ROYAUME DU MAROC UNIVERSITE MOHAMED V
FACULTE DE MEDECINE ET DE PHARMACIE RABAT
Neuropathies périphériques associées aux
Gammapathies monoclonales
(Neuropathies dysglobulinémiques)
Service de neurophysiologie clinique Hôpital des spécialités, Rabat
MEMOIRE DE FIN DE SPECIALITE EN NEUROLOGIE
: Sous la direction du :
Présenté par
Dr. HARBOUZ SOUMIYA Pr. BIROUK NAZHA
Sommaire
Liste des abréviations ... 4
Liste des figures ... 5
Liste des tableaux ... 6
INTRODUCTION ... 8
RAPPEL DES IMMUNOGLOBULINES ... 9
A) Définition des Immunoglobulines : ... 10
B) Structure des Immunoglobulines : ... 10
C) Les classes des Immunoglobulines : ... 11
ETUDE THEORIQUE ... 13
A) Définition et cadre nosologique : ... 14
1) Gammapathies monoclonales de signification indéterminée : ... 15
2) Gammapathies monoclonales malignes : ... 16
2-1) Myélome multiple ou « maladie de Kahler » : ... 16
2-2) Maladie de Waldenström (MW) : ... 18
2-3) POEMS syndrome : ... 18
2-4) Lymphomes : ... 19
2-5) L’amylose AL : ... 19
3) Gammapathies monoclonales associées à une pathologie non lymphoïde : ... 20
B) Epidémiologie : ... 21
C) Mécanismes physiopathologiques : ... 22
D) Neuropathies dysglobulinémiques : ... 23
1-1) Neuropathies associées à MGUS à IgM avec activité anti-MAG : ... 24
1-2) Neuropathies péripheriques associées au SYNDROME CANOMAD ... 27
1-3) Neuropathies associées à la macroglobulinémie de Waldenström : .. 27
1-4) Neuropathies associées au lymphome : ... 28
2) Neuropathies dysglobulinémiques à type IgG/ IgA : ... 28
2-1) Neuropathies associées à une gammapathie monoclonale à signification indéterminée à type IgG et IgA : ... 28
2-2) Neuropathies périphériques associées au syndrome de POEMS :... 28
2-3) Neuropathies associées au Myélome multiple : ... 29
2-4) Neuropathies associées à l’amylose AL : ... 30
2-5) Neuropathies associées aux Cryoglobulinémies : ... 30
E) Stratégies diagnostiques : ... 32 F) Prise en charge : ... 35 ETUDE PRATIQUE ... 39 A) Observations cliniques : ... 40 B) Discussion : ... 47 CONCLUSION ... 51 RÉFÉRENCES BIBLIOGRAPHIQUES ... 52
Liste des abréviations :
CANOMAD : Chronic Ataxic Neuropathy with Ophtalmoplegia, M-protein Agglutination and Disialosyl antibodies.
CRAB : Calcium level elevated, renal failure, anemia, bone lesions
DADS : Distal acquired demyelinating neuropathy. EFNS : European Federation of Neurological Societies
GM : Gammapathie monoclonale.
GMSI : Gammapathie monoclonale de signification indéterminée. LMNH : Lymphome malin non hodgkinien.
MAG : Myelin Associated Glycoprotein.
MGUS : Monoclonal gammopathy of undetermined significance. MM : Myélome multiple.
MW : Maladie de Waldenstrom.
NO : Non obtenu
PGAM : Potentiel global d'action musculaire.
PIDC : Polyneuropathies inflammatoires démyélinisantes chroniques.
POEMS : Polyneuropathy, Organomégaly, Endocrinopathy, Monoclonal gammapathy, Skin changes.
Liste des figures
Figure 1 : Structure de base des Ig [1] ... 10 Figure 2 : Critères diagnostic de myélome multiple de 2014 [6] ... 17 Figure 3: Les principales caractéristiques électrophysiologiques des
neuropathies associées à IgM-MGUS [28] ... 26 Figure 4: Electrophorèse des protéines sériques montrant un pic monoclonal migrant dans la zone Gamma [58]. ... 33 Figure 5: Algorithme de Stratégie de la gestion des neuropathies
dysglobulinémiques selon la Mayo Clinic [79] ... 38 Figure 6 : Electrophorèse des protéines sériques montrant un pic monoclonal . 42
Figure 7 :
ongles blancs à gauche et lésions cutanées papuleuses diffusesListe des tableaux
Tableau 1 : Critères diagnostiques de POEMS syndrome [10] ... 19
Tableau 2 : Classification des neuropathies dysglobulinémiques ... 23
Tableau 3 : Phénotypes clinique et électrophysiologiques des neuropathies dysglobulinémiques ... 31
Tableau 4 : conduction nerveuse motrice à l’ENMG ... 41
Tableau 5 : conduction nerveuse sensitive à l’ENMG ... 41
Tableau 6 : Résultats de la conduction motrice de l’EMG ... 44
INTRODUCTION
- Les gammapathies monoclonales constituent un spectre hétérogène d’affections allant de la gammapathie monoclonale de signification indéterminée dite bénigne (GMSI ou MGUS en anglais), aux hémopathies malignes (tels qu’un myélome multiple, une maladie de Waldenström ou un lymphome sécrétant) et à certaines affections systémiques comme l’amylose AL ou la cryoglobulinémie.
- Les neuropathies périphériques associées à une gammapathie monoclonale appelées également neuropathies dysglobulinémiques forment un groupe complexe de neuropathies périphériques notamment en raison d’une importante hétérogénéité clinique et électrophysiologique.
- L'objectif de notre travail est d'analyser le profil clinique et électrophysiologique des neuropathies dysglobulinémiques et étudier les différentes stratégies diagnostiques et thérapeutiques de ces neuropathies en se basant sur les données de la littérature et des cas cliniques illustrant des neuropathies périphériques révélant une gammapathie monoclonale.
A) Définition des Immunoglobulines :
Les Immunoglobulines (ou des anticorps) sont des glycoprotéines membranaires ou solubles, sécrétées par les plasmocytes (des cellules dérivées des lymphocytes B) et impliquées dans la défense immunitaire pour détecter et neutraliser les agents pathogènes de manière spécifique.
B) Structure des Immunoglobulines :
La structure de base des anticorps est en forme de « Y » et est constituée de quatre chaînes d’acides aminés [1] :
+ 2 Chaînes polypeptidiques dites légères ou L ("Light") identiques. Il existe 2 types de chaînes L : Kappa κ ou Lambda λ.
+ 2 Chaînes polypeptidiques dites lourdes ou H ("Heavy") identiques
+ Les chaînes polypeptidiques H et L sont reliées par des ponts disulfure intra-chaînes et inter-intra-chaînes
- les chaînes lourdes et les chaînes légères peuvent être divisées en deux régions basées sur la variabilité des séquences en acides aminés. Ce sont :
1. Les régions Variables (V) ou Fab ("fragment, antibody binding") :(environ 100 - 130 acides aminés) qui reconnaissent l’antigène.
2. Les régions Constantes (C) ou Fc ("fragment, crystalline") : (environ 100 - 130 acides aminés) qui interagissent avec les récepteurs à la surface des cellules.
C) Les classes des Immunoglobulines :
- Les anticorps humains sont répartis en cinq classes ou encore en cinq isotypes : IgG, IgM, IgA, IgD et IgE
- Chaque catégorie d’anticorps est caractérisée par sa chaîne lourde [2] : – IgG : chaîne lourde de type γ (Gamma)
– IgA : chaîne lourde de type α (Alpha) – IgM : chaîne lourde de type µ (Mu) – IgD : chaîne lourde de type δ (Delta) – IgE : chaîne lourde de type ε (Epsilon)
• Les immunoglobulines G (IgG) constituent 75 à 80% de nos anticorps circulants. Elles sont produites en réaction à un antigène, et protègent ainsi l’organisme contre les bactéries, virus et certaines toxines. Elles fixent le complément et jouent un rôle dans la réponse mémoire, qui permet la vaccination. Elles peuvent traverser la barrière placentaire et apporter ainsi une immunité passive au fœtus. • Les immunoglobulines A (IgA) forment une barrière empêchant la majorité des pathogènes de se lier aux cellules des muqueuses et de l’épiderme. Elles sont présentes dans la salive, les larmes, le lait maternel, les sécrétions nasales, gastro-intestinales et du tractus respiratoire.
• Les immunoglobulines M (IgM) sont présentes à la surface des lymphocytes B natifs, et peuvent ensuite être sécrétés par les plasmocytes. Un taux sanguin élevé signe une infection en cours.
