Versatile synthesis of chiral 6-oxoverdazyl radical
ligands
– new building blocks for multifunctional
molecule-based magnets
†
Atena B. Solea,
aTobie Wohlhauser,
aParisa Abbasi,
bYvan Mongbanziama,
aAurelien Crochet,
cKatharina M. Fromm,
cGhenadie Novitchi,
dCyrille Train,
dMelanie Pilkington*
band Olimpia Mamula
*
aA versatile synthetic methodology to access the first family of chiral verdazyl N,N’-chelate ligands is described and exemplified by N,N’-dimethyl-, N,N’-di-isopropyl- and N,N’-diphenyl oxoverd-azyls bearing two isomers of the pinene-pyridine functional group. Their physical properties were probed by X-band EPR spec-troscopy, cyclic voltammetry and DFT calculations. Preliminary reactivity studies show they can act as N,N’-chelate ligands affording a chiral 1 : 1 complex (3b) with CuCl2, which was
charac-terized by single-crystal X-ray diffraction. Variable temperature EPR studies on (3b) confirm the presence of antiferromagnetic interactions between the spins of the Cu(II) ion and the verdazyl
radical.
The unique properties associated with chiral molecules and their interaction with light inter alia has a rich history that today plays a central role in many areas of chemistry that include organometallic, coordination compounds, metal nano-particles and molecule-based materials.1The marriage of chir-ality with free radical chemistry has also found applications in diverse fields ranging from enantioselective catalysis2 to the development of chiral radicals as building blocks for chiral organic, or hybrid organic/inorganic magnets.3 In the latter case, crystallization of chiral molecules into non-centro-symmetric space groups can confer unique physical phenom-ena that include ferroelectric, piezoelectric, second harmonic generation (SHG) and/or non-linear optical (NLO) properties.4
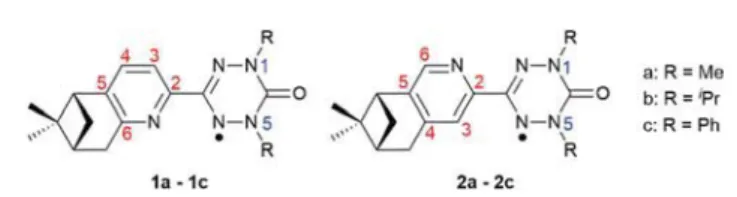
Furthermore, in select examples, when magnetic chirality co-exists with nuclear chirality, the spatial and temporal symmetry is simultaneously broken affording a range of magneto-optical properties that includes magneto-chiral dichroism (MChD).5 In the field of single molecule magnets (SMMs), a promising strategy to increase the blocking temperatures is to employ radical bridging ligands to couple the large magnetic an-isotropy of 4f ions.6 The introduction of chirality into such linkers could therefore not only facilitate strong magnetic exchange, but also afford novel multifunctional compounds where interplay between two or more physical properties may be achieved.7The development of versatile synthetic routes to chiral radicals is therefore an important milestone towards multifunctional SMMs. Although a range of different stable organic radicals are known,8 the majority of progress in this field has focussed on synthetic routes to chiral nitronyl nitrox-ides.9 Over the past decade, verdazyl (vd) radicals have been prepared and exploited as attractive ligands for the develop-ment of molecular magnets, coordinating to a range of both d-and f-block ions.10However, in contrast to the nitronyl nitrox-ides, methodologies to incorporate chirality into the vd unit have not been explored, with only one example reported in the literature to-date.11In the current paper we describe the versa-tile synthesis and characterization of two isomeric series of enantiopure N,N′-chelate vd radical ligands bearing (−)-pinene-pyridyl functionality, i.e. the (−)-5,6 series, 1a–1c and the (−)-4,5 series, 2a–2c, bearing different substituents at the 1,5 positions of the oxoverdazyl radical in order to modify the stereo-electronics, (Fig. 1). We also show proof-of-principle studies for future coordination chemistry through the
Fig. 1 Molecular structures of two families of chiral verdazyl radicals.
