Targeting IRAK1 in T-Cell acute lymphoblastic leukemia
Texte intégral
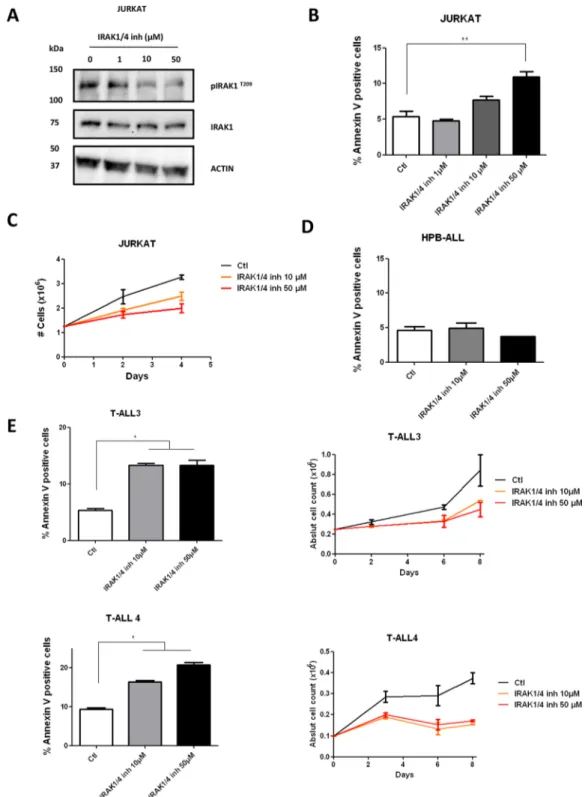
Figure

Documents relatifs
The projects were Human Resource Development for the Mekong Region (1993-2007) on vegetable research; Social Forestry Support Programme (1994-2002) for development of five
Conclusions: Hypoxia is a factor in leukemia cell resistance and for two conventional chemotherapies modulates cell death signaling pathways without affecting total cell density or
Encouraged by these promising results, we evaluated cell death induced by PKHB1 (the first- described serum- stable CD47- agonist peptide) on CEM and MOLT- 4 human
Handegord, Ridging, Shrinkage and Splitting of Built-up Roofing Membranes, National Research Council of Canada, Divi- sion of Building Research, Building Research Note 112,
Sterman, John, Business Dynamics: Systems Thinking and Modeling for a Complex World Sussman, Joseph, Introduction to Transportation Systems. Tenner, Edward, Why Things Bite Back:
C’est donc dans l’arrêt ici commenté, celui du 4 février 2020, que le Tribunal fédéral a appliqué concrètement pour la première fois la théorie de l’imprévision : «
This gives us a notion of negative type, that is a function type whose range type is ⊥, which can be made distinct from positive types (which could be anything other than a
Sœur Thérèse-Marguerite Notter, directrice de la Maison Chappuis à Soyhières, tient à préciser que même si la garderie Sainte-Léonie ne bénéficie d’aucune subvention, elle