• Les immunoglobulines D (IgD) sont presque toujours attachées à la surface des lymphocytes B où elles fixent les antigènes.
• Les immunoglobulines E (IgE) sont reliées à deux types de globules blancs (mastocytes et granulocytes basophiles). La capture d’un antigène par cette immunoglobuline entraîne la sécrétion de produits, dont l’histamine, engendrant une réaction inflammatoire et éventuellement allergique.
A) Définition et cadre nosologique :
- Une gammapathie monoclonale ou dysglobulinémie se définit par la présence d’une immunoglobuline monoclonale (appelée aussi protéine monoclonale M) dans le sang et/ou dans les urines secondaire à la prolifération d’un clone lympho-plasmocytaire produisant en quantité excessive des immunoglobulines (Ig) de même idiotype : même chaîne lourde (IgG, A, D, E ou M), même chaîne légère (lambda ou kappa).
- Les étiologies des gammapathies monoclonales sont très variées mais on peut les classer en 3 catégories :
+ Gammapathies monoclonales dites bénignes, de signification indéterminée (GMSI ou MGUS en anglais).
+ Gammapathies monoclonales malignes : qui comportent essentiellement le myélome multiple, la macroglobulinémie de Waldenstrom, le lymphome et le syndrome POEMS (Polyneuropathie, Organomégalie, Endocrinopathie, Pic de protéines M et Manifestations cutanées).
1) Gammapathies monoclonales de signification indéterminée :
- Elle se traduise par la présence dans le sang d’une immunoglobuline (Ig) monoclonale sérique, en concentration modérée, sans anomalie biologique, ou manifestation clinique faisant évoquer une hémopathie maligne dysglobulinémique [3].
- Le diagnostic de MGUS est basé sur plusieurs arguments biologiques qui sont : 1) L’existence d’un pic monoclonal sérique d’Ig de concentration inférieure à 30 g/l.
2) Une plasmocytose médullaire inférieure à 10%.
3) Une protéinurie de Bence-Jones négative ou inférieure à 1g/24 h.
4) Absence de signes de prolifération maligne (encore appelés critères CRAB) : lésions osseuses lytiques, anémie, hypercalcémie ou insuffisance rénale en rapport avec la dysglobulinémie [4].
+ C pour hypercalcémie : ≥ 115 mg /l ou ≥ 2,65 mmol/l.
+ R pour insuffisance rénale : clairance ≤ 50ml/mn sans autres causes ou créatinémie >177μmol/L.
+ A pour anémie (taux d’hémoglobine < 10 g/dl ou plus de 2g/dl en dessous de la limite inférieure de la normale)
+ B pour lésions osseuses lytiques (Bone lesions en Anglais). 5) Stabilité dans le temps du pic monoclonal à l’électrophorèse des Protéines sériques. En effet, l’évolution vers une pathologie maligne,
Principalement le myélome multiple, peut apparaître après plusieurs années. - Il existe trois principaux types de MGUS selon le type de protéine M sécrétée : + IgM MGUS : est associée à un risque de progression vers la macroglobulinémie de Waldenstrom ou lymphome.
+ Non-IgM MGUS (comprend IgG MGUS et IgA MGUS) : risque d’évolution vers le myélome multiple.
+ MGUS à chaîne légère : est une entité nouvellement découverte qui est associée à un risque de progression vers le type de chaîne légère de MM.
- Toutes les formes de MGUS peuvent évoluer vers l’amylose AL. 2) Gammapathies monoclonales malignes :
- La gammapathie monoclonale peut être secondaire à une prolifération maligne d’un clone lymphoplasmocytaire (lignée lymphocytaire B), souvent rencontrée dans des hémopathies malignes à type de myélome multiple, macroglobulinémie de Waldenström, leucémie lymphoïde chronique ou lymphome malin non hodgkinien.
- On distingue des gammapathies monoclonales malignes à type IgG et IgA représentées par essentiellement par le myélome multiple et à type IgM rencontrées dans la maladie de Waldenström et lymphomes.
2-1) Myélome multiple ou « maladie de Kahler » :
- C’est une hémopathie maligne due à la prolifération tumorale de plasmocytes. - Les signes cliniques les plus fréquents sont : La fatigue, douleurs osseuses, infections récurrentes [5].
- Le diagnostic de myélome multiple est basé sur la présence de ces 2 critères suivants [6] :
+ Plasmocytose médullaire clonale ≥ 10% ou plasmocytome osseux ou extra médullaire prouvé à la biopsie.
+ Un ou plusieurs des critères suivants et liés au myélome :
* Mise en évidence d'une ou plusieurs atteintes d’organe (critères de CRAB) attribuables à la prolifération plasmocytaire
* Plasmocytose médullaire clonale ≥ 60%
* Rapport chaînes légères libres (FLC) impliquées/non impliquées ≥ 100 (mais il faut que le taux de FLC soit ≥ 100 mg/L)
* Plus qu’une lésion focale à l'IRM (de diamètre supérieur à 5 mm)
Figure 2 : Critères diagnostic de myélome multiple de 2014 [6]
Remarque :
- La présence d’un pic monoclonal n’est pas nécessaire pour le diagnostic de myélome mais Elle est utilisée pour classer le myélome en type secrétant ou non sécrétant.
Myélome indolent (Asymptomatique) :
-Il est défini par un taux excessif de pic monoclonal IgG ou IgA supérieur à 3 g/dl et un taux de plasmocytes médullaires supérieur à 10 % sans manifestation systémique du myélome multiple.
2-2) Maladie de Waldenström (MW) :
- La maladie de Waldenstrom est caractérisée par une prolifération lymphoplasmocytaire au niveau de la moelle osseuse et les tissus lymphoïdes associée à une production d’IgM sérique.
- La MW a un large spectre clinique dépendant du degré d’infiltration tumorale et de la production et des dépôts d’IgM dans les tissus. Les manifestations principales incluent l’hépatomégalie, la splénomégalie, la lymphadénopathie, l’hyperviscosité et la fatigue liée à l’anémie [7].
- Les critères diagnostiques définis lors du deuxième workshop sur la MW sont les suivants [8] :
• IgM monoclonale sérique quelle que soit la concentration ;
• Infiltration médullaire par de petits lymphocytes avec différenciation plasmocytaire ;
• Infiltration à la biopsie médullaire souvent diffuse ;
• Phénotype des cellules tumorales : IgM+ , CD5–/+ , C10– , CD19+ , CD20+ , CD22+ , CD23– , CD25+ , CD27+ , FMC7+ , CD103.
2-3) POEMS syndrome :
- C’est un acronyme de Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal gammapathy, Skin changes.
- Le tableau clinique le plus fréquent est celui d'une neuropathie périphérique progressive associée à une chaîne légère monoclonale très souvent de type λ. Il y a souvent une endocrinopathie, une hépatosplénomégalie, des lésions osseuses condensantes, une thrombocytose ou une polyglobulie [9].
- Le diagnostic de POEMS syndrome est confirmé devant la présence de : 2 critères majeurs obligatoires, un seul parmi les 3 autres critères majeurs et au moins un critère mineur (Critères de Dispenzieri [10]).
Tableau 1: Critères diagnostiques de POEMS syndrome [10]
2-4) Lymphomes :
Les lymphomes sont des tumeurs malignes développées à partir des lymphocytes le plus souvent B. Ils s’expriment par des masses tumorales au niveau des organes lymphoïdes (adénopathies, splénomégalie) et au niveau extra-ganglionnaire (estomac, système nerveux central, rein, os, testicules). Les lymphomes sont classés en lymphomes Hodgkiniens et non hodgkiniens (LMNH).
2-5) L’amylose AL :
-L’amylose AL est une maladie multi systémique caractérisée par la présence de dépôts amyloïdes formés à partir de chaines légères d’une immunoglobuline monoclonale dans les tissus de plusieurs organes : rein, cœur, foie, peau et nerf périphérique.
-Le diagnostic repose sur la mise en évidence de dépôts d’amylose sur les examens anatomopathologiques (biopsie de glande salivaire accessoire, biopsie rectale, biopsie de graisse sous-cutanée, biopsie de nerf, objectivée par la coloration rouge Congo) ainsi que la mise en évidence d’une chaîne légère au sein
de la substance amyloïde et la présence des signes de dyscrasie plasmocytaire monoclonale (protéine monoclonale sérique ou urinaire, rapport kappa-lambda anormal [11].
3) Gammapathies monoclonales associées à une pathologie non lymphoïde : Maladies infectieuses : infections virales aigues Bénignes (cytomégalovirus,
virus d’Epstein-Barr, rougeole, etc.) ou infection à VIH qui s’accompagne d’une gammapathie monoclonale dans 3 à 5% des cas [12].