†Electronic supplementary information (ESI) available: Syntheses,
crystallo-graphic data, analytical/spectroscopic data, DFT studies. CCDC
1532176–1532178 and 1811236. For ESI and crystallographic data in CIF or other
electronic format see DOI: 10.1039/c8dt00840j
aDepartment of Chemistry, University of Applied Sciences Western Switzerland
(HES-SO), HEIA-FR, Pérolles 80, CH-1700 Fribourg, Switzerland. E-mail: olimpia.mamulasteiner@hefr.ch
bDepartment of Chemistry, Brock University, 1812 Sir Isaac Brock Way,
St Catharines, Ontario L2S3A1, Canada. E-mail: mpilkington@brocku.ca
cDepartment of Chemistry, University of Fribourg, Ch. du Musée 9,
CH-1700 Fribourg, Switzerland
dLaboratoire National des Champs Magnétiques Intenses, UPR CNRS 3228,
25, rue des Martyrs, B.P. 166 38042 Grenoble, France
http://doc.rero.ch
Published in "Dalton Transactions 47(14): 4785–4789, 2018"
which should be cited to refer to this work.
synthesis and structural characterization of the 1 : 1 complex 3b prepared from the isopropyl radical ligand 2b and CuCl2.
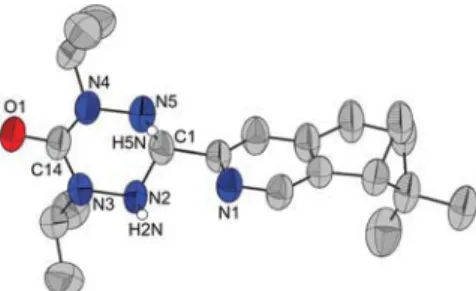
An important approach to the design of chiral radicals is to utilize a range of readily accessible precursors drawn from a chiral library. In this context we targeted the preparation of chiral aldehydes, which prove to be useful precursors for a range of stable radicals including both nitronyl nitroxides12 and verdazyls.8,13The chiral (−)-5,6- and (−)-4,5-aldehyde inter-mediatesP1 and P2 respectively (Scheme 1) were synthesized in three steps from (−)-myrtenal or (−)-alpha-pinene according to the procedure proposed by Bernhard et al.,14and their struc-tures confirmed by X-ray diffraction (see S2 of the ESI†). Subsequent condensation of these aldehydes with the appro-priate dialkyl- or diphenyl-carbonylhydrazide in methanol afforded the corresponding tetrazinanone intermediates T1 and T2 in 45–85% recovered yields (Scheme 1). Although synthetic approaches to diisopropyl- and diphenyl-carbonyl-hydrazines have been reported previously,15 we found that Boc protection of their hydrazine precursors followed by reac-tion with phosgene and subsequent deprotecreac-tion afforded the best yields. The solid state structure of 1,5-diisopropyl-3-(4,5-pinenepyridine) tetrazinanone, T2b was determined by X-ray diffraction as a representative member of this series (Fig. 2).
