Bicarbonate Is Not a General Acid in
Au-Catalyzed CO₂ Electroreduction
The MIT Faculty has made this article openly available.
Please share
how this access benefits you. Your story matters.
Citation
Wuttig, Anna et al. “Bicarbonate Is Not a General Acid in
Au-Catalyzed CO₂ Electroreduction.” Journal of the American Chemical
Society 139, 47 (November 2017): 17109–17113 © 2017 American
Chemical Society
As Published
http://dx.doi.org/10.1021/jacs.7b08345
Publisher
American Chemical Society (ACS)
Version
Author's final manuscript
Citable link
http://hdl.handle.net/1721.1/118297
Terms of Use
Article is made available in accordance with the publisher's
policy and may be subject to US copyright law. Please refer to the
publisher's site for terms of use.
Bicarbonate is Not a General Acid in Au-Catalyzed CO
2Electroreduc-tion
Anna Wuttig, Youngmin Yoon, Jaeyune Ryu, Yogesh Surendranath
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA, 02139, USA Supporting Information Placeholder
ABSTRACT: We show that bicarbonate is neither a general acid nor a reaction partner in the rate-limiting step of CO2 reduction ca-talysis mediated by planar polycrystalline Au surfaces. We formu-late microkinetic models and propose diagnostic criteria to distin-guish the role of bicarbonate. Comparing these models with the ob-served zero-order dependence in bicarbonate and simulated inter-facial concentration gradients, we conclude that bicarbonate is not a general acid co-catalyst. Instead, it acts as a viable proton donor past the rate-limiting step and a sluggish buffer that maintains the bulk but not local pH in CO2-saturated aqueous electrolytes.
The electroreduction of CO2 would enable the storage of renew-able sources of electricity in energy-dense carbonaceous fuels.1,2 CO2 reduction reactions (CO2RRs) require multiple proton-coupled electron transfer (PCET) steps, and thus control over fuel-for-mation rates and selectivities necessitates an understanding of the PCET pathways3 and the identification of the proton donor(s). CO2RR is most practically conducted in aqueous electrolytes, where HCO3– exists by necessity because it is the product of CO2 hydration.4–7 There is a consensus that HCO
3–(and CO32–) is not directly reduced at the electrode surface,8,9 leaving open the ques-tion of whether bicarbonate acts as: (1) a general acid co-catalyst that directly participates in the rate-limiting step (RLS); and/or (2) simply a participant in electrolyte acid-base equilibria. Herein, we address this question in the context of the two-electron, two-proton reduction of CO2 to CO catalyzed by polycrystalline Au surfaces.
The equilibrium values and reaction rates that govern (2) are well-established. CO2 hydration to form bicarbonate presents an operative pH restriction for catalysis– only intermediate pH values are viable for CO2RR in aqueous electrolytes due to the buffering point established by the CO2-water-bicarbonate equilibrium,4 Fig.
1. Stopped-flow studies of CO2 hydration and bicarbonate dehydra-tion5 combined with femtosecond time-resolved infrared studies of carbonic acid generation6 demonstrate that while the rate of CO
2 hydration (Fig. 1, k2 = 2.9×10–2 s–1) is slow, all other equilibria,
including carbonic acid dissociation (Fig. 1, k3 ≈ 3×106 s–1), car-bonic acid formation (Fig. 1, k–3 ≈ 1×1010 M–1s–1), and carbonic acid dehydration (Fig. 1, k–2 = 1.8×10 s–1), are relatively fast. These
rate constants suggest that CO2 and bicarbonate equilibrate to buffer the pH in the bulk electrolyte. In addition, provided that the rate of CO2RR catalysis does not outpace that of carbonic acid for-mation (k–3[H+][HCO3–]) and dehydration (k–2[H2CO3]), CO2 can be replenished within the reaction diffusion layer proximal to the electrode surface via equilibrium proton transfer (PT) from buffer constituents10 (Fig. 1, k
–3 and k–2). Indeed, a recent report using
in-frared and mass spectroscopies9 support CO
2 replenishment via these equilibria on the timescale faster than gas-liquid CO2 mixing.
The answer to question (1), whether bicarbonate is an explicit reaction partner in the RLS of catalysis, remains the subject of de-bate. Computational11–14 and experimental15 studies invoke mech-anisms for CO production that proceed via concerted proton elec-tron transfer (CPET) to CO2 (Fig. 1, k7) to form surface-bound COOH, but do not specify the identity of the proton donor, imply-ing that H3O+ and/or H2O act as general acids in the mechanistic sequence. In contrast, bicarbonate has been invoked as a general acid in mediating CO formation on oxide-derived polycrystalline Au electrodes.16 Conversely, for planar polycrystalline Au elec-trodes, electrokinetic studies17,18 point to rate-limiting electron transfer (ET) and adsorption of CO2 (Fig. 1, k6) followed by rapid PT, suggesting that CO production is subject to neither general nor specific acid co-catalysis. Further complicating the picture, bicar-bonate has been recently proposed to not serve as a proton donor, but instead, as a reactant in a new equilibrium9 (Fig. 1, k
5 and k–5) to form an anionic adduct of water, CO2 and bicarbonate, denoted as I–. The adductis proposed9 to be directly reduced at the electrode surface to form an adsorbed COOH intermediate (Fig. 1, k8). Ad-ditionally, studies of the role of bicarbonate are sometimes con-ducted in the presence of additional buffering ions,9 which them-selves can promote or inhibit the reaction. The wide-ranging per-spectives on the role of bicarbonate in fuel-forming catalysis moti-vate further mechanistic investigations.