Maladies auto-immune : Polyarthrite rhumatoïde, lupus Érythémateux aigu disséminé, les polymyosites, la sclérodermie, l’hépatite chronique active [13]. Néoplasie (carcinome des voies biliaires, de la vessie, du Sein, du foie, du
poumon, de l’ovaire, de la prostate, de l’utérus, mélanome malin, angiosarcome) [14].
B) Epidémiologie :
- Afin de comprendre l’épidémiologie de neuropathies dysglobulinémiques, il faut savoir que la gammapathie monoclonale de signification indéterminée (GMSI) représente plus de 60% des gammapathies monoclonales [15] et elle est relativement courante dans la population générale, avec une prévalence de 3 à 4% chez les sujets plus de 50 ans et augmentant à 8 à 9% à l'âge de 80 ans [16]. La neuropathie périphérique affecte 2 à 4% de la population générale, passant à 8 à 0% avec l'âge avancé [17].
- La prévalence de la neuropathie périphérique dans les gammapathies monoclonales de signification indéterminée (GMSI) est d'environ 5% en IgG, 15% en IgA et pouvant aller jusqu'à 30 à 50% en IgM GMSI [18 ; 19 ; 20 ; 21]. - L’existence d’un lien de causalité entre la gammapathie monoclonale et la neuropathie périphérique reste toujours un défi à relever pour montrer que l’association n’est pas simplement fortuite.
- La gammapathie monoclonale GM est généralement découverte au cours du bilan étiologique d’une neuropathie périphérique. Environ 10% des patients avec une polyneuropathie de cause inconnue ont une gammapathie monoclonale [22]. - les neuropathies périphériques surviennent également au cours des hémopathies malignes et peuvent parfois être révélatrices.
Certains médicaments utilisés pour traiter ces hémopathies peuvent être neurotoxiques et peuvent être la véritable cause des symptômes.
C) Mécanismes physiopathologiques :
Plusieurs mécanismes peuvent être impliqués dans la physiopathologie des neuropathies dysglobulinémiques :
Soit par un mécanisme auto-immun : Activité directe de l’immunoglobuline monoclonale sur un constituant du nerf comme dans les neuropathies anti-MAG. La protéine MAG a été identifiée pour la première fois en 1982 par l’équipe de Braun [23]. Il s’agit d’une glycoprotéine membranaire ayant pour rôle le maintien de l’intégrité de la structure de la myéline. La présence d’anticorps anti-MAG entraîne une “décompaction” des lamelles de myéline.
Soit secondaire à des dépôts d’immunoglobulines de chaînes légères monoclonales dans l’endonèvre comme dans l’amylose AL [24].
Soit par le biais d’une vascularite (exemple : Cryoglobuline) [25].
Soit par l’infiltration lymphomateuse du nerf périphérique (Neurolymphomatose) [26].
Soit par une activation anormale des cellules endothéliales par le VEGF, qui est surexprimé dans les nerfs des patients atteints du syndrome POEMS, induisant ainsi des changements microvasculaires et une altération de la perméabilité vasculaire [27]. Ces altérations entraînent une microangiopathie ischémique secondaire et des lésions axonales chroniques des fibres nerveuses par altération de la barrière hémato-nerveuse [27].
D) Neuropathies dysglobulinémiques :
Il n’existe pas à l’heure actuelle de classification internationale des neuropathies dysglobulinémiques à cause de leurs aspects hétérogènes et complexes.
La classification de ces neuropathies dysglobulinémiques doit prendre en considération plusieurs facteurs (Selon l’EFNS 2010) [28] :
Le pattern clinique et électrophysiologique de ces neuropathies, Le type des Immunoglobulines,
Association ou non à une activité anti-MAG, La présence ou non de malignité,
Et le mécanisme physiopathologique de leur survenue.
Dans ce chapitre, on va classer les neuropathies dysglobulinémiques en fonction de la protéine M exprimée : Soit à type IgM ou à type IgG/ IgA et dans ces sous-groupes, on va les différencier en fonction de la présence ou non de malignité.
On distingue alors :
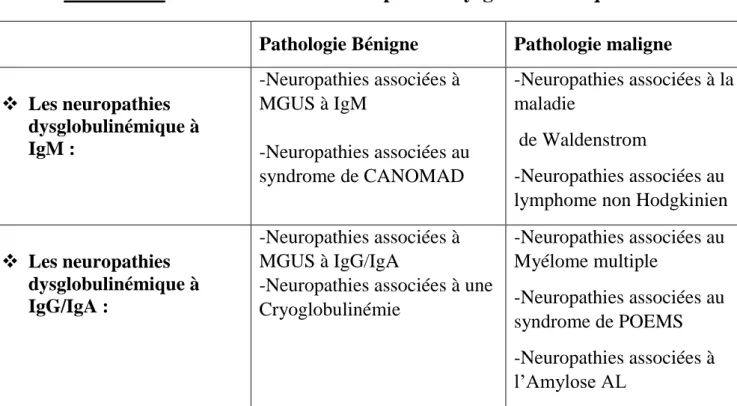
Tableau 2 : Classification des neuropathies dysglobulinémiques
Pathologie Bénigne Pathologie maligne
Les neuropathies dysglobulinémique à IgM : -Neuropathies associées à MGUS à IgM -Neuropathies associées au syndrome de CANOMAD -Neuropathies associées à la maladie de Waldenstrom -Neuropathies associées au lymphome non Hodgkinien
Les neuropathies
dysglobulinémique à IgG/IgA :
-Neuropathies associées à MGUS à IgG/IgA
-Neuropathies associées à une Cryoglobulinémie -Neuropathies associées au Myélome multiple -Neuropathies associées au syndrome de POEMS -Neuropathies associées à l’Amylose AL
On va essayer de décrire les principales caractéristiques cliniques et électrophysiologiques de ces neuropathies en fonction de cette classification. 1) Neuropathies dysglobulinémiques à type IgM :
1-1) Neuropathies associées à MGUS à IgM avec activité anti-MAG :
Les neuropathies associées aux anticorps anti-MAG (anti-myelin Associated glycoprotein) appartiennent au groupe des DADS (Distal acquired
demyelinating neuropathy), qui constitue une variante de polyneuropathies
inflammatoires démyélinisantes chroniques (PIDC). Ce sont des neuropathies démyélinisantes distales, associées à une gammapathie monoclonale d’isotype M et des anticorps anti-MAG.
Les neuropathies à anti-MAG sont souvent associées au MGUS mais elles peuvent associer aussi à la maladie de Waldenström ou lymphome non hodgkinien [38].
Pattern clinique :
- La présentation clinique habituelle des neuropathies anti-MAG est celle d’une neuropathie de DADS.
Elle est caractérisée donc par : + Une évolution chronique,
+ Une topographie distale et symétrique,
+ Une prédominance sensitive : paresthésies des extrémités avec hypoesthésie à tous les modes, douleurs neuropathiques et surtout une ataxie proprioceptive. (Au contraire de la PIDC qui est proximale et distale avec atteinte motrice prédominante) [29].
+ Des Réflexes ostéo-tendineux abolis.
+ Un déficit moteur souvent tardif et modéré (steppage, chez environ 30% des patients).
- On n’a pas souvent d’atteinte des paires crâniennes, ni de dysautonomie. Pattern EMG :
Certains paramètres électrophysiologiques permettent de distinguer la neuropathie anti-MAG d’une PIDC et cela nécessite une analyse approfondie et appropriée de l’EMG.
Le caractère démyélinisant des neuropathies anti-MAG est présent chez presque 90% des cas [31].
La démyélinisation est à prédominance distale, elle se manifeste par un allongement important des latences distales motrices par rapport aux vitesses de conduction motrice proximale avec une atteinte clinique motrice discrète ou au second plan (discordance clinico-électrophysiologique). Plusieurs indices ont été créés pour différencier entre les polyneuropathies
anti-MAG et les PIDC, le plus classique est l’indice de latence terminale (Terminal Latency Index : TLI), qui représente un signe de démyélinisation distale lorsqu’il est inférieur à 0,25 sur au moins deux nerfs (nerfs médians et ulnaires).
-C’est un outil qui permet de comparer la vitesse de conduction motrice distale à la vitesse de conduction motrice plus proximale. Il a été décrit pour la première fois par l’équipe de Kaku [32].
-On le calcule à partir de la formule suivante :
𝑇𝐿𝐼 =d (distance entre la stimulation distale et l’électrode de réception) VCM proximale x Latence distale motrice
- Il permet ainsi de distinguer les neuropathies anti-MAG des PIDC. Ces Derniers ont un TLI normal ou augmenté [32, 33].