The compound T2b crystallizes in the chiral space group P212121, with one independent molecule in the asymmetric
unit. The four N atoms and the carbonyl C atom of the tetrazi-nanone heterocycle are close to co-planarity with the carbonyl atom C14 showing the largest displacement from the ring plane (0.174 Å), while the sp3 hybrid C atom (C1) adopts a tetrahedral geometry and is displaced out of the ring plane by 0.571 Å. The displacement of the two C atoms from the ring plane generates a“screw boat” conformation with a puckering parameter (θ = 68.7(7)°)16similar to achiral pyridine and pyra-zole substituted tetrazinanones.17,18Previous studies on tetra-zinanone derivatives place the C-substituent either equatorial17 or axial,17,18 with equatorial positions favoured for systems capable of undergoing intramolecular hydrogen bonding. In the case ofT2b, the pyridine substituent at C1 adopts an equa-torial position with respect to the tetrazinanone and is rotated almost perpendicular to the tetrazinanone ring (angle between the two planes: 82.55°), permitting the pyridyl N atom (N1) to form a bifurcated set of intramolecular N⋯H–N hydrogen bonds to N2 and N5 (N1⋯N2 at 2.820(8) Å and N1⋯H–N2 angle of 104(4)°; N1⋯N5 at 2.916(8) Å and N1⋯H–N5 angle of 109(4)°). Oxidation of these tetrazinanones with benzoquinone in toluene afforded the two series of 5,6- and 4,5-substituted vd radicals1a–1c and 2a–2c respectively, with final yields after
column chromatography in the range of 10–40%. In the case of1c, the radical could not be separated from benzoquinone and has been attributed to formation of a stable charge-trans-fer salt with hydroquinone reflected in a broad charge transcharge-trans-fer band19centred around 430 nm in the UV-Vis spectrum which is conspicuously absent from the other derivatives in the series (see S3.3 ESI†). Although these radicals failed to provide X-ray diffraction quality crystals under a range of solvent combi-nations, their molecular structures were confirmed by CHN, HRMS, UV-Vis and X-band EPR spectroscopy and their chiral-ity was confirmed by optical rotation studies. The EPR spectra of2a–2c (Fig. 3) illustrate the differing coupling patterns due to the R substituents along this series. Notably, the isomeric form of the pinene-pyridyl substituent does not affect the hyperfine coupling patterns.
The corresponding spectra for the 5,6-series 1a–1c are pro-vided in the S-3 of the ESI.† Although previous studies have established that the redox properties of vd radicals are sensi-tive to the nature of the bridging carbon (e.g., CvO vs. CH2) as
well as the R substituents on the two N atoms,20we were inter-ested to investigate if the substitution pattern of the chiral pinene heterocycle had any direct effect on the redox chem-istry of the radicals. For this we chose to carry out electro-chemical studies on the dimethyl radicals1a and 2b, as resentative examples from each series. As demonstrated pre-viously, vd radicals can be reduced to the 8-π electron-contain-ing anion and oxidized to the 6-π electron-containelectron-contain-ing cation.20
From our studies, radical 1a displays a quasi-reversible oxi-dation potential at +0.26 V and a quasi-reversible reduction potential at−1.5 V (vs. Fc/Fc+) (Fig. 4, left), and radical2b dis-plays the same quasi reversible oxidation at +0.26 V (vs. Fc/Fc+), but the reduction at−1.5 V is irreversible (Fig. 4, right).
In this context, both radicals are fairly good electron donors; their oxidation potentials fall at the lower end of the
Scheme 1 Synthetic route to verdazyl radicals 1 and 2.
Fig. 2 Crystal structure of intermediate tetrazinanone T2b with thermal ellipsoids at 50% probability. The H atoms bound to C atoms have been removed for clarity.
