismes sont les membres de communautés complexes, composés de cent à des milliers de taxa différents3. Les outils de métagénomique ont été développés notamment pour décrire et comprendre la composition et la structure microbienne des environnements4. C’est une approche nouvelle et puissante. L’enjeu de cette approche est de permettre d’analyser les génomes de tous les microorganismes d’une niche écologique, même de ceux, largement majoritaires, qui ne peuvent pas être cultivés. Elle se fait par l’échantillonnage aléatoire des génomes d’un petit sous-ensemble des micro-organismes présents dans un environnement5. Cependant le séquençage complet des génomes d’un environnement est couteux en temps, en argent et en ressources humaines et informatiques. L’approche de la métagénomique par amplicons est donc une bonne alternative car plus accessible. Elle se base sur l’amplification et le séquençage d’une petite partie du génome, chez les bactéries. Il est fréquent de s’intéresser à certaines régions des gènes de l’ARNr 16S.
1.1.2 Le séquençage d’amplicons
Le séquençage d’amplicons est une technique fondamentale en métagénomique6. Elle se fait par l’amplification par PCR d’un gène ou d’une partie d’un gène ubiquitaire, dont le taux évolutif est proche de celui de l’espèce7
. C’est la raison pour laquelle il est souvent ciblé, chez les bactéries, le gène de l’ARNr 16S. Le séquençage d'amplicons permet l’analyse de données en écologie microbienne à grande profondeur et permet ainsi la recherche d’espèces faiblement présentes dans l’environnement8,9
. Bien que quelques problèmes techniques existent, tels que des biais de PCR et d'efficacité d'extraction de l'ADN, ces techniques aident à une meilleure compréhension du monde microbien et sont largement utilisées9.
1.1.3 Les bases de données et les outils d’analyse
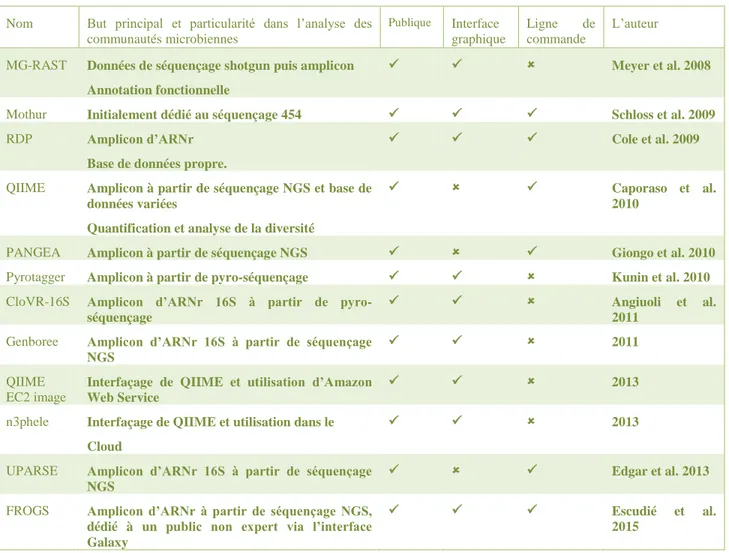
À la suite du séquençage les biologistes doivent analyser et affilier leurs données. Ils ont pour ça le choix d’un assez grand nombre d’outils (Tableau 1) et de bases de données (Tableau 2). Les bases de données les plus abondantes et qui sont les références dans le domaine pour les gènes d’ARNs ribosomiques sont Greengenes10
, SILVA11,12 et RDP-II15. Les outils d’analyse les plus utilisés sont : MG-RAST13
, mothur14, RDP15, QIIME16, PANGEA17, Pyrotagger18, CloVR-16S19, Genbore (2011), QIIME EC2 image (2013), n3phele (2013), UPARSE20 et FROGS21.
2
Nom But principal et particularité dans l’analyse des communautés microbiennes Publique Interface graphique Ligne de commande L’auteur
MG-RAST Données de séquençage shotgun puis amplicon Annotation fonctionnelle
Meyer et al. 2008
Mothur Initialement dédié au séquençage 454 Schloss et al. 2009
RDP Amplicon d’ARNr
Base de données propre.
Cole et al. 2009
QIIME Amplicon à partir de séquençage NGS et base de données variées
Quantification et analyse de la diversité
Caporaso et al.
2010
PANGEA Amplicon à partir de séquençage NGS Giongo et al. 2010
Pyrotagger Amplicon à partir de pyro-séquençage Kunin et al. 2010
CloVR-16S Amplicon d’ARNr 16S à partir de
pyro-séquençage
Angiuoli et al.
2011
Genboree Amplicon d’ARNr 16S à partir de séquençage
NGS
2011
QIIME EC2 image
Interfaçage de QIIME et utilisation d’Amazon Web Service
2013
n3phele Interfaçage de QIIME et utilisation dans le Cloud
2013
UPARSE Amplicon d’ARNr 16S à partir de séquençage
NGS
Edgar et al. 2013
FROGS Amplicon d’ARNr à partir de séquençage NGS,
dédié à un public non expert via l’interface Galaxy
Escudié et al.
2015
Tableau 1 : Résumé des outils d’analyse des données de communautés microbiennes
Nom Description référence
Greengenes Base de données des gènes d’ARN ribosomiques 16S sans chimère. Dernière mise à jour : mai 2013
DeSantis et al. 2006
SILVA Base de données des gènes d’ARN ribosomiques de Bactéries, d’Archaeas et d’Eucaryotes
Dernière mise à jour : septembre 2016
Pruesse et al. 2007 Quast et al. 2013
RDP-11 Base de données des gènes d’ARN ribosomiques 16S de Bactéries, d’Archaeas. ARNr 28S Fungi : contrôlées, alignées, annotées.
Dernière mise à jour : septembre 2015
Cole et al. 2003 Cole et al. 2007 Cole et al. 2014
3
1.2 D
EVELOPPEMENT D’
OUTIL SOUSG
ALAXY LIEE A LA METAGENOMIQUE AMPLICON 1.2.1 La plateforme GalaxyGalaxy22,23 est une plateforme développée pour la bioinformatique dédiée notamment pour les analyses en génomique. Galaxy met à disposition des outils et des « workflows » pour les biologistes qui n’ont alors pas besoin de compétences en informatique pour les utiliser. Dans Galaxy, la plupart des formats de données biologiques sont définis et la conversion entre les différents types de format est également prise en charge. L’interface web de Galaxy permet de suivre le dérouler des traitements de fichiers. Un des avantages notable de Galaxy est que l’on peut réutiliser les « workflows » déjà fonctionnels dans le cadre d’un travail de routine dans un objectif de reproductibilité du traitement des données. Enfin, avec l’interface graphique, on peut facilement paramétrer les différents outils et visualiser les résultats (les tables, les textes, les graphiques).
1.2.2 FROGS: Find, Rapidly, OTUs with Galaxy Solution
Le séquençage haut-débit d’amplicons d'ARN 16S/18S a ouvert de nouveaux horizons dans l'étude des communautés microbiennes. Avec le séquençage à haut débit, les chaines de traitements actuels peinent à s'exécuter rapidement et les solutions les plus efficaces sont souvent dédiées aux spécialistes. Ces outils offrent aux biologistes une table d'abondance contenant les OTUs (unités taxonomiques opérationnelles) et leur affiliation taxonomique. Dans ce contexte, mes équipes d’accueil ont développé le pipeline FROGS : « Find, Rapidly, OTUs with Galaxy Solution » mis au point pour la plateforme Galaxy (Figure 1). Les données prises en charge par FROGS sont de type amplicons d’ARNr 16S, 18S ou 23S.