Les blocs de conduction sont très rares, contrairement aux PIDC [32-34]. Vitesses de conduction motrice très diminuées surtout aux MI [34].
D’autres indices de démyélinisation ont été proposés comme : latences résiduelles des nerfs ulnaires et la durée du PGAM distale. Ces indices restent peu sensibles.Les latences résiduelles des nerfs ulnaires représentent la différence entre la latence motrice distale mesurée et la latence motrice distale théorique. Une latence résiduelle supérieure à 4 ms sur les nerfs ulnaires est fortement suggestive du diagnostic de neuropathie anti-MAG [35, 36].
Les potentiels sensitifs sont habituellement diminués et souvent même absents aux membres inférieurs, en particulier pour le nerf sural [37]. L’EFNS a publié en 2010 les principales caractéristiques
électrophysiologiques des neuropathies associées à une gammatpathie monoclonale de type IgM (voir Figure 3).
Figure 3 : Les principales caractéristiques électrophysiologiques des neuropathies associées à IgM-MGUS [28]
: Neuropathies péripheriques associées au SYNDROME CANOMAD 2)
-1
Le syndrome CANOMAD est l’acronyme de Chronic Ataxic Neuropathy with Ophtalmoplegia, M-protein Agglutination and Disialosyl antibodies.
La neuropathie du syndrome de CANOMAD est une polyneuropathie sensitive ataxiante chronique associée à une ophtalmoplégie et gammapathie monoclonale IgM dirigée contre les gangliosides disialylés des gaines de myéline des nerfs périphériques (GD3, GQ1b, GD1b et GT1b) avec activité d’agglutinines froides [39].
Le tableau clinique ressemble à celui d’un syndrome de Miller Fisher (ophtalmoparésie, aréflexie et ataxie), mais avec une évolution chronique. Son diagnostic repose sur la détection d’anticorps anti gangliosides disialylés
dans le sérum des patients.
1-3) Neuropathies associées à la macroglobulinémie de Waldenström :
La prévalence d’une neuropathie périphérique chez les patients présentant une Maladie de Waldenström est de 47% [40].
Le phénotype clinique de ces neuropathies est identique à celui des neuropathies à IgM-MGUS (Les DADs).
Les symptômes les plus observés sont les paresthésies au niveau des pieds et l’ataxie proprioceptive.
La dysautonomie est limitée à une atteinte légère du sudomoteur (Transpiration).
Le tremblement est moins fréquent.
Sur le plan électrophysiologiques, les neuropathies associées à la maladie de Waldenstrom sont souvent de type axonale contrairement aux neuropathies anti-MAG (27% des patients ont Le caractère démyélinisant a été trouvé chez 27% des patients avec MW en comparant à 62% dans les IgM-MGUS) [41].
1-4) Neuropathies associées au lymphome :
Elles se manifestent souvent par une mononeuropathie multiple évoluant en une neuropathie diffuse aux caractéristiques d’atteinte axonale [42].
2) Neuropathies dysglobulinémiques à type IgG/ IgA :
2-1) Neuropathies associées à une gammapathie monoclonale à signification indéterminée à type IgG et IgA :
Elles sont moins fréquentes par rapport aux neuropathies associées à IgM-MGUS [43]. Elles ressemblent sur le plan clinique et électrophysiologique à une polyneuropathie inflammatoire démyélinisante chronique PIDC avec un début subaigu, surtout chez les patients âgés.
On trouve un déficit sensitivo-moteur modéré à sévère avec une bonne réponse aux immunomodulateurs, ce qui suggère une pathogenèse dysimmune.
Il n’existe pas à l’heure actuelle des anticorps spécifiques des neuropathies à IgA ou à IgG.
Elles peuvent se présenter également sous forme d’une axonopathie sensorielle chronique à prédominance distale progressive lente survenant chez les patients âgés. Les symptômes sont généralement légers, mais la réponse aux immunosuppresseurs est faible [44, 45].
2-2) Neuropathies périphériques associées au syndrome de POEMS :
-Le syndrome de POEMS se définit par la présence d’une neuropathie périphérique (P), une anomalie des plasmocytes (M), et d’autres manifestations paranéoplasiques, les plus fréquentes d’entre elles étant l’organomégalie (O), l’endocrinopathie (E), les altérations cutanées (S pour skin).
- La neuropathie périphérique est souvent le premier symptôme de la maladie. Elle se traduit par une polyneuropathie démyélinisante sensitivomotrice
ascendante, révélée initialement par un déficit sensitif distal au niveau des pieds, suivi par une atteinte motrice sévère, d’évolution subaigüe.
- La neuropathie est souvent confondue avec une PIDC (Tableau de PRN à prédominance motrice subaiguë). La douleur touche 75% des patients atteints de POEMS, prédominant au niveau des membres inférieures et elle est plus rapportée chez les patients atteints de POEMS par rapport à ceux atteints de PIDC [46, 47]. - Malgré que l’ENMG objective essentiellement une polyneuropathie démyélinisante, les études neurophysiologiques chez les patients atteints de POEMS montrent un degré plus élevé de la perte axonale longueur-dépendant avec des amplitudes réduites des potentiels d'action musculaire (PGAM) et des potentiels de fibrillation plus élevés par rapport à la PIDC [48].
- Un ralentissement plus prononcé de la conduction nerveuse dans les segments
intermédiaires que dans les segments nerveux distaux a été signalé, tandis que la démyélinisation dans la PIDC montre un modèle multifocal impliquant à la fois les segments distaux et intermédiaires [49].
- L'examen du liquide céphalo-rachidien et l'imagerie des racines nerveuses peuvent montrer également des anomalies, mais elles ne sont pas spécifiques du syndrome POEMS et peuvent également être observées dans les PIDC (Proteinorachie, épaississement des racines nerveuses) [50].
- La présence d'une gammapathie monoclonale IgA ou IgG de type λ est obligatoire pour le diagnostic du syndrome POEMS.
2-3) Neuropathies associées au Myélome multiple :
+ La prévalence de survenue des neuropathies au cours des myélomes multiples non traités est de 5 à 20%, avec l’existence des anomalies à l’EMG chez des patients asymptomatiques, Elle est augmentée à 39% [51].
Cliniquement, les neuropathies associées au MM sont des neuropathies sensitives ou sensitivo-motrices longueurs dépendant. Les douleurs neuropathiques et les signes dysautonomiques sont rares.
+ L’EMG montre souvent : Des potentiels moteurs (PGAM) faibles ou abolis avec légère diminution des vitesses de conduction motrice, potentiels sensitifs qui sont également faibles ou absents, faisant évoquer une neuropathie axonale.
+ Il existe des cas rares de neuropathies associées au MM qui se présentent comme des ganglionopathies sensorielles avec une ataxie proprioceptive ou comme des polyradiculoneuropathies motrices avec atteinte faciale, bulbaire et respiratoire [52].
2-4) Neuropathies associées à l’amylose AL :
- Les neuropathies associées à l’amylose AL sont des polyneuropathies sensitivomotrices, à prédominance sensitive, qui touchent surtout les petites fibres, avec dysautonomie précoce (constipation, impuissance ou hypotension orthostatique) [53].
- La neuropathie périphérique est une manifestation courante de l’amylose AL, avec des symptômes sensoriels distaux, notamment une dysesthésie douloureuse, une perte de toucher léger et une sensation de température, et une atteinte autonome marquée étant la principale caractéristique clinique [54, 55]. Une faiblesse symétrique distale se développe pendant la progression de la AL.
- Les patients atteints de l’amylose AL ont des caractéristiques électrophysiologiques compatibles avec une polyneuropathie axonale sensorimotrice, fréquemment associée à une neuropathie autonome et au syndrome du canal carpien [56].
- Le diagnostic repose sur la mise en évidence de dépôts amyloïdes dans les tissus obtenus à partir des organes impliqués [55]. Les résultats de la moelle osseuse et le taux plasmatique ou sérique de VEGF sont fondamentaux pour distinguer le syndrome POEMS de l'amylose AL [57].
2-5) Neuropathies associées aux Cryoglobulinémies :
- Elles sont souvent associés à des atteintes rénales, un purpura ou à un phénomène de Raynaud.
- L’atteinte nerveuse peut se présenter de manière variée, comme une neuropathie distale sensitive ou sensitivo-motrice symétrique ou une atteinte de type mononévrite multiple.