range for previously reported pyridyl oxoverdazyls which suggests that they are easier to oxidize, most likely since the electron withdrawing ability of their pyridine rings are compro-mised by the electron donating nature of their fused pinene heterocycles. The quasi reversibility in their oxidation poten-tials is also not unusual and has been observed for other dimethyl substituted vd radicals measured at low scan rates. The reduction potentials of both radicals occur at negative potentials and are on the lower end of the range (−1.14 to −1.33 V) reported previously for other oxoverdazyl radicals.20
The irreversibility observed in the reduction process of 2a is also not unusual and was reported previously for a related dimethyl pyridyl oxoverdazyl radical.19
DFT and TD-DFT calculations based on geometry optimized UB3LYP-6311G structures of2a and 2b reveal that in both com-pounds the unpaired electron density is primarily located on
the vd moiety (S-4, ESI†). The presence of the pyridyl ring clearly leads to a breaking of the symmetry and a slight asym-metry in the spin distribution within both molecules. Nevertheless, each radical can be considered as having two pairs of near-equivalent N atoms with each pair bearing ca. 39% and 19.5% spin density. This is consistent with the experimental EPR spectra which reveal coupling to two sets of two chemically distinct 14N nuclei (I = 1), with additional hyperfine coupling to the methyl-H atoms arising through hyper-conjugation effects (S-3, ESI†).13b
Previous studies have shown that the chirality of pyridine ligands bearing pinene substituents can be successfully trans-ferred to their d/f coordination complexes.21In order to probe the coordination chemistry of this series of radicals the reactiv-ity of2b with CuCl2in EtOH was examined, leading to the 1 : 1
complex (3b). This complex crystallizes in the non-centro-symmetric space group P21with two crystallographically
inde-pendent molecules in the asymmetric unit. This structure is very similar to the previously reported vd-CuCl2 complex
described by Hicks and co-workers.22 However, in that example a long Cu⋯N intermolecular contact (3.889 Å) linked two units together affording a centrosymmetric dimer. In the current case, aggregation appears inhibited by the steric bulk of the pinene-pyridyl moiety. In both molecules of 3b, the copper ion adopts an intermediate geometry (τ4 = 0.67
and 0.59 for Cu1 and Cu2 respectively), between square planar (τ4= 0) and tetrahedral (τ4= 1),23consistent with a Jahn–Teller
distortion away from idealized tetrahedral coordination (Fig. 5).
For an idealized square planar geometry, the π-magnetic orbital of the vd radical is mutually orthogonal to the Cu2+ dx2−y2magnetic orbital and thus is expected to lead to a
ferro-magnetic interaction. Conversely, under tetrahedral symmetry an antiferromagnetic interaction is expected due to the non-zero overlap of the two magnetic orbitals. The intermediate coordination geometry is therefore challenging to predict, but the emergence of non-zero overlap integrals typically leads to a rapid dominance of antiferromagnetic interaction. In the structurally related [CuCl2(vd)]2 system the coordination
geo-metry is pseudo-5-coordinate with CuCl2and CuN2ring planes
twisted by 47.8° and a strong antiferromagnetic exchange
Fig. 3 Room temperature solution EPR spectra of the (−)-4,5-series (top) 2a, R = CH3; (middle) 2b, R =iPr and (bottom) 2c, R = Ph in CH2Cl2.
Experimental spectra in black and simulations in red. Simulation para-meters are provided in S3.5 of the ESI.†
Fig. 4 Cyclic voltammetry measurements of a 1 mM solution of 1a (left) and 2a (right) in CH3CN. The redox event centered at 0 mV corresponds
to the ferrocene/ferrocenium redox couple (ferrocene added as an internal reference). Both CVs were performed in MeCN solution at a scan rate of 100 mV s−1with 0.1 M Bu4NBF4electrolyte.
Fig. 5 Crystal structure of one of the crystallographically independent molecules of 3b with the thermal ellipsoids at 50% probability. The H atoms have been removed for clarity.
coupling ( J = −204 cm−1) was observed, but with a strong exchange term to account for exchange between CuCl2(vd)
sub-units (zJ′ = −260 cm−1).22
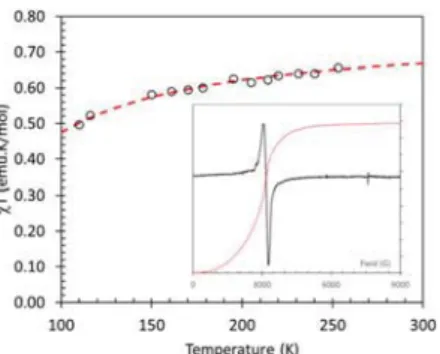
Variable temperature EPR studies on 3b were undertaken and the EPR signal intensity examined (Fig. 6) in the range 100–300 K as a function of temperature (the double integral of the first derivative EPR spectrum is proportional to the mag-netic susceptibility).24A fit to the Bleaney–Bowers equation25
afforded J/k = −60 K for this system.