Figure 1: FROGS – une nouvelle suite logicielle pour analyser les données de séquençages amplicons
La Figure 2 illustre les principales étapes de FROGS. Les données d’entrées peuvent être démultipléxées ou non et sont alors prises en charge par l’outil FROGS_demultiplex. L’outil FROGS_Preprocess permet le prétraitement des données, i.e. contigage de séquences pairées Illumina avec flash24, nettoyage des données avec cutadapt25, suppression des chimères avec VSEARCH26 et déréplication des séquences avec un script python maison. L'outil de clusterisation, FROGS_clustering fonctionne avec SWARM27 qui utilise un seuil de clustering local et non un seuil de clustering global comme le font la plupart des autres logiciels. L’étape de filtration, FROGS_filter, permet l’élimination d’OTUs ne répondant pas aux requêtes de filtres, tels que les OTUs trop faiblement abondants afin qu’ils ne puissent être confondus avec des séquences artéfactuelles. L'outil d'affiliation, FROGS_affiliation_OTU retourne l’affiliation taxonomique de chaque OTU en utilisant RDPClassifier28 ou NCBI blast+ 29 sur les banques de données SILVA 16S, 18S, 23S11,12, greengenes10, Midas30 ou d’autres banques personnelles de certains laboratoires. Tout au long des traitements de nombreux graphiques et statistiques sont générés afin que l’utilisateur suive facilement le traitement de ses données. En sortie de FROGS,
4 une table d’abondance d’OTUs est produite. L’analyse de cette table au regard des métadonnées associées (pH, température, poids, traitement, etc.) nécessite de faire appel à des méthodes statistiques dédiées aux données de type compositionnelles.
Figure 2: Diagramme des étapes principales de FROGS (http://frogs.toulouse.inra.fr)
1.2.3 Les besoins en analyses statistiques
La table d’abondance d’OTUs qui est produite par FROGS contient les informations d’abondance de chaque OTU pour chaque échantillon ainsi que leur affiliation taxonomique. Or les questions telles que « Quelle est le nombre d’espèces dans chaque échantillon, i.e. la richesse ? » et « Quelles sont les OTUs abondants et rares de mes échantillons, i.e. la structure ? », ou encore « Comment varie la communauté microbienne selon telle ou telle métadonnées ? » ne peuvent se baser que sur le calcul de distances, d’indices et les statistiques descriptives. FROGS était dépourvu jusqu’à lors d’outils permettant ces calculs. Donc, afin que les utilisateurs réfractaires à la ligne de commande puissent poursuivre le traitement de leurs données dans le cadre de Galaxy, j’ai développé et mis en place ces outils dans FROGS.
Les priorités de développement se sont tournées sur les outils de l’étude de la diversité microbienne dans chaque communauté i.e. la diversité alpha et entre les communautés i.e. la diversité bêta. Enfin, parce que les conditions expérimentales des données microbiennes sont diverses, les tests compositionnels, tel que MANOVA, sont indispensables à l’analyse de ces données et ont dues donc être mises à disposition des utilisateurs. D’autre part, certaines méthodes statistiques sont basées sur la phylogénie des espèces présentes. J’ai donc également développé un outil sous Galaxy pour la reconstruction d’un arbre phylogénétique à partir des OTUs, FROGS_Tree. Il existe déjà de multiples méthodes statistiques appliquées à l’étude des
5 communautés d’écologie microbienne dont la plupart sont disponibles sous R31. On peut citer distory32, phangorn33, picante34, et le package Phyloseq35. J’ai pour ma part utilisé PhyloSeq.
2 MATERIELS
ET
METHODES
2.1 D
EMARCHE METHODOLOGIQUE POUR DEVELOPPEMENT D’
OUTIL SOUSG
ALAXY 2.1.1 GalaxyGalaxy est un logiciel libre sous la licence « Academic Free Licence » version 3.0 de l’Université de l’état de Pennsylvanie. Le code source est accessible à tous et est déposé sur GitHub « https://github.com/galaxyproject/galaxy/ ». Les scripts sources de Galaxy sont écrits en langage Python 2.7. Le fichier de base de configuration des outils qui sont implémentés dans Galaxy est écrit en XML et nommé tool_conf.xml. L’ajout de nouveaux outils se fait soit par le « Tool Shed » (bibliothèque de partage d’outils avec installation automatique), soit par le développement de nouvelles applications, ce que j’ai fait pendant mon stage. Les configurations par défaut des outils se font via le fichier «config/galaxy.ini».
2.1.2 Les langages de programmation
J’ai écrit en Python 2.7 les applications gérant l’interopérabilité des logiciels utilisés. Par exemple, pour la reconstruction d’arbre phylogénétique, nous lançons trois logiciels différents et donc les connexions entre ces logiciels se font par des fichiers d’entrées et de sorties adaptés à chacun. La génération et le formatage de ces fichiers sont faits à l’aide de scripts Python.
J’ai également développé Rscript écrit en langage R 3.4 permettant le lancement des scripts se chargeant des analyses statistiques. Pour générer les résultats sous forme de fichier html directement visualisable sous Galaxy, j’ai utilisé Rmarkdown (un package R). Rmarkdown a aussi été combiné à du JavaScript afin que les fichiers html gagnent en ergonomie.
Le langage XML est le langage de l’interface de Galaxy. Pour chaque outil, j’ai écrit un fichier XML permettant de générer l’interface de l’outil. Ce fichier XML permet à la fois la configuration de l’outil, le lancement des scripts de traitement des données, le passage des entrées et la définition des sorties et la création du formulaire dédié à l’utilisateur lui permettant de paramétrer l’outil (i.e. les champs de formulaire, les menus déroulants, les cases à cocher, l'aide, etc.).
J’ai également codé en JavaScript. C’est un langage de programmation qui est généralement inclus dans les pages web et qui indique à cette page comment elle doit réagir. Je m’en suis particulièrement servi pour mettre à jour des éléments de la page sans la recharger. Le code JavaScript sert donc à donner du dynamisme à la page. Pour le développement html j’ai également utilisé du CSS (Cascading Style Sheets) qui est le langage utilisé pour la mise en forme des sites web. Il m’a permis de mettre en forme les informations et éléments affichés avec html.
6
2.2 D
EMARCHE METHODOLOGIQUE POUR LA RECONSTRUCTION D’
ARBREPHYLOGENETIQUE
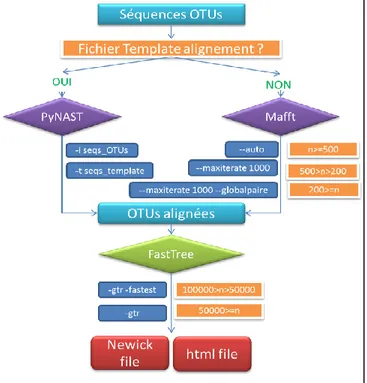
Il y a deux étapes principales pour la reconstruction d’arbre phylogénétique (Figure 3). Premièrement, on effectue un alignement des séquences, dans notre cas les séquences représentatives des OTUs issues de FROGS et la seconde étape est la reconstruction de l’arbre en lui-même. Concernant l’alignement multiple des séquences, deux outils ont été choisis : PyNAST36 et MAFFT v.737,38. Si l’utilisateur a récupéré ou construit un fichier de séquences alignées « template » alors PyNAST sera utilisé, sinon, c’est MAFFT qui est utilisé. Pour reconstruction d’arbre phylogénie, nous avons choisi FastTree239.