Le tableau suivant résume les principales caractéristiques cliniques et électrophysiologiques des neuropathies dysglobulinémiques :
Tableau 3 : Phénotypes clinique et électrophysiologiques des neuropathies dysglobulinémiques Type de gammapathie monocloname Type des Ig Phénotype clinique de neuropathie périphérique Phénotype Electrophysiologique
IgM-MGUS IgM Neuropathie ataxiante distale de type DADs
Démyélinisant avec allongement de LDM IgG/IgA-MGUS IgG/IgA Neuropathie de type
PIDC avec début subaigu
Démyélinisant
Maladie de Waldenstrom
IgM Idem IgM-MGUS Axonal++> Démyélinisant avec allongement de LDM Myélome multiple IgG>IgA Polyneuropathie sensitive
ou sensitivo-motrice longueur-dépendant Axonal Syndrome de POEMS IgG/IgA Lambda
PIDC like Démyélinisant
Amylose AL Lambda Polyneuropathie
sensitivo-motrice avec dysautonomie
E) Stratégies diagnostiques :
Etape 1 : Déterminer le phénotype clinique et électrophysiologique de la neuropathie périphérique :
- L’examen neurologique couplé à l’électroneuromyogramme est essentiel pour déterminer le phénotype clinique et électrique de la neuropathie périphérique : - Interrogatoire : Identifier les signes fonctionnels et leur chronologie.
- Examen clinique : préciser la nature et la sévérité des troubles sensitifs (Atteinte des petites ou grosses fibres ou les deux), la présence d’un déficit moteur, la recherche de signes dysautonomiques (hypotension orthostatique), l’examen des réflexes ostéo-tendineux et la présence ou non d’un tremblement d’attitude. Cet examen permettra de typer la neuropathie en sensitive, sensitivomotrice ; longueur dépendante, petites fibres ou toutes les fibres.
- L’ENMG est très utile pour identifier le caractère démyélinisant ou axonal de la neuropathie. Chercher les signes de démyélinisation distale déjà détaillé dans le chapitre des neuropathies anti-MAG.
Etape 2 : Eliminer les autres étiologies de la neuropathie périphérique : + ATCD de diabète, alcoolisme, insuffisance rénale, médicaments neurotoxique, maladie systémique, maladie néoplasique, etc ….
+ Réaliser les examens complémentaires en fonction de type de la neuropathie (Caractère démyélinisant ou axonal).
: Si neuropathie démyélinsiante quelque soit le mode de Ponction lombaire
début ou neuropathie aiguë quel que soit le type démyélinisant ou axonal. A la recherche de dissociation albumino-cytologique ou de méningite.
- Bilan biologique : Vitesse de sédimentation, CRP, urée, créatinine, ASAT, ALAT, bilan phosphocalcique, bilan thyroïdien, Hémoglobine glyquée et les Sérologies VIH, Hépatite B et C.
-Autres : Scanner thoraco-abdominal, IRM des racines, Bilan immunologique :
Ac anti-nucléaire, Ac anti-ADN natif, Ac anti-phospholipides, biopsie nerveuse. Etape 3 : Confirmer la présence d’une gammapathie monoclonale, déterminer le type de la dysglobulinémie et affirmer ou non le caractère malin :
- Examen général complet : recherche d’hépatomégalie, splénomégalie, adénopathies, lésions cutanées, douleurs osseuses, macroglossie, signes hémorragiques…
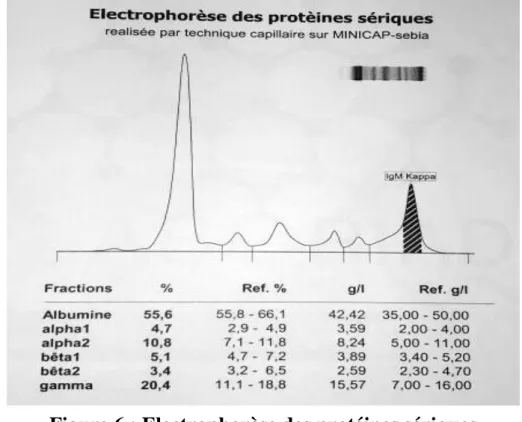
- L’électrophorèse des protéines sériques (EPS) est un examen biologique simple qui permet une appréciation quantitative mais également qualitative (sur l’aspect du tracé) des principales composantes protéiques du plasma (supérieure au gramme/litre en quantité). Les protéines sont analysées par migration dans un champ électrique et déposées en fonction de leur poids et de leur charge électrique. L’EPS permet de mettre en évidence et de caractériser le pic monoclonal. Celui-ci se situe le plus souvent dans la zone gamma (figure 4), plus rarement dans la zone bêta [58].
Figure 4 : Electrophorèse des protéines sériques montrant un pic monoclonal migrant dans la zone Gamma [58].
- L’immunofixation : est un test immunologique réalisé sur les protéines sériques ou urinaires. Il permet de poser le diagnostic de dysglobulinémie monoclonale. Elle confirme la clonalité du pic visualisé sur l’électrophorèse en déterminant l’isotype de la chaine lourde (G, A, M, D, E, rare pour les deux derniers) et/ou de la chaine légère (kappa ou lambda) (12). Les chaines lourdes et légères sont des chaines polypeptidiques. Elles sont les principales composantes (2chaines lourdes et 2chaines légères) des immunoglobulines, glycoprotéines douées d’une fonction anticorps.
- Dosage pondéral des immunoglobulines : Il permet un dosage précis des immunoglobulines G, A et M. Il ne permet pas de quantifier le composant monoclonal.
- Bilan urinaire : L’électrophorèse des protéines urinaires fait partie, avec la protéinurie des 24h et la recherche des chaines légères libres urinaires (protéinurie de Bence Jones), du bilan urinaire devant être réalisé devant une situation clinico-biologique évocatrice de dysglobulinémie monoclonale.
- Numération formule sanguine : Examen capital qui permet de mettre en évidence une anémie (généralement macrocytaire) ou une thrombopénie.
L’examen du frottis met en évidence des rouleaux érythrocytaires, témoins du composant monoclonal sérique [58].
- Myélogramme ou biopsie ostéo-médullaire : pour quantifier la prolifération lymphoplasmocytaire de la moelle osseuse.
+ Bilan radiologique : Radiographies de squelette complet (à la recherche d’une lyse osseuse ou d’ostéocondensations) et un scanner thoraco-abdomino-pelvien. + Dosage des Ac anti-MAG : en cas de gammapathie monoclonale à IgM. + Autres examens : pourront être demandés en fonction de l’orientation clinique : bilan immunologique, cryoglobulinémie, VEGF en cas de suspicion du syndrome de POEMS.
Etape 4 : Etablir le lien de causalité entre la neuropathie et la gammapathie monoclonale
- Le lien causal entre la gammapathie monoclonale et la neuropathie est plus probable avec IgM-MGUS ou maladie de Wladenstrom qu’avec une IgG ou IgA-MGUS [28].
- Si la protéine M n'est pas de type IgM, les données sur l'association causale avec la neuropathie et les données sur l'efficacité du traitement sont très limitées.
F) Prise en charge :
- La prise en charge thérapeutique des neuropathies périphériques associées aux gammapathies monoclonales dépend de l’âge du patient, de type de la dysglobulinémie identifiée, de la gêne fonctionnelle et de l’évolutivité de la neuropathie.
Neuropathies avec gammapathie monoclonale à IgM (MGUS/ Maladie de Waldenstrom) :
- Des recommandations de prise en charge ont été établies par l’EFNS en 2010, avec notamment la préconisation d’une abstention thérapeutique en cas de neuropathies associées à IgM-MGUS sans handicap fonctionnel avec un suivi régulier chez l’hématologue (car risque de transformation maligne) [28].
- Il n’y a pas de preuve fiable pour recommander un traitement par immunothérapie pour les neuropathies à IgM-MGUS non évolutives avec ou sans anticorps d’anti-MAG. Le traitement est justifié lorsque les symptômes sont invalidants pour éviter l’installation d’un handicap. Plusieurs thérapeutiques ont été proposées : traitements immunomodulateurs incluant les immunoglobulines et les échanges plasmatiques, les corticostéroïdes, le Chlorambucil, le Cyclophosphamide, , l’Azathioprine, la Fludarabine, la Cladiribine, l’Interferon alpha, l’Adriamycine, le Mephalan et les biothérapies [75] .
- Les anticorps monoclonaux anti-CD20 (Rituximab) ont été essayés également
Waldenstrom. Le protocole de traitement dans la plupart de ces études était hebdomadaire : des perfusions de 375 mg / m2 de rituximab sur 4 semaines. Les résultats de ces études suggèrent un effet bénéfique limité chez certains patients [59-61]. Des études ont rapporté une diminution de taux d’IgM avec traitement au Rituximab et amélioration des symptômes en cas de maladie de waldenstrom associées aux Ac anti-MAG [60]. Tandis que d’autres études montrent une aggravation de la neuropathie à IgM associée à la maladie de Waldenstrom [62-63].