In summary we have described the versatile synthesis of two chiral aldehydes from which we have prepared novel enantiopure verdazyl ligands bearing pinene-pyridyl substitu-ents in which the stereo-electronic effects at the 1,5-positions of the verdazyl radical can be controlled. Preliminary studies reveal that these ligands can coordinate d-block metals, which offers the potential for magnetic exchange coupling. This augurs well for the future development of high spin chiral mole-cules and further studies are underway to explore their coordi-nation chemistry with magnetically interesting 3d and/or 4f metal ions.
Con
flicts of interest
There are no conflicts to declare.
Acknowledgements
OM, AS, TW, YM acknowledge the undergraduate students who participated in this project during their second year lab-oratory courses or Bachelor work (S. Agrebi, M. Poretti, A. Spörri, F. Miserez, N. Beretta) and ChemTech for financial support. MP would like to thank NSERC DG and PA acknowl-edges the Ontario Trillium Scholarship program for financial support. AC and KMF thank the Fribourg Center for Nanomaterials FRIMAT for support. CT and GN acknowledge the ANR (ANR-13-BS08-0016-01 project).
Notes and references
1 (a) M. Dutartre, J. Bayardon and S. Juge, Chem. Soc. Rev., 2016,45, 5771; (b) Y. Wang, J. Xu, Y. Wang and H. Chen, Chem. Soc. Rev., 2013,42, 2930; (c) E. C. Constable, Chem. Soc. Rev., 2013,42, 1637.
2 (a) T. Hashimoto, Y. Kawamata and K. Maruoka, Nat. Chem., 2014,6, 702; (b) J. G. Stack, D. P. Curran, S. V. Geib, J. Rebek Jr. and P. Ballester, J. Am. Chem. Soc., 1992,114, 7007. 3 (a) J. R. Gallan-Mascaros, E. Coronado, P. A. Goddard,
J. Singleton, A. I. Coldea, J. D. Wallis, S. J. Coles and A. Alberola, J. Am. Chem. Soc., 2010, 132, 9271; (b) M. Minguet, D. B. Amabilino, I. Mata, E. Molins and J. Veciana, Synth. Met., 1999,103, 2253.
4 (a) C. Train, M. Gruselle and M. Verdaguer, Chem. Soc. Rev., 2011, 40, 3297; (b) M. Minguet, D. Luneau, C. Paulsen, E. Lhotel, A. Gorski, J. Waluk, D. B. Amabilino and J. Veciana, Polyhedron, 2003, 22, 2349; (c) P. Sarbadhikary, S. Shil, A. Panda and A. Misra, J. Org. Chem., 2016,81, 5623. 5 (a) G. L. J. A. Rikken and E. Raupach, Nature, 1997, 390,
493; (b) J. R. Galan-Mascaros, Nat. Phys., 2014, 11, 7; (c) R. Sessoli, M.-E. Boulon, A. Caneschi, M. Mannini, L. Poggini, F. Wilhelm and A. Rogalev, Nat. Phys., 2015,11, 69; (d) C. Train, R. Gheorghe, V. Krstic, L.-M. Chamoreau, N. S. Ovanesyan, G. L. J. A. Rikken, M. Gruselle and M. Verdaguer, Nat. Mater., 2008,7, 729.
6 (a) C. A. Gould, L. E. Darago, M. I. Gonzalez, S. Demir and J. R. Long, Angew. Chem., Int. Ed., 2017, 56, 10103; (b) S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546; (c) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011,3, 538.
7 H. Kumagai and K. Inoue, Angew. Chem., Int. Ed., 1999,38, 1601. 8 See for example. Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, J Wiley & Sons, 2010.
9 (a) S. Shimono, R. Tamura, N. Ikuma, T. Takimoto, N. Kawame, O. Tamada, N. Sakai, H. Matsuura and J. Yamauchi, J. Org. Chem., 2004,69, 475; (b) M. Minguet, D. B. Amabilino, K. Wurst and J. Vecciana, J. Solid State Chem., 2001,159, 440.