2.2.1 PyNAST
PyNAST est basé sur la méthode NAST : Nearest Alignment Space Termination, « Py » étant pour python. NAST40 est
particulièrement adapté au traitement des séquences produites lors d’analyse de communautés en écologie microbienne. PyNAST demande en fichiers d’entrée le fichier contenant les séquences au format fasta et le fichier d’alignement de séquences connues appelé « template ». La ligne de commande de Pynast utilisé dans FROGS est « pynast -i my_input.fasta -t my_template.fasta –l my_min_length –p 75.0 –m uclust » avec –i le fichier de séquences à aligner au format fasta, -t le template au format fasta (celui basé sur le 16S de la banque de données GreenGenes est
téléchargeable ici:
https://github.com/biocore/qiime-default-reference/blob/master/qiime_default_reference/gg_13_8_otus/rep_set_aligned/85_otus.pynast.fa sta.gz et celui pour la base de données SILVA est disponible ici : https://www.arb-silva.de/fileadmin/silva_databases/qiime/Silva_128_release.tgz) –l la longueur minimale des séquence à aligner que nous calculons automatiquement d’après le calcul de la médiane des longueurs des séquences du fichier de séquences fasta en entrée, –m la méthode d’alignement de séquences par paires ici uclust. Le fichier de sortie de Pynast est un fichier multifasta des séquences alignées qui est utilisé par la suite par FastTree.
2.2.2 MAFFT
Si l’utilisateur ne peut pas fournir de fichier « template », alors les séquences sont alignées avec MAFFT v.737. MAFFT est un logiciel d’alignement de séquences multiples rapide et efficient qui a été éprouvé par mes directeurs de stages avant mon arrivée. Selon le nombre de séquences du fichier fasta donné en entrée, trois groupes d’options différentes sont utilisés pour MAFFT :
Figure 3: Logigramme de l’outil de reconstruction d’arbre phylogénétique.
7 (i) mafft --auto méthode automatique pour aligner plus de 500 séquences, (ii) mafft --maxiterate 1000 méthode itérative avec 1000 itérations maximum pour aligner de 200 à 500 séquences ou (iii) mafft --maxiterate 1000 --globalpair méthode itérative avec 1000 itérations maximum pour aligner de manière globale moins de 200 séquences à l’aide de l’algorithme de Needleman and Wunsch41. Ces seuils de taille de fichiers ont été ajustés selon des critères de temps d’exécution de MAFFT que j’ai obtenu après avoir testé MAFFT sur des fichiers contenant des nombres de séquences de 20, 100, 200, 500 et 1000 espèces différentes. Moins, il y a de séquences à aligner plus on utilise un algorithme précis et lent et inversement (Annexe 1).
2.2.3 FastTree2
Le logiciel FastTree2 est utilisé pour la reconstruction d’un arbre phylogénétique en se basant sur le fichier des séquences alignées (le résultat de Pynast ou Mafft). FastTree a été choisi car il est extrêmement rapide et est tout à fait adapté à la reconstruction phylogénétique basée sur l’ADNr 16S39. La ligne de commande de FastTree pour les séquences nucléotidiques alignées mise en place dans FROGS est FastTree -gtr -nt alignment_file > tree_file avec –gtr demandant l’utilisation de FastTree avec le modèle évolutif nucléotidiques, « generalized time-reversible » et –nt l’application de FastTree sur des données nucléotidiques.
2.3 D
EMARCHE METHODOLOGIQUE POUR L’
ANALYSE STATISTIQUE LIEE A LA METAGENOMIQUE2.3.1 Phyloseq
Phyloseq35 est un package spécifique pour les données du séquençage de métagénomique, plus précisément du séquençage d’amplicon. Il regroupe une suite de fonctions pour la manipulation de la table d’abondance et des métadonnées associées en sous échantillonnant ou en agglomérant les données sur des critères d’abondances, de conditions expérimentales ou encore de rang taxonomique. Il permet également de normaliser les données. Phyloseq est dédié à l’analyse de la composition et de la structure des communautés au sein des échantillons. Il propose des méthodes pour le calcul des indices de diversités alpha (intra échantillon) et bêta (inter échantillons), pour l’ordination des échantillons.
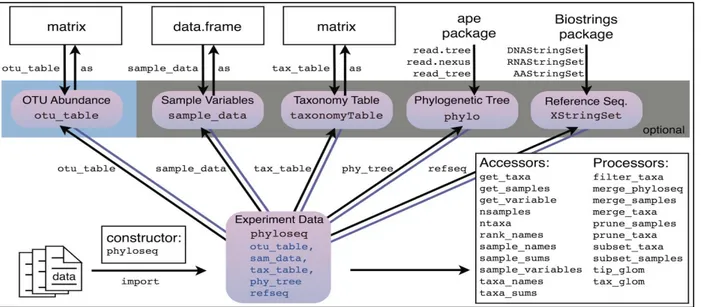
Phyloseq est un package orienté objet, c’est-à-dire qu’il construit un objet regroupant toutes les informations produites par le pipeline de reconstruction et quantification d’OTUs (dans notre cas FROGS). Ainsi l’objet Phyloseq contient une table d’abondance contenant le nombre de chaque OTU présent dans chaque échantillon, les métadonnées associées contenant les conditions expérimentales de chaque échantillon, l’affiliation taxonomique des OTUs, un arbre phylogénétique construit à partir de ces OTUs au format Newick et les séquences de références des OTUs (Figure 4). Seule la table d’abondance est obligatoire, mais les informations de métadonnées des échantillons et d’affiliation taxonomique permettent d’obtenir une analyse plus fine. Lors de la construction de l’objet Phyloseq, ces trois premiers éléments peuvent être dans des fichiers indépendants ou dans un seul fichier de type biom44.
8
Figure 4: Représentation de la structure d’un objet Phyloseq
Le package R Phyloseq donne accès à un ensemble de méthode de calculs d’indice ou de distance. Pour l’alpha diversité, on peut choisir parmi les indices Observed, Chao1, Shannon, InvSimpson, Simpson, ACE ou Fisher. Pour la bêta diversité, Phyloseq supporte plus de quarante méthodes comme Bray-Cutis, Jaccard ou Unifrac et weighted Unifrac (si un arbre phylogénétique est disponible pour ces deux dernières). Phyloseq inclus également des fonctions permettant d’analyser la structure des échantillons telles que CA (Correspondance Analysis), DCA (Detrended Correspondance Analysis), PCA (principal componants analysis), DPCoA (double principle coordinates analysis) et MDS/PCoA (multidimentional scaling).
Figure 5: Schéma résumant les fonctions principales de Phyloseq (* fonctions R extérieures s’interconnectant à l’objet Phyloseq)
L’objet Phyloseq peut être utilisé pour réaliser des tests de MANOVA (Multivariate Analysis Of Variance) grâce aux fonctions du package vegan42, adonis et capscale. Pour chaque résultat
9 numérique, Phyloseq propose des fonctions de représentation graphique spécifique grâce à ggplot243. Premièrement, on peut visualiser directement les compositions avec des plots comme plot_tree pour les arbres ou plot_bar et plot_composition pour des histogrammes. Ensuite, pour étudier les diversités en OTUs, Phyloseq va calculer des indices de diversité alpha et les représenter avec plot_richness en particulier sous forme de boxplot, et pour la bêta diversité, plot_heatmap génère des heatmaps, plot_ordination des nuages de points et hclust un arbre (Figure 5). À noter, la fonction hclust de R est facilement utilisable avec l’objet Phyloseq. Cette fonction propose différentes méthodes qui construisent les liens entre les échantillons qui sont clusterisés de manière hiérarchique. Ces méthodes sont notamment, single linkage, complete linkage ou Ward linkage.