Neuropathies du CANOMAD
Le traitement de ces neuropathies n’est pas encore codifié. Il fait appel aux Immunoglobulines intraveineuses, à un traitement par anticorps monoclonal anti-CD20 (Rituximab) et à chimiothérapie.
Neuropathies à MGUS à IgG ou IgA :
Les Neuropathies périphériques associées à MGUS à IgG ou IgA doivent être traité comme les PIDC dans les formes sévères et/ou évolutives ; il fait appel aux corticoïdes, aux IgIV, aux plasmaphérèses et aux immunosuppresseurs (chloraminophène, cyclophosphamide) [73,74]. Aucune étude randomisée n’a permis de préciser le meilleur choix thérapeutique.
Neuropathies associées au syndrome de POEMS :
- Le traitement du syndrome POEMS dépend de taux des plasmocytes monoclonaux et dépend de l'étendue de la maladie et de l'atteinte de la moelle osseuse ainsi que l’âge du malade.
- Chez les patients présentant deux lésions ou moins de plasmacytome, la radiothérapie est la thérapie de première intention. Les patients avec des lésions sclérotiques diffuses ou une atteinte médullaire disséminée, doivent être traités par thérapie systémique [65, 66]. Notamment, la plasmaphérèse et les immunoglobulines intraveineuses utilisées dans les neuropathies à médiation immunitaire semblent être inefficaces [67].
- La greffe de cellules souches autologues conditionnées par chimiothérapie à haute dose est le traitement de référence actuel chez les sujets jeunes pour le syndrome POEMS, montrant un bon contrôle hématologique, une réponse neurologique, avec une amélioration de la neuropathie [68-69].
Neuropathies de l’Amylose AL
Un régime combiné de Melphalan à haute dose et de greffe autologue de cellules souches reste actuellement une option uniquement pour les patients atteints de maladie focale (20-25% des patients), ce qui entraîne une augmentation du taux de survie à 10 ans à 43-53% [70-71].Pour les autres patients, des essais de schémas chimiothérapeutiques comprenant du melphalan, des corticostéroïdes (Dexaméthasone), de la thalidomide, du lénalidomide et du bortézomib devraient être envisagés [72].
Neuropathies associées au myélome multiple :
Les traitements actuels du MM comprennent la greffe autologue de cellules souches et diverses options chimiothérapeutiques, notamment la thalidomide, le bortézomib, le lénalidomide et le pomalidomide. Certaines chimiothérapies peuvent provoquer ou exacerber une neuropathie existante chez jusqu'à 65% des patients [76]. Les médicaments les plus fréquemment incriminés sont le bortézomib et la thalidomide, qui peuvent produire une polyneuropathie axonale dépendante de la longueur qui est principalement sensorielle [77] tandis que le lénalidomide est moins neurotoxique [78].
A) Observations cliniques :
Observation 1 :
- Nous rapportons le cas de Monsieur M.M âgé de 57 ans, ayant un antécédent de néo du rectum en 1992 traité par chirurgie et radio-chimiothérapie avec rémission complète.
- Le début de la symptomatologie remonte à 2009, marquée par l'apparition progressive des paresthésies associées à des douleurs neuropathiques avec une distribution en chaussettes. Le tableau clinique s’est aggravé en 2012 par l’apparition des troubles de l’équilibre et des paresthésies des mains.
- L’examen clinique a objectivé : une ataxie proprioceptive, une hypoesthésie superficielle en chaussette et proprioceptive aux membres inférieurs et un tremblement d’attitude des mains. Les réflexes ostéo-tendineux étaient abolis et il n’y avait pas d’atteinte cutanée, ni d’organomégalie.
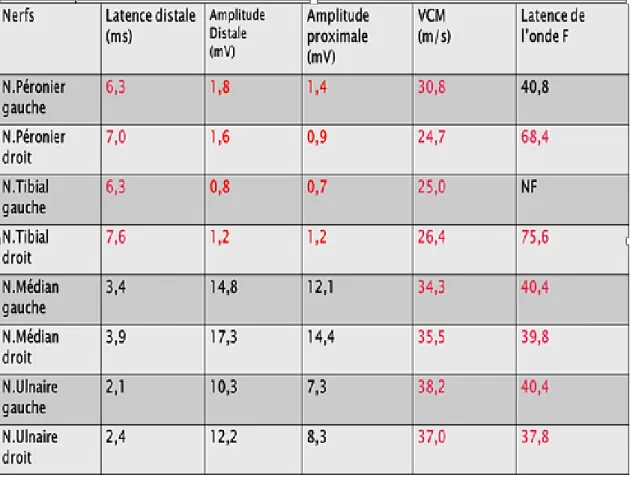
- L’ENMG a montré une polyneuropathie démyélinisante sensitivo-motrice avec ralentissement diffus de la conduction nerveuse prédominant sur les segments distaux (Voir Tableau 4 et 5).
- L’examen du LCR a montré une dissociation albumino-cytologique. - Le bilan biologique a objectivé une gammapathie monoclonale à IgM
Kappa (Voir Figure 5).
- Le scanner thoraco-abdomino-pelvien, la biopsie ostéo-médullaire et la scintigraphie osseuse sont revenues normales.
- La recherche des Ac anti-MAG est revenue positive > 700000 (BTU). - Le diagnostic d’une neuropathie à IgM-MGUS avec activité anti-MAG a
Tableau 4 : conduction nerveuse motrice à l’ENMG
- Le malade a reçu au début un traitement à base de corticoïdes associé à un traitement symptomatique des douleurs neuropathiques (Gabapentine). - Devant l’aggravation de la symptomatologie, un traitement par Rituximab
a été instauré en raison de 375 mg/m2 chaque semaine pendant 4 semaines. - L’évolution fut marqué par une nette amélioration avec un bon score ONLS (Overall Neuropathy Limitation Scale) et une réduction du taux d’IgM kappa de 12 %.
Figure 6 : Electrophorèse des protéines sériques montrant un pic monoclonal
Observation 2 :
- Il s’agit d’une patiente âgée de 57 ans, ayant comme antécédent : + Diabète type 2, bien équilibré, sous anti-diabétiques oraux, + Hypertension artérielle,
+ Opéré pour thyroïdectomie en 2012, sous Levothyrox. + Troubles obsessionnels compulsifs traités
+ Dégénérescence maculaire précoce avec baisse sévère de l’acuité visuelle (origine Héréditaire).
- Elle présentait depuis 2 ans une lourdeur avec des douleurs neuropathiques au niveau des membres inférieurs (MI).
- L’examen clinique a objectivé une marche ataxique, une paraparésie, Une hypoesthésie tactile et algique avec anesthésie vibratoire aux MI. Les réflexes ostéo- tendineux étaient abolis aux MI et présents aux MS.
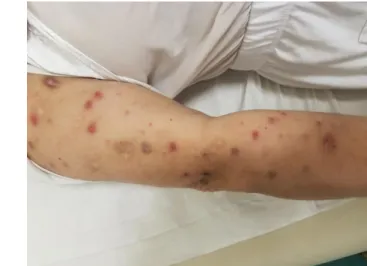
L’examen général a trouvé des lésions cutanées papuleuse diffuses récidivantes et ongles blancs (Figure7) avec un syndrome de Raynaud.
Figure 7 : ongles blancs à gauche et Lésions cutanées papuleuses diffuses nécrosées et récidivantes à droite
- L’examen ENMG a montré les résultats suivants (Tableau 6 et Tableau 7) :
Allongement des Latences distales motrices des nerfs péroniers et tibiaux avec diminution de leurs amplitudes motrices
Ralentissement des VCM au niveau des 4 membres
Allongement de latence de l’onde F des nerfs tibial et péronier droits et des nerfs médians et ulnaires
Diminution des amplitudes sensitives des potentiels sensitifs des nerfs médians et ulnaires.
Ralentissement des VCS au niveau des 4 membres.
- On conclut donc à une polyneuropathie démyélinisante sensitivomotrice. Tableau 6 : Résultats de la conduction motrice de l’EMG
Tableau 7 : Résultats de la conduction sensitive de l’EMG
- La ponction lombaire a objectivé une dissociation albumino-cytologique. Le bilan biologique, comportant l’ionogramme sanguin, bilan rénal, hépatique et thyroïdien, est revenu normal.
- L’mmunoélectrophorèse des protides a révélé une gammapathie monoclonale minime à type IgA lambda.
- Le bilan inflammatoire, comportant la CRP, la Vitesse de sédimentation et la Numération formule sanguine, était négatif. L’hémoglobine glyquée était également normale. Les sérologies virales HVB, HVC, HIV et syphilitique sont revenues négatives, ainsi que les anticorps anti-nucléaire.