10 (a) B. D. Koivisto and R. G. Hicks, Coord. Chem. Rev., 2005, 249, 2612; (b) R. G. Hicks, B. D. Koivisto and M. T. Lemaire, Org. Lett., 2004,6, 1887; (c) M. Chahma, X. S. Wang, A. van der Est and M. Pilkington, J. Org. Chem., 2006, 71, 2750; (d) G. Novitchi, S. Shova, Y. Lan, W. Wernsdorfer and C. Train, Inorg. Chem., 2016, 55, 12122; (e) L. Norel, F. Pointillart, C. Train, L.-M. Chamoreau, K. Boubekeur, Y. Journaux, A. Brieger and D. J. R. Brook, Inorg. Chem., 2008,47, 2396. 11 T.-N. Le, H. Grewal, V. Changoco, V. Truong and
D. J. R. Brook, Tetrahedron, 2016,72, 6368.
12 (a) E. F. Ullman, J. H. Osiecki, D. G. B. Boocock and R. Darcy, J. Am. Chem. Soc., 1972,94, 7049; (b) C. Stroh, M. Mayor and C. von Hanisch, Tetrahedron Lett., 2004,45, 9623.
13 (a) F. A. Neugebauer, H. Fischer and R. Siegel, Chem. Ber., 1988, 121, 515; (b) E. C. Pare, D. J. R. Brook, A. Brieger, M. Badik and M. Shinke, Org. Biomol. Chem., 2005,3, 4258.
Fig. 6 Temperature dependence ofχT for 2b (circles) determined from EPR spectroscopy (scaled to that expected for exchange coupling between two S = 1/2 ions) with fit to the Bleaney–Bowers model (dashed line); (inset) Solid state EPR spectra of 2b at 150 K with double integral of the EPR spectrum.
14 A. L. Sauers, D. M. Ho and S. Bernhard, J. Org. Chem., 2004, 69, 8910.
15 (a) Y. Masuda, M. Kuratsu, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, Polyhedron, 2009, 28, 1950; (b) D. Matuschek, S. Eusterwiemann, L. Stegemann, C. Doerenkamp, B. Wibbeling, C. G. Daniliuc, N. L. Doltsinis, C. A. Strassert, H. Eckert and A. Studer, Chem. Sci., 2015,6, 4712. 16 J. C. A. Boeyens, J. Cryst. Mol. Struct., 1978,8, 317.
17 O. Oms, L. Norel, L.-M. Chamoreau, H. Rousselière and C. Train, New J. Chem., 2009,33, 1663.
18 M. J. Plater, J. P. Sinclair, S. Kemp, T. Gelbrich, M. B. Hursthouse and C. J. Gómez-García, J. Chem. Res., 2006,8, 515.
19 C. L. Barr, P. A. Chase, R. G. Hicks and M. T. Lemaire, J. Org. Chem., 1999,64, 8893.
20 J. B. Gilroy, S. D. J. McKinnon, B. D. Kovisto and R. G. Hicks, Org. Lett., 2007,9, 4837.
21 (a) M. Lama, O. Mamula, G. S. Kottas, F. Rizzo, L. De Cola, A. Nakamura, R. Kuroda and H. Stoeckli-Evans, Chem. – Eur. J., 2007,13, 7358; (b) O. Mamula and A. von Zelewsky, Coord. Chem. Rev., 2003,242, 87.
22 J. B. Gilroy, B. D. Koivisto, R. McDonald, M. J. Ferguson and R. G. Hicks, J. Mater. Chem., 2006,16, 2618.
23 L. Yang, D.R. Powell and R.P. Houser, Dalton Trans., 2007, 955.
24 P. E. Richardson and R. W. Kreilick, J. Magn. Reson., 1978, 29, 285.
25 B. Bleany and K.D. Bowers, Proc. R. Soc. London, Ser. A, 1952,214, 451.