2.3.2 DESeq2
Pour détecter les espèces d’abondances différentielles à partir de données de métagénomique amplicon, il faut tenir compte des différences de taille des librairies de séquençage et de la variabilité biologique en utilisant un modèle de mélange approprié44. Nous avons choisi d’intégrer dans notre outil Galaxy DESeq245, un package R, pour l’analyse d’abondance différentielle. DESeq2 est un des programmes les plus utilisés dans la communauté pour répondre à cette question. En effet, comme certains autres packages sous R comme la package edgeR46, DESeq2 est dédié à l’analyse d’expression différentielle, problématique proche de l’abondance différentielle. Il est basé sur un modèle de distribution binomiale négative47
. DESeq2 permet de comparer les abondances entre elles en comparant les deux modalités d’une même variable expérimentale. Une fonction Phyloseq permet la conversion d’un objet Phyloseq en un objet DESeq2 (Figure 6).
Figure 6: DESeq2 pour d’analyse de l’abondance différentielle
DESeq2 effectue quatre étapes de calculs: (i) l’estimation des facteurs de taille, (ii) l’estimation de la dispersion, (iii) l’estimation des paramètres du modèle de binomial négatif, en fonction des résultats précédents, et (iv) la comparaison du modèle vis-à-vis de l’hypothèse nulle. DESeq2 va générer un tableau indiquant pour chaque OTU des mesures statistiques comme le log2fold-change, la p-value, ou encore la p-value ajustée. Pour visualiser ces résultats d’une façon plus détaillée, DESeq2 propose des fonctions comme plot_Volcano, plot_MA, et plot_Heatmap.
3 RESULTATS
Dans cette partie je vais décrire les 10 outils que j’ai développés pour la suite logicielle FROGS. Les analyses avec le logiciel R sont compliquées pour les biologistes qui ont peu de connaissances en statistiques et en informatique. L’utilisation de Galaxy facilite ainsi ces
10 analyses. Chaque outil a été conçu pour être facile à utiliser et convivial. Nous avons mis l’accent sur le fait de demander peu de paramètres l’utilisateur, de produire une aide détaillée pour chaque outils et de générer de nombreuses sorties graphiques pour accompagner les utilisateurs dans l’interprétation de leurs analyses. Les outils sont présentés dans un ordre logique avec des noms clairs, liés directement aux buts de l’outil. Les paramètres demandés sont expliqués clairement, les autres paramètres sont fixés ou calculés automatiquement pour minimiser la complexité d’utilisation.
3.1 FROGS
T
REE:
L’
OUTIL POUR LA RECONSTRUCTION D’
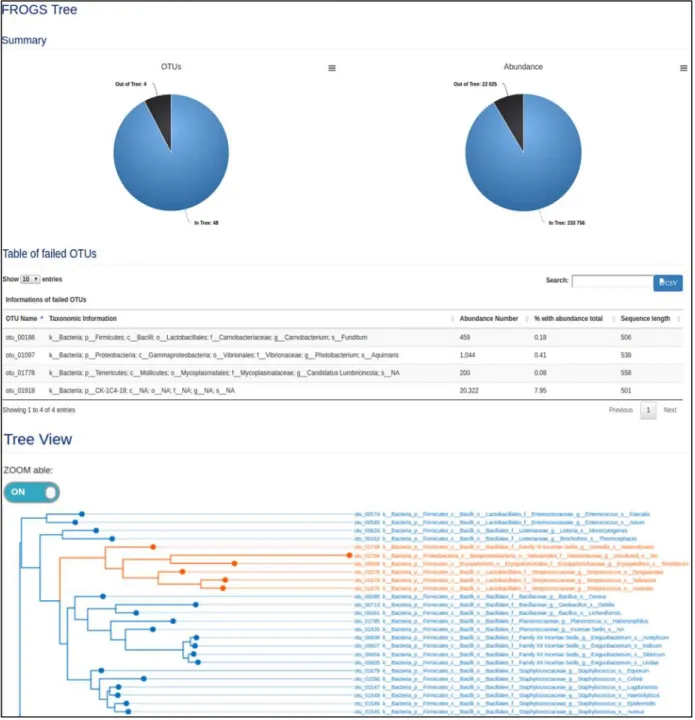
ARBRES PHYLOGENETIQUESL’outil FROGS Tree a été développé pour la reconstruction d’arbres phylogénétiques des séquences d’OTUs issues de FROGS. L’utilisateur utilise l’outil via l’interface de Galaxy. Cette fenêtre de paramétrage permet à l’utilisateur de soumettre les fichiers d’entrée demandés ainsi que de paramétrer l’option avec ou sans fichier « template » (cf. MATERIELS ET METHODES). Il y trouve également les définitions/explications de chaque champ à remplir ainsi que les formats des fichiers attendus (Figure 7). En détails, l’outil a besoin de deux fichiers obligatoires, le fichier des séquences d’OTUs et le fichier qui contient les informations des OTUs au format biom. En option, on peut donner un fichier d’alignement des séquences références déjà connues. Avec cet outil, deux fichiers de sortie sont générés (i) un fichier contenant l’arbre phylogénétique au format Newick, et (ii) un fichier html pour visualiser le résumé des résultats. Ce dernier montre (i) le pourcentage des OTUs présents dans l’arbre et leur abondance à l’aide deux diagrammes circulaires, (ii) un graphique interactif d’arbre phylogénétique et (iii) un tableau contenant la liste des OTUs absents de l’arbre phylogénétique (Figure 8). En effet dans le cas d’utilisation de Pynast, seuls les OTUs avec un pourcentage d’identité supérieur à 75 avec au moins une séquence de référence sont alignés et donc dans l’arbre final.
11
Figure 8: captures d’écran du rapport html délivré par l’outil FROGS Tree
3.2 FROGS
P
HYLOSEQ:
LES OUTILS SOUSG
ALAXY POUR L’
ANALYSE STATISTIQUE DES DONNEES D’
ECOLOGIE MICROBIENNE3.2.1 Import data
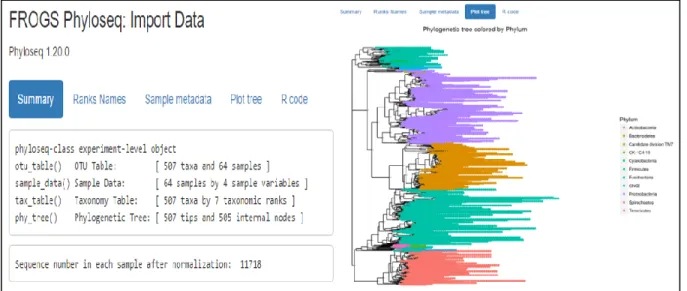
Le but de cet outil est de produire une classe d’objet Phyloseq destinée à être utilisé par la suite par l’ensemble des autres outils. Pour construire l’objet Phyloseq, il est demandé en entrée : le fichier biom qui contient les informations des abondances et des affiliations taxonomiques des OTUs, le fichier tabulé qui contient les métadonnées des échantillons c’est-à-dire qui décrit les
12 conditions expérimentales, et en option, le fichier d’arbre phylogénétique Newick. Si le nom et le nombre de rangs taxonomiques n’est pas standard il est possible, dans le quatrième champ, de les modifier. Les analyses de diversité ont besoin de comptages normalisés, c’est pourquoi, l’outil permet en option de normaliser les abondances des OTUs dans chaque (Figure 9).