- La biopsie des glandes salivaires était normale. Le scanner thoraco-abdomino-pelvien est revenu sans particularité.
- Le diagnostic de PIDC a été posé sur les arguments cliniques, électroneuromyographiques et la dissociation albumino-cytologique sur l’étude du LCR.
- La malade a reçu des cures des immunoglobulines (IgIV) avec corticoïdeset un traitement symptomatique à base de prégabaline. Une amélioration des symptômes a été remarquée chez le malade après avoir reçu la première cure des IgIV. Lors de sa 2ième cure, on notait une recrudescence des symptômes, principalement les douleurs neuropathiques. La malade a
Même présenté, lors de sa 3ième cure, une thrombose de l’aorte abdominale et des artères iliaques traitées chirurgicalement.
- Ensuite, elle a bénéficié d’un PET scan, qui a montré une hépatosplénomégalie et une 2ième immunoéléctrophorèse des protides qui a confirmé la gammapathie monoclonale à IgA lambda.
- Le POEMS syndrome a été suspecté devant l’existence des lésions cutanées papuleuse diffuses récidivantes et ongles blancs (Image 1) avec un syndrome de Raynaud et a été confirmé par un taux élevé de VEGF à 4918 pg/ml.
- La malade s’est nettement améliorée sous chimiothérapie (Revlimid + Dexaméthazone).
B) Discussion :
Observation 1 :
Notre première observation illustre un cas de neuropathie périphérique associée à une IgM anti-MAG, qui touche en particulier les hommes après 50 ans, comme chez notre patient [80].
Le tableau clinique initial observé chez notre malade est celui d’une polyneuropathie distale, symétrique, prédominant aux membres inférieurs, sensitive chronique, qui touche les petites et les grosses fibres sensitives, associée à un tremblement d’attitude des mains, ressemblant à une neuropathie à type de DADS. Notre cas était conforme à la description clinique classique des neuropathies à IgM anti-MAG qui se présentent principalement sous forme d’une neuropathie chronique symétrique à prédominance sensitive [29,30].
Le profil électroneuromyographique (ENMG) est celui d’une polyneuropathie démyélinisante avec des caractéristiques spécifiques similaires à celles observées chez notre malade [31] :
Prédominance distale des anomalies de la conduction motrice associée à un allongement des latences motrices distales de façon disproportionnée par rapport au ralentissement de la vitesse de conduction motrice (Tableau 4), Ces anomalies électriques de la conduction motrice sont discordantes avec l’atteinte clinique motrice [34].
Index de latence terminale (ILT) inférieur à 0,25 dans au moins deux nerfs (surtout au niveau des nerfs médian et ulnaire) [32,33]. Il permet de comparer la conduction motrice dans le segment distal par rapport à celle des segments proximaux pour différencier les neuropathies anti-MAG des PIDC.
Atteinte sensitive précoce prédominant aux MI [37].
On note également une absence de bloc de conduction, ce qui est comparable aux données de la littérature [32-34].
Sur le plan biologique, la mise en évidence de la gammapathie monoclonale repose sur l’électrophorèse des protéines sériques avec immunofixation,ainsi que le dosage quantitatif des Ac anti-MAG. Il s’agit d’une IgM monoclonale avec, dans la très grande majorité des cas, une chaîne légère kappa [31]. l’étude du liquide céphalorachidien montre le plus souvent une dissociation albuminocytologique avec absence de réaction cellulaire et hyperprotéinorachie modérée, comme le cas de notre malade [31]. L’IgM monoclonale anti-MAG peut être mise en évidence dans le liquide céphalo-rachidien (LCR), elle n’a pas été recherchée dans notre cas [81].
Sur le plan thérapeutique, notre malade a reçu, au début, un traitement à base de corticoïdes, qui s’est avéré inefficace, voir même une aggravation des douleurs neuropathiques et des troubles de l’équilibre. Certaines études ont démontrés que les corticostéroïdes étaient inefficaces en monothérapie, mais lorsqu'ils étaient administrés en association avec d'autres thérapies (comme le cyclophosphamide par exemple), ils produisaient une réponse chez environ la moitié des patients présentant un taux élevé d'IgM anti-MAG [82-83].
Plusieurs médicaments immunomodulateurs ou immunosuppresseurs (incluant entre autre les immunoglobulines intraveineuses, les plasmaphérèses et l’azathioprine) ont été testés chez les malades qui ont une neuropathie associée à IgM anti-MAG mais sans preuve fiable démontrée et les conclusions des données étaient insuffisantes pour recommander un protocole spécifique [88].
Le Rituximab est un anticorps monoclonal IgG chimérique homme/souris dirigé contre la protéine membranaire CD20 des lymphocytes B conduisant à une baisse des IgM et C’est grâce à ces propriétés que le rituximab a été proposé dans les neuropathies anti-MAG et il a été essayé chez notre malade. Il a été utilisé dans plusieurs études avec des résultats contradictoires. Il a été administré dans plusieurs essais à dose de 375 mg/m2 hebdomadaire pendant quatre semaines avec des résultats encourageants [84-85]. Un avantage
supplémentaire était observé lorsqu’une double dose de rituximab est utilisée [86]. Dans d’autres cas, une aggravation de la neuropathie ou aucune amélioration n'a été rapportée [87]. Chez notre malade, une amélioration spectaculaire a été remarquée après l’administration du rituximab.
Plusieurs facteurs pronostiques de la réponse au traitement par rituximab ont été identifiés dans diverses études : déficit moteur proximal, évolution subaiguë [89], un taux élevé d’anticorps anti-MAG (comme chez notre patient) [90] et un taux bas du B-cell Activating Factor (BAFF), acteur de l’homéostasie des cellules B, avant le traitement [91].
Observation 2 :
Notre 2ième observation illustre un cas de neuropathie périphérique qui a révélé un syndrome de POEMS.
Sur le plan clinique, la malade a présenté un tableau de polyneuropathie sensitivomotrice d’évolution chronique touchant les membres inférieurs. Des douleurs neuropathiques intenses ont prédominé ce tableau clinique, ce qui rejoint les données de la littérature, qui affirment que la douleur touche 75% des patients atteints de syndrome de POEMS [46, 47].
L’ENMG a confirmé le caractère démyélinisant de la neuropathie périphérique. Le diagnostic d’une PIDC a été évoqué au début, vu la présence des caractéristiques clinico-électriques évocateurs avec l’existence d’une dissociation albumino-cytologiques. Notre malade a reçu initialement des cures des immunoglobulines en intraveineuse et des corticoïdes, mais sans bénéfice prouvé, elle a même présenté une thrombose de l’aorte abdominale et des artères iliaques lors de sa 3ième cure des IgIV, ayant été traitées chirurgicalement. La thrombose artérielle, qui fait partie des effets indésirables des IgIV, a probablement été précipité par les troubles de la crase sanguine, décrits dans le syndrome de POEMS.
D’autres examens paracliniques ont été demandés, y compris un 2ième prélévement sanguin de l’éléctrophorèse des immunoglobulines avec
immunofixation qui a confirmé la présence d’une gammapathie monoclonale à IgA Lambda, un PET Scan, qui a objectivé la présence d’une hépato-splénomégalie et le dosage de VEGF, qui est revenue franchement positive. L’examen général joue également un rôle primordial pour l’orientation du diagnostic et chez notre malade, il y avait des lésions cutanées papuleuse diffuses récidivantes et ongles blancs avec un syndrome de Raynaud. On aurait pu évoquer le diagnostic au début devant les lésions dermatologiques qu’elle a présenté.
Notre malade remplissait donc tous les critères diagnostiques du Syndrome de POEMS (Tableau 1), elle répond à deux critères majeurs obligatoires, qui sont la présence des Igb monoclonales à type IgA lambda et la polyneuropathie périphérique, elle a aussi un critère majeur, qui est le VEGF élevé et 3 critères mineurs : l’hépato-splénomégalie, l’endocrinopathie (Diabète) et les anomalies cutanées. Ensuite, elle a reçu un traitement à base de chimiothérapie, débuté par l’hématologue, vu la présence des lésions systémiques avec une bonne évolution [65, 66].
CONCLUSION
Les neuropathies dysglobulinémiques présentent un spectre hétérogène des neuropathies périphériques, souvent avec certains phénotypes cliniques et électrophysiologiques caractéristiques et la détection de dysplasies sanguines distinctes. Elles peuvent parfois révéler une gammapathie monoclonale non connue auparavant, comme l’illustrent nos 2 observations ci-dessus.