Figure 9: Interface graphique Galaxy de la fenêtre de paramétrage de l’outil FROGS Phyloseq Import Data
Cet outil génère deux fichiers (i) un fichier RData contenant l’objet Phyloseq, et (ii) un fichier html (Figure 9) contenant les informations liées à l’objet Phyloseq comme le nombre d’OTUs, le nombre d’échantillons, le nombre et les noms des rangs taxonomiques et le nombre de variables expérimentales ainsi que leurs valeurs. Si l’option de normalisation a été choisie alors il est également indiqué dans ce fichier l’abondance totale de séquence dans chaque échantillon. On y trouve également une représentation aux branches colorées par Phylum de l’arbre phylogénétique soumis. Ceci a été fait à l’aide de la fonction plot_tree de Phyloseq. Finalement, nous délivrons également à l’utilisateur les lignes de commande R qui ont été lancées à travers cet outil permettant ainsi aux utilisateurs plus expérimentés de réutiliser ces lignes de commandes et de les modifier/compléter via un interpréteur de R. Ce dernier onglet est présent dans chaque sortie html des outils FROGS Phyloseq et FROGS DESeq2.
13
Figure 9: capture d’écran du 1er onglet du rapport html délivré par l’outil FROGS Phyloseq Import Data
3.2.2 Visualisation des compositions
Le but de l’outil FROGS Phyloseq Composition Visualization est de visualiser la composition des échantillons c’est à dire en connaitre le nombre d’OTUs et leurs abondances en séquences selon différentes conditions. Nous avons utilisé pour cela les fonctions plot_bar et plot_composition du package R Phyloseq. La fonction plot_bar représente les OTUs en fonction de leur abondance dans chaque échantillon. Ils sont colorés en fonction d’un rang taxonomique. La fonction plot_composition est une
fonction personnalisée pour visualiser les OTUs les plus abondants dans un rang taxonomique, i.e. un taxon d’intérêt. Elle répond à la question « quelle est l’abondance d’un taxon et la répartition des sous rangs taxonomiques les plus abondants ? ».
L’outil demande un fichier RData en entrée contenant l’objet Phyloseq créé précédemment par FROGS Phyloseq Import Data. L’utilisateur doit indiquer le taxon d’intérêt via son rang et son nom, puis indiquer le nombre de sous rangs taxonomiques qu’il souhaite visualiser : par exemple pour connaître les 9 Phylums les plus abondants parmi le Kingdom Bacteria. L’outil demande également le nom d’une variable expérimentale pour subdiviser le graphique en fonction de cette variable. (Figure 10)
Figure 10: Interface graphique Galaxy de la fenêtre de paramétrage de l’outil FROGS Phyloseq composition
14 Les sorties graphiques de cet outil sont incluses dans un fichier résultats html. Le résultat de plot_bar est une image que l’on peut sauvegarder facilement à l’aide d’un simple clic sur un bouton, cette fonctionnalité est d’ailleurs présente pour tous les graphiques et tableaux produits par les outils que nous avons développés. Le résultat de plot_composition est un graphique interactif qui utilise package R plotly (Figure 12). Enfin, le code R est représenté dans un dernier onglet.
Figure 11: Le plot bar et le plot composition du fichier html issus de l’outil FROGS Phyloseq Visualize Data
3.2.3 L’alpha diversité
Le but de cet outil est de construire le tableau des indices de diversité alpha de chaque échantillon. En utilisant la fonction plot_richness on permet de visualiser un ou plusieurs indices choisis en fonction d’une variable expérimentale. Pour cela, on demande en entrée le fichier RData qui contient l’objet Phyloseq et le nom de la variable expérimentale. L’utilisateur peut choisir plusieurs indices de l’alpha diversité parmi Observed, Chao1, Shannon, InvSimpson, Simpson, ACE et Fisher (Figure 12).
Cet outil produit également les courbes de raréfaction qui représentent le nombre cumulé d'espèces nouvellement trouvées pour chaque échantillon. L’outil génère deux fichiers : un fichier tabulé qui contient le tableau des indices de diversité alpha, et un fichier html contenant les graphiques produits. La richesse peut être représentée à l’aide de boîtes à moustaches. Comme pour les autres outils le code R est disponible (Figure 13).
15
Figure 12: Interface graphique Galaxy de la fenêtre de paramétrage de l’outil FROGS Phyloseq Alpha Diversity
Figure 13: Les plots produits par l’outil FROGS Phyloseq Alpha Diversity Visualization Composition Visualization
3.2.4 Diversité bêta
Cet outil calcule la matrice de distances basées sur la diversité bêta. L’outil demande en entrée un fichier RData qui contient l’objet Phyloseq construit précédemment et le nom de la variable expérimentale que l’on veut étudier. Les utilisateurs peuvent choisir une ou plusieurs méthodes de diversité bêta (Figure 14). En sortie, l’outil génère autant de fichiers que de méthodes choisies
16 pour le calcul des distances, et un fichier html contenant un graphique de type heatmap par méthodes choisies (Figure 15). Comme pour les autres outils le fichier contient également le code R.
Figure 14: Interface graphique Galaxy de la fenêtre de paramétrage de l’outil FROGS Phyloseq Beta Diversity
17
3.2.5 Visualisation de structure
Le but de cet outil est de visualiser la structure de données en fonction d’une variable expérimentale. L’outil demande en entrée: un fichier RData qui contient l’objet Phyloseq et un fichier tabulé qui contient la matrice des distances basées sur la bêta diversité issue de l’outil FROGS Phyloseq Beta Diversity. L’utilisateur doit entrer le nom d’une variable expérimentale d’intérêt et d’une méthode d’ordination pour représenter les échantillons dans un espace à deux dimensions conservant au mieux les distances de diversité bêta (Figure 16).
Figure 17: Exemples de graphiques produits par l’outil FROGS Phyloseq Structure Visualization Figure 16: L’interface Galaxy de la fenêtre de paramétrage de l’outil FROGS Phyloseq
18 En sortie, l’outil génère un fichier html contenant les résultats graphiques des fonctions plot_ordination et plot_heatmap de Phyloseq, ainsi que le code R. Afin d’améliorer l’interprétation des images, nous donnons la possibilité aux utilisateurs d’ajouter des ellipses regroupant les points de chaque condition ou les noms des échantillons sur les graphiques (Figure 17).
3.2.6 Regroupement hiérarchique des échantillons
Le but de cet outil est de regrouper les échantillons entre eux selon une variable expérimentale. Il est demandé en entrée l’objet Rdata, la matrice de distances basée sur la bêta diversité ainsi que le nom de la variable expérimentale (Figure 18).
Figure 18: L’interface Galaxy de la fenêtre de paramétrage de l’outil FROGS Phyloseq Sample Clustering
Ce regroupement des échantillons se fait grâce à la méthode hclust de R selon trois constructions différentes : lien simple, lien complet et lien de Ward. L’outil génère un fichier html contenant les graphiques illustrant ces regroupements, colorés en fonction de la variable expérimentale choisie (Figure 19). Ce fichier inclut également le code R généré.
Figure 19: Exemple de graphique produit par l’outil FROGS Phyloseq Sample Clustering selon la méthode Ward.D2
19
3.2.7 L'analyse de variance multivariée (MANOVA)
Le but de cet outil est l'analyse de variance multivariée. Comme pour les deux outils précédents, Il est demandé en entrée l’objet R.data, la matrice de distances basée sur la bêta diversité produite par l’outil FROGS Phyloseq Beta Diversity ainsi que le nom de la variable expérimentale (Figure 20).
Figure 20: L’interface Galaxy de l’outil FROGS Phyloseq Multivariate Analysis Of Variance
L’outil génère un fichier html contenant les résultats des tests MANOVA portés par la fonction adonis et capscale du package vegan de R (Figure 21).
Figure 21: Capture d’écran du rapport html délivré par l’outil FROGS Phyloseq Multivariate Analysis of Variance
20
3.2.8 FROGS DESeq2: deux outils pour l’analyse différentielle des abondances
3.2.8.1 DESeq2 Preprocess
Le but de cet outil est de générer les calculs préparatoires pour les analyses différentielles des abondances par le package R DESeq2. Ces calculs correspondent aux étapes d’estimation des facteurs de taille, de la dispersion et des paramètres du modèle de binomial négative. Le facteur de taille, pour chaque échantillon, va permettre de prendre en compte les différences de profondeur de séquençage entre échantillon. La dispersion, pour chaque OTU, est une valeur modérée entre la dispersion individuelle de l’OTU et la dispersion globale des OTUs ayant une abondance similaire. L’outil demande en entrée le fichier RData contenant un objet Phyloseq non normalisé puisque DESeq2 estime les facteurs de taille, et le nom des variables expérimentales (une ou deux) que l’on veut étudier (Figure 22).
Figure 22: L’interface Galaxy de la fenêtre de paramétrage de l’outil FROGS DESeq2 Preprocess
En écologie, les tables des abondances générées contiennent généralement beaucoup de valeurs égales à zéro, donc afin de pouvoir utiliser DESeq2, nous avons transformé notre matrice en ajoutant 1 à chaque abondance. La sortie de cet outil est simplement un fichier RData qui contient le jeu de données i.e. un objet DESeq2.
3.2.8.2 DESeq2 Visualisation
Le but de cet outil est de visualiser graphiquement les analyses différentielles des abondances des OTUs en utilisant le package DESeq2. L’intérêt est de proposer l’application de fonctions différentes offrant à l’utilisateur différentes sorties graphiques de ses analyses différentielles. Un objectif important est l’analyse différentielle des échantillons selon une variable expérimentale et selon une ou deux conditions. Ainsi il est demandé en entrée les fichiers RData contenant l’objet Phyloseq sans la normalisation et l’objet RData issus de l’outil FROGS DESeq2 Preprocess. L’utilisateur doit entrer le nom de la variable expérimentale d’intérêt ainsi que le nom d’une ou de deux variables qui conditionnent analyse différentielle. Cet outil permet aussi de choisir la valeur maximale de p-value ajustée48 qui va être utilisée dans les tests (paramètre optionnel) (Figure 23). En sortie, l’utilisateur obtient un fichier html contenant les résultats des fonctions R
21 DESeq2 à l’aide d’un tableau interactif, rendu possible grâce au package « formattable » (Figure 24).
Figure 23: L’interface Galaxy de la fenêtre de paramétrage de l’outil FROGS DESeq2 Visualization
Figure 24: Le tableau contenant les résultats des analyses différentielles. Par exemple, avec l’OTU otu_0026, les
résultats des tests DESeq2 (log2 Fold Change est 1.7 avec p-value ajustée est faible 0.0498), c’est-à-dire cette OTU est différentielle d’abondance dans 2 conditions qu’on a testé, en détail, il est surexprimé en condition première de
22 Ce fichier contient également l’ensemble des graphiques produits aidant à l’interprétation des données. Ces derniers sont produits avec la fonction plot_Pie utilisée avec le package googleVis le rendant interactif, la fonction plot_Volcano, ainsi que plot_MA, et plot_Heatmap (rendu interactif à l’aide du package plotly) (Figure 25).
23
4 DISCUSSION
Au cours de mon stage, j’ai développé des outils sous Galaxy pour la reconstruction d’arbres phylogénétiques et pour les analyses statistiques de composition et de structure des communautés microbiennes. Ce travail fait suite au développement logiciel FROGS, un pipeline dédié à l’identification et à la quantification relative des communautés microbiennes à partir de séquençage d’amplicons par analyse métagénomique.
FROGS s’interface avec Galaxy, afin de permettre aux utilisateurs non-experts en bioinformatique de faire leurs analyses en toute autonomie. Les outils existants de FROGS, permettent d’identifier les OTUs, de les quantifier et de les affilier taxonomiquement. L’ensemble de ces résultats est stocké dans une table d’abondances. Les nouveaux outils développés lors de mon stage dialoguant également avec Galaxy, permettent d’analyser ces tables d’abondances grâce aux statistiques. Ces analyses sont toutes développées en R et se basent notamment sur l’exploitation des packages Phyloseq et DESeq2. Certaines analyses statistiques ont besoin d’un arbre phylogénétique comme le calcul de la bêta diversité en se basant sur les méthodes Unifrac ou Weighted Unifrac. C’est la raison pour laquelle, j’ai développé un outil qui permet de construire cet arbre à partir des résultats obtenus avec FROGS. Les reconstructions phylogénétiques, vraies, sont complexes à obtenir pour les non-initiés et sont, en plus, gourmandent en temps de calculs. Il était donc nécessaire de créer un outil simple d’utilisation et optimisé. Avec le premier outil, FROGS_Tree, la reconstruction de l’arbre phylogénétique des OTUs se fait en deux étapes. La première étape utilise différentes méthodes d’alignement multiple, optimisée selon le temps de calcul : Pynast ou MAFFT, et la seconde étape utilise la méthode FastTree pour construire l’arbre.
Pynast est une des références en outil d’alignement de séquences amplicon 16S. En basant l’alignement sur des séquences connues, il permet de produire un alignement de qualité en quelques minutes. Cependant Pynast nécessite un alignement de référence, c’est-à-dire un alignement généralement créé par des experts. Donc si les espèces d’intérêts ne se trouvent pas dans cet alignement référence, les séquences absentes de cet alignement le seront également de l’arbre phylogénétique reconstruit. Il était donc nécessaire d’offrir une alternative à nos utilisateurs. MAFFT permet de créé un alignement de novo. J’ai étudié et testé les paramètres de MAFFT afin de l’adapter aux séquences d’amplicons et pour obtenir un alignement de qualité, dans un temps des calculs raisonnable (Annexe 1). FastTree2, un logiciel particulièrement adapté aux données d’ARN ribosomiques, utilise une méthode de maximum de vraisemblance pour construire un arbre phylogénétique de qualité avec un temps de calculs qui est 100 à 1000 fois plus rapide que des logiciels de haut niveau tels que PhyML 3.0 ou RAxML.739,49. La phylogénie demande normalement un travail approfondi sur l’alignement et sur la reconstruction d’arbre. Notre idée n’est pas de fournir le meilleur outil de phylogénie, mais de fournir un outil intermédiaire permettant d’avoir un arbre de qualité correcte (limitation des incongruences) dans un temps de calculs raisonnables. C’est pourquoi nous avons limité l’utilisation de FROGS_tree à un nombre d’OTUs inférieur à 10 000.