L’approche diagnostique de ces neuropathies commence d’abord par examen clinique minutieux incluant aussi bien l’examen neurologique, que l’examen général des malades et l’importance de l’examen dermatologique qui peut être la clé du diagnostic. Ensuite, il faut déterminer le phénotype clinique et électrophyqiologique de ces neuropathies afin de guider le bilan et les explorations paracliniques.
Le lien de causalité entre la gammapathie monoclonale et la neuropathie périphérique reste toujours un défi à relever puisque leur prévalence est élevée dans la population générale.
Devant tout tableau de PIDC, l’existence de signes systémiques, du bilan biologique perturbé et de la mauvaise réponse au traitement doit faire suspecter le POEMS syndrome et doser le VGEF.
Le traitement de neuropathies dysglobulinémiques nécessite souvent une collaboration entre plusieurs spécialités, y compris l'hématologie, la neurologie, la radio-oncologie, la chirurgie, la thérapie physique, l’orthophonie et la rééducation.
Un soutien psychologique est souvent nécessaire pour les malades et surtout pour les hémopathies malignes.
RÉFÉRENCES BIBLIOGRAPHIQUES
[1] Schroeder, H. W., and L. Cavacini, 2010, Structure and function of immunoglobulins.: J Allergy Clin Immunol, v. 125, p. S41-52.
[2] Bengtén, E., M. Wilson, N. Miller, L. W. Clem, L. Pilström, and G. W. Warr, 2000, Immunoglobulin isotypes: structure, function, and genetics.: Curr Top Microbiol Immunol, v. 248, p. 189-219.
[3] Touzeau C, Moreau P. Gammapathies monoclonales de signification indéterminée. EMC - Hématologie 2012 ; 7(2) :1-6 [Article 13-014-F-10].
[4] Kyle R.A., Durie B.G., Rajkumar S.V., Landgren O., Blade J., Merlini G et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management Leukemia 2010; 24: 1121- 1127 [cross-ref].
[5] Rajkumar SV, Kyle RA. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc 2005; 80(10):1371Y1382.
[6] Rajkumar SV, Dimopoulos MA, Palumbo A, et al: International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 15 :e538-e548, 2014.
[7] Dimopoulos MA, Panayiotidis P, Moulopoulos LA, Sfikakis P, Dalakas M (2000) Waldenström’s macroglobulinemia: clinical features, complications, and management. J Clin Oncol 18:214–226.
[8] Owen RG, Treon SP, Al-Katib A, Fonseca R, Greipp PR, McMaster ML, et al. Clinicopathological definition of Waldenström’s macroglobulinaemia: consensus panel recommendations from the second international workshop on Waldenström’s macroglobulinaemia. Semin Oncol 2003; 30:110-5.
[9] Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway GD, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine (Baltimore). 1980; 59: 311-22.
[10] Dispenzieri A. et al. Diagnostic criteria for POEMS syndrome (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal gammopathy, Skin changes). . Am. J. Hematol. 89:214-23, 2014. {3] D’Souza A, Hayman SR. [11] Duston MA, Skinner M, Anderson J, Cohen AS (1989) Peripheral neuropathy as an early marker of AL amyloidosis. Arch Intern Med 149:358–360. [12] P. Chaïbi, L. Merlin, C. Thomas, F. Piette. Les gammapathies monoclonales de signification indéterminée. Ann. Med. Interne, 2002 ; 153(7) :459-466.
[13] Zandecki M, Genevieve F, Jego P, Grosbois B. Gammapathies monoclonales de signification indéterminée. Rev Med Interne 2000; 21(12):1060-74.
[14] Kyle R.A. Monoclonal gammopathy of undetermined significance and solitary plasmocytoma. Implications for progression to overt multiple myeloma. Hematol Oncol Clin N Am 1997; 11: 71-87 [crossref].
[15] Decaux O, Rodon P, Ruelland A, et al. Épidémiologie descriptive des gammapathies monoclonales. Comparaison de l’expérience d’un centre hospitalier général et d’un service de Médecine interne de centre hospitalier et universitaire. Rev Med Interne 2007 ; 28(10) :670- 676.
[ 16
] Kyle R.A. Therneau, T.M., Rajkumar, S.V., Lar-son, D.R., Plevak, M.F., Offord, J.R., Dispen-zieri, A., Katzmann, J.A. & Melton, L.J. 3rd (2006) Prevalence of monoclonal gammopathyof undetermined significance. New England Jour-nal of Medicine, 354, 1362–1369. (2006)
[17] Martyn, C.N. & Hughes, R.A. (1997) Epidemiologyof peripheral neuropathy. Journal of Neurology,Neurosurgery and Psychiatry, 62, 310–318
[18] Nobile-Orazio, E., Latov, N., Hays, A.P., Takatsu,M., Abrams, G.M., Sherman, W.H., Miller, J.R.,Messito, M.J., Saito, T., Tahmoush, A., Lovelace,R.A. & Rowland, L.P. (1984) Neuropathy andanti-MAG antibodies without detectable serumM-protein. Neurology, 34, 218–221.
[19] Yeung KB, Thomas PK, King RHM, Waddy H, Will RG, Hughes RAC, et al. The clinical spectrum of peripheral neuropathies associated with benign monoclonal IgM, IgG and IgA paraproteinaemia: Comparative clinical, immunological and nerve biopsy findings. Journal of Neurology. 1991 ; 238(7) :383-91.
[20] Gosselin S, Kyle RA, Dyck PJ. Neuropathy associated with monoclonal gammopathies of undetermined significance. Annals of Neurology. juillet 1991 ; 30(1) :54-61.
[21] Kissel, J.T. & Mendell, J.R. (1996) Neuropathiesassociated with monoclonal gammopathies.Neuromuscular Disorders, 6, 3–18.issel, J.T. & Mendell, J.R. (1996) Neuropathies associated with monoclonal gammopathies. Neuromuscular Disorders, 6, 3 –18.
[22] Kelly J. J, Kyle RA, O’Brien PC, Dyck PJ. Prevalence of monoclonal protein in peripheral neuropathy. Neurology. 1981.
[23] Braun PE, Frail DE, Latov N. Myelin-Associated Glycoprotein Is the Antigen for a Monoclonal IgM in Polyneuropathy. Journal of Neurochemistry. nov 1982 ; 39(5) :1261-5
[24] Adams D, Lozeron P, Theaudin M, Denier C, Fagniez O, Rerat K, et al. Varied patterns of inaugural light-chain (AL) amyloid polyneuropathy: a monocentric study of 24 patients. Amyloid. juin 2011 ; 18(sup1) :98-100.
[25] Nemni R, Corbo M, Fazio R, Quattrini A, Comi G, Canal N. Cryoglobulinaemic neuropathy: a clinical, morphological and immunocytochemical study of 8 cases. Brain. 1988: 111(3): 541-52.
[26] Diaz-Arrastia R, Younger DS, and Hair L. Neurolymphomatosis: a clinicopathologic syndrome reemerges. Neurology. 1992.
[27] Scarlato M, Previtali SC, Carpo M, Pareyson D, Briani C, Del Bo R, Nobile-Orazio E, Quattrini A, Comi GP (2005) Polyneuropathy in POEMS syndrome: Role of angiogenic factors in the pathogenesis. Brain 128(8):1911–1920.
[28] Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society - First Revision. Journal of the Peripheral Nervous System. mars 2010 ; 15(1) :1‑9.
[29] Steck AJ,Stalder AK, Renaud S. Anti-myelin-associated glycoprotein neuropathy. Curr Opin Neurol 2006; 19:458-63.
![Figure 1 : Structure de base des Ig [1]](https://thumb-eu.123doks.com/thumbv2/123doknet/15063290.699480/10.892.182.720.661.1072/figure-structure-base-des-ig.webp)
![Tableau 1: Critères diagnostiques de POEMS syndrome [10]](https://thumb-eu.123doks.com/thumbv2/123doknet/15063290.699480/19.892.113.779.151.558/tableau-critères-diagnostiques-de-poems-syndrome.webp)
![Figure 4 : Electrophorèse des protéines sériques montrant un pic monoclonal migrant dans la zone Gamma [58]](https://thumb-eu.123doks.com/thumbv2/123doknet/15063290.699480/33.892.263.657.790.1060/figure-electrophorèse-protéines-sériques-montrant-monoclonal-migrant-gamma.webp)
![Figure 5 : Algorithme de Stratégie de la gestion des neuropathies dysglobulinémiques selon la Mayo Clinic [79]](https://thumb-eu.123doks.com/thumbv2/123doknet/15063290.699480/38.1262.126.1137.130.723/figure-algorithme-stratégie-gestion-neuropathies-dysglobulinémiques-mayo-clinic.webp)