Avec les outils d’analyses statistiques d’étude des communautés microbiennes, chaque outil correspond à un groupe de fonctions du package Phyloseq de R. On peut étudier la diversité des
24 communautés microbiennes dans chaque échantillon ou entre les différents échantillons et les sorties graphiques contenues dans les fichiers html que nous produisons, aide l’utilisateur dans ses analyses. En comparant nos outils mis à disposition et ces mêmes outils sous R, le paramétrage et les possibilités d’analyses sont plus restreints dans notre cas (ce qui rend les outils plus facile à utiliser). Mais pour les utilisateurs qui veulent pousser leurs analyses, nous mettons, systématiquement, à disposition le code R dans un onglet du fichier html. Ainsi, ils peuvent s’inspirer de ces codes pour faire des analyses plus détaillées en R directement. Par exemple, il est parfois intéressant de maîtriser la palette de couleurs pour illustrer les échantillons et ainsi visualiser une deuxième variable expérimentale. Ceci n’est pas permis dans notre outil, car cela est valable pour un nombre restreint de protocoles expérimentaux, nous sortons donc ici du cadre général d’utilisation pour lequel cet outil a été développé. Mais avec le code R à disposition, les utilisateurs peuvent s’inspirer des commandes fournies et utiliser par eux-mêmes une autre palette de couleurs.
Toujours dans un souci de convivialité des outils, nous produisons des graphiques qui sont interactifs en utilisant la fonction ggplotly du package plotly. Avec ce type de graphique, on peut visualiser plus en détails et obtenir des informations supplémentaires. Par exemple, le graphique de type heatmap de l’outil FROGS_Phyloseq_Structure_Visualization permet de visualiser l’abondance des OTUs dans les échantillons et de mettre en évidence des structures par bloc. Le fait d’être interactif, permet de zoomer sur une région d’intérêt et de connaître quels OTUs sont inclus dans ce bloc, dans quels échantillons et à quelles abondances. Cependant les graphiques interactifs rendent les fichiers html volumineux et peuvent faire ralentir leur affichage à l’écran. Par conséquent, nous avons choisi de rendre interactifs seulement les graphiques le plus intéressants comme celui de composition et de heatmap. Une des solutions possible aurait été l’utilisation de Shiny de R qui permet l’affichage, sans problème, de graphiques interactifs. Mais Shiny ne dialogue par encore correctement avec Galaxy nous n’avons donc pas choisi cette solution et privilégié la génération de fichier html.
Pour les analyses différentielles des abondances nous avons choisi d’utilisé les fonctions portées par le package R DESeq2 car c’est l’une des références pour l’analyse d’expression différentielle des gènes. Avec les données de métagénomique, la table d’abondance contient beaucoup de valeurs à zéro représentant l’absence d’un OTU dans un échantillon. Ceci est spécifique à la métagénomique, car en analyse d’expression des gènes on assume qu’une grande majorité des gènes soient exprimés dans toutes les conditions. Ainsi pour pallier cette différence, j’ai ajouté 1 à chaque abondance, ainsi les données modifiées se rapprochent des hypothèses du modèle et sont toutes non-nulles et donc deviennent utilisables par DESeq2. Nous avons choisi de conserver DESeq2, car il n’y a pas aujourd’hui de méthode de référence spécifique à la métagénomique. Certains scientifiques assument que l’abondance différentielle peut être analysée comme l’expression différentielle44
25
5 CONCLUSION
ET
PERSPECTIVES
Durant mon stage j’ai développé des outils sous Galaxy pour faire suite aux analyses du pipeline FROGS. Tout d’abord, l’outil FROGS_Tree qui permet la reconstruction d’arbre phylogénétique et deux groupes d’outils utilisant soit les fonctions R du package Phyloseq (7outils) soit celles du package DESeq2 (2 outils) qui permettent les analyses statistiques des communautés microbiennes. Le groupe d’outils FROGS_Phyloseq permet de caractériser la table d’abondance et les échantillons au travers d’indices de diversité, de calcul d’ordination ou encore de clusterisation et de tester l’impact d’une variable expérimentale sur la diversité des échantillons. FROGS_Phyloseq c’est aussi une suite d’outils illustrant ces analyses sous des formes graphiques très diverses : histogrammes, boites à moustache, répartition dans un plan ou dans un arbre ou encore via des heatmaps. Les deux outils du groupe FROGS_DESeq2, quant à eux, permettent de faire les analyses d’abondance différentielle.
Avec ces nouveaux outils, FROGS devient un pipeline complet et les biologistes peuvent effectuer seuls les analyses les plus importantes en métagénomique amplicon via l’interface de Galaxy i.e. de la construction des OTUs, aux analyses de composition et de structure ainsi que d’abondance différentielle (Figure 27). Bien que FROGS ait intégré de nombreux outils statistiques à son analyse initiale en bioinformatique, plusieurs questions biologiques restent encore en suspens, par exemple, l’annotation fonctionnelle des communautés microbiennes qui peut être faite par des outils tel que PICRUSt50 , Tax4Fun51 ou PAPRICA52 ou encore des travaux plus complexes comme la sélection des données intérêts en utilisant les fonctions d’agrégation d’échantillon, de filtre sur une co-variable.
Figure.27 : Processus de FROGS pour l’analyse des données de séquençage d’amplicon
N.B. : En Annexes 2 et 3, sont décrits l’environnement de travail de mon stage ainsi que le bilan personnel que j’ai tiré de ces 6 derniers mois.
6 R
EFERENCES
1. Prosser, J. I. et al. The role of ecological theory in microbial ecology. Nat. Rev. Microbiol. 5, 384–392 (2007).
2. Falkowski, P. G., Fenchel, T. & Delong, E. F. The Microbial Engines That Drive Earth’s Biogeochemical Cycles. Science 320, 1034–1039 (2008).
3. Lozupone, C. A. & Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci.
104, 11436–11440 (2007).
4. Vieites, J. M., Guazzaroni, M.-E., Beloqui, A., Golyshin, P. N. & Ferrer, M. Metagenomics approaches in systems microbiology. FEMS Microbiol. Rev. 33, 236–255 (2009).
5. Tringe, S. G. Comparative Metagenomics of Microbial Communities. Science 308, 554–557 (2005).
6. Lundberg, D. S., Yourstone, S., Mieczkowski, P., Jones, C. D. & Dangl, J. L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 10, 999–1002 (2013). 7. Ram, J. L. et al. Reproduction and genetic detection of veligers in changing Dreissena
populations in the Great Lakes. Ecosphere 2, art3 (2011).
8. Bartram, A. K., Lynch, M. D. J., Stearns, J. C., Moreno-Hagelsieb, G. & Neufeld, J. D. Generation of Multimillion-Sequence 16S rRNA Gene Libraries from Complex Microbial Communities by Assembling Paired-End Illumina Reads. Appl. Environ. Microbiol. 77, 3846– 3852 (2011).
9. Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012).
10. DeSantis, T. Z. et al. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
11. Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188– 7196 (2007).
12. Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
13. Meyer, F. et al. The metagenomics RAST server a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386 (2008). 14. Schloss, P. D. et al. Introducing mothur: Open-Source, Platform-Independent,
Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
15. Cole, J. R. et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145 (2009).
16. Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
17. Giongo, A. et al. PANGEA: pipeline for analysis of next generation amplicons. ISME J.
4, 852–861 (2010).
18. Kunin, V. & Hugenholtz, P. PyroTagger : A fast , accurate pipeline for analysis of rRNA amplicon pyrosequence data. Open J. 1–8 (2010).
19. Angiuoli, S. V. et al. CloVR: a virtual machine for automated and portable sequence analysis from the desktop using cloud computing. BMC Bioinformatics 12, 356 (2011).
20. Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
21. Escudie, F. et al. FROGS: Find Rapidly OTU with Galaxy Solution. (2016).
22. Blankenberg, D. et al. Galaxy: A Web-Based Genome Analysis Tool for Experimentalists. in Current Protocols in Molecular Biology (eds. Ausubel, F. M. et al.) (John Wiley & Sons, Inc., 2010).
23. Giardine, B. et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455 (2005).
24. Magoc, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
25. Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, pp–10 (2011).
26. Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
27. Mah?, F., Rognes, T., Quince, C., de Vargas, C. & Dunthorn, M. Swarm: robust and fast clustering method for amplicon-based studies. PeerJ 2, e593 (2014).
28. Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol.
73, 5261–5267 (2007).
29. Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
30. McIlroy, S. J. et al. MiDAS: the field guide to the microbes of activated sludge. Database
31. Ihaka, R. R: Past and future history. Comput. Sci. Stat. 392–396 (1998). 32. Chakerian, J., Holmes, S. & Holmes, M. S. Package ‘distory’. Space (2010). 33. Schliep, K. et al. Package ‘phangorn’. (2017).
34. Kembel, S. W. & Kembel, M. S. W. Package ‘picante’. (2014).
35. McMurdie, P. J. & Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 8, e61217 (2013).
36. Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267 (2010).
37. Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780 (2013).
38. Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059– 3066 (2002).
39. Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 5, e9490 (2010).
40. DeSantis, T. Z. et al. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34, W394–W399 (2006).
41. Needleman, S. B. & Wunsch, C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443–453 (1970). 42. Oksanen, J. et al. Package ‘vegan’. Community Ecol. Package Version 2, (2013).
43. Wickham, H. A Layered Grammar of Graphics. J. Comput. Graph. Stat. 19, 3–28 (2010). 44. McMurdie, P. J. & Holmes, S. Waste not, want not: why rarefying microbiome data is
inadmissible. PLoS Comput Biol 10, e1003531 (2014).
45. Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, (2014).
46. Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
47. Jain, G. C. & Consul, P. C. A Generalized Negative Binomial Distribution. SIAM J. Appl. Math. 21, 501–513 (1971).
48. Wright, S. P. Aujusted P-Values for Simultaneous Inference. Biometrics 48, 1005–1013 (1992).
49. Stamatakis, A. The RAxML 7.0. 3 Manual. Exelix. Lab Heidelb. Inst. Theor. Stud. Heidelb. Httpwww Trex Uqam CadocumentsRAxML-Man. 7, (2008).
50. Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
51. Aßhauer, K. P., Wemheuer, B., Daniel, R. & Meinicke, P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data: Fig. 1. Bioinformatics 31, 2882–2884 (2015).
52. Bowman, J. S. & Ducklow, H. W. Microbial Communities Can Be Described by Metabolic Structure: A General Framework and Application to a Seasonally Variable, Depth-Stratified Microbial Community from the Coastal West Antarctic Peninsula. PLOS ONE 10, e0135868 (2015).
7 A
NNEXE
1
Résumé des résultats de tests de temps de calculs des alignements fais avec MAFFT et PyNast :
Graphique des temps de calcul des différentes méthodes d’alignement selon différents paramétrages
8 A
NNEXE
2
Présentation de la structure d’accueil :
Ce stage s’est effectué dans l’unité UMR 1313 - Génétique Animale et Biologie Intégrative (GABI) de l’INRA de Jouy-en-Josas au sein de deux équipes PSGEN et SIGENAE.
Le centre Inra Île-de-France-Jouy-en-Josas oriente ses recherches autour de l’aliment, l’animal et le microbe, en s’appuyant sur des recherches en mathématiques. C’est le plus grand centre INRA en terme de personnel : 820 titulaires Inra, 430 personnels rattachés aux structures partenaires (AgroParisTech, Institut de l’élevage, CNRS, …) et plus de 250 contractuels. Sept des treize départements scientifiques de l’Inra sont représentés sur le centre dont le département de Génétique Animale.
L'unité GABI est structurée en neuf équipes: trois équipes dédiées à trois espèces d’animaux d’élevage (bovin, cheval, poisson) ce qui permet de maintenir des liens forts avec les filières animales; cinq équipes sont structurées autour de disciplines, de fonctions ou de thématiques transversales et une équipe est dédiée aux plateformes d’appui à la recherche. Au sein de l’unité GABI, j’ai intégré l’équipe PSGEN: Population, Statistique et Génome. Cette équipe a pour vocation de conduire des recherches méthodologiques en génétique des populations et en biostatistiques, ces recherches sont ensuite mises en applications pour répondre à des problématiques concrètes.
L’équipe SIGENAE : Système d'information pour l'Analyse des Génomes des Animaux d'Elevage est une plateforme de bioinformatique au service des biologistes, spécialisé sur 6 espèces (bovin, poulet, porc, lapin, mouton, et truite). Cette équipe a la particularité d’être présente sur plusieurs centres INRA (1,5 titulaire à Jouy en Josas affiliés à l’équipe PSGEN, et 5 titulaires à Toulouse) dans différentes unités de recherche appartenant à différents départements scientifiques (Génétique Animal, Physiologie Animal et Système d’Elevage, et Santé Animale). L'équipe SIGENAE accompagne les biologistes dans leurs projets en faisant des traitements et des analyses à façon, comme par exemple la détection et sélection des polymorphismes responsables d’un phénotype d’intérêt ou encore la mise en place d'outils spécifiques notamment sur la plateforme Galaxy qui permet aux biologistes plus d’autonomie.
J’ai également été encadrée par Mahendra Mariadassou, chargé de recherche dans l’équipe StatInfOmics de l’unité MaIAGE sur le centre de Jouy en Josas et Géraldine Pascal, ingénieur de recherche dans l’équipe NED de l’unité GenPhySE du centre INRA de Toulouse. L’unité MaIAGE est structurée en quatre équipes de recherche dont le but est de développer des méthodes mathématiques et informatiques autours de problèmes biologique et agro-écologique. L’unité GenPhySE étudient la structure et le fonctionnement du génome des espèces domestiques, identifient les zones du génome influençant les principaux caractères d’intérêt agronomique et développent des méthodes pour l’amélioration et la gestion génétique des populations animales.
9 A
NNEXE
3
Bilan personnel du stage :J’ai réalisé mon stage dans l’unité UMR 1313 - Génétique Animale et Biologie Intégrative (GABI) de l’INRA de Jouy-en-Josas sur le sujet « Développement d’outil sous Galaxy pour la reconstruction phylogénétique et pour l’analyse statistique liée à la métagénomique » sous la responsabilité de Maria Bernard, Mahendra Mariadassou et Géraldine Pascal.
La réalisation de ce stage a été pour moi une occasion de mettre en application et développer les connaissances acquises au cours des deux années de master Bioinformatique et Génomique de Rennes I. J’ai pu ainsi apprendre à employer différents langages informatiques, logiciels bioinformatiques et analyses statistiques pour répondre aux problématiques soulevées par mon sujet de stage. Ce travail m’a permis d’avoir une expérience concrète d’application de la bioinformatique dans le domaine de la recherche en métagénomique amplicon.
C’est mon deuxième stage réalisé en France, durant lequel j’ai travaillé six mois sur un sujet s’intégrant dans un programme de recherche réel. Tout au long de ce stage, j’ai bénéficié de l’aide et des conseils de mes encadrants et plus particulièrement du suivi et du soutien continu de Maria Bernard, avec qui j’ai interagi presque quotidiennement et dont j’ai beaucoup appris. Ce stage m’a également permis de voir comment fonctionne une équipe de recherche et d’avoir des contacts et des discussions avec les autres membres du laboratoire (étudiants et enseignants-chercheurs).
Cette expérience dans le domaine de la recherche a été très enrichissante pour ma formation et je l’espère pour le laboratoire d’accueil. Elle me sera très utile dans mon processus de formation professionnelle. C’est une expérience qui me suivra dans tous mes travaux futurs.