Herein, we formulate diagnostic criteria for a series of possible mechanistic sequences that explicitly involve bicarbonate in or pre-ceding the RLS of CO2RR to form CO. We use electrokinetic data and stochastic reaction modeling to parse these competing mecha-nistic models and arrive at several viable possibilities.
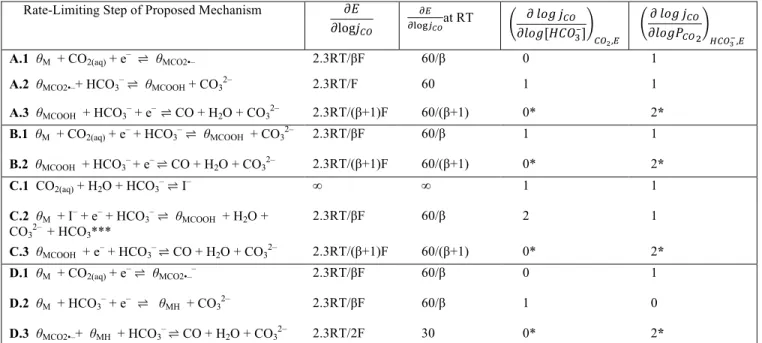
In order to distinguish between various mechanistic pathways for CO production, we derived the expected current-potential (Tafel) behavior, CO2 reaction order, and HCO3–reaction order for all previously proposed reaction sequences (Table 1). These theo-retical Tafel slopes and reaction orders provide diagnostic criteria19 that can be compared with experimental data to down-select among viable mechanistic models. In order to thoroughly examine the pos-sible role of HCO3–, all mechanistic sequences deliberately invoke this anion as the proton donor in all PT and CPET steps. Analogous
Figure 1: Possible pathways for involvement of bicarbonate anion -for CO2 electroreduction on an electrode surface up to the first elec-tron-transfer (ET) or concerted proton electron transfer (CPET) step. A recently proposed new equilibrium (reference 9), which forms an anionic adduct of water, CO2, and bicarbonate (I–), is also included.
tables that invoke water and hydronium as proton donors in PT and CPET steps are shown in Table S1 and S2, respectively. Pathway A (Tables 1, S1-S2, elementary steps A.1 – A.3) invokes stepwise ET/PT activation of CO2 to generate an adsorbed COOH16–18 spe-cies that is further reduced in an CPET step to generate the CO product. Pathway B (Tables 1, S1-S2, steps B.1 – B.2) invokes a sequence similar to A, but with substitution of the initial ET/PT activation of CO2 (steps A.1 and A.2) with a CPET step11–15,20 (step B.1). Pathway C (Tables 1, S1-S2, steps C.1 – C.3) is similar to pathway B, but incorporates formation of the proposed9 anionic ad-duct I– (step C.1) and its direct reduction at the surface (step C.2). Pathway D (Tables 1, S1-S2, steps D.1 – D.3) summarizes a dis-tinct pathway in which an adsorbed H reacts with an adsorbed CO2 species21 to generate CO.
For each sequence in Tables 1, S1, and S2 the expected Tafel slope and reaction orders were derived assuming that the elemen-tary reaction step in question was the rate limiting step (RLS) with all other steps in quasi-equilibrium (derivations are in the SI). The transfer coefficient, β, is included for sequences involving an ET or CPET RLS. Importantly, this analysis assumes that the reaction is occurring under activation control: the current is not limited by the mass transfer of reactants to the surface. Additionally, for se-quences that involve pre-equilibrium PT prior to a RLS involving
HCO3– (A.3, B.2, C.3, D.3), the reaction will display an inverse first order dependence in pH, leading to an apparent bicarbonate order of 0 because pH and [HCO3–] are directly correlated under conditions of CO2-saturation. In addition, we assume that all ad-sorption reactions can be described by a Langmuirian isotherm and that the surface coverage of kinetically competent adsorbates is low – i.e. that vacant metal active sites (θM) comprise the catalyst rest-ing state.22 This assumption of low coverage is required in order to explain the widely observed first order dependence in the partial pressure of CO2 (PCO2)8,9,16–18. Furthermore, we assume that the re-action is not limited by gas-liquid mixing (Fig. 1, k1/k–1), i.e. the reaction occurs under CO2-saturation conditions. Finally, we as-sume that CO desorption is not rate-limiting and exists in major equilibrium, in line with the zero order dependence for CO evolu-tion observed as a funcevolu-tion of the partial pressure of CO (PCO).18
In examining Table 1, we observe that assessing the role of bi-carbonate prior to or in the RLS of CO2RR to CO requires experi-mental identification of its reaction order coupled with the Tafel slope. Specifically, the electrochemical reaction order (ρ) in bicar-bonate is defined by the partial derivative,
𝜌 = # $%& '()
#$%&[+,-.–] ,-1,3,
which characterizes the dependence of the CO production rate or partial current density (jCO) on the concentration of bicarbonate ([HCO3–]) in the electrolyte. For an irreversible electrochemical re-action, the reaction order is measured at a constant potential relative to a pH-independent reference electrode (E).22–24
We note that the reaction order can also be measured at constant overpotential (η), defined as the difference in E and the reversible potential (Erev). However, CO evolution on polycrystalline Au foils occurs far from Erev, and, therefore, the overall reaction is irreversi-ble under all conditions sampled. Thus, the rate expressions for the reaction can be most straightforwardly derived with respect to E, not η (see SI). Under these irreversible conditions, reaction orders measured at constant η are convoluted by the Tafel slope because the exchange-current density (j0) in the Bulter-Volmer equation is dependent not only on the concentration of species but also on the transfer coefficient for the overall reaction and Erev.22 Furthermore, in most experiments, Erev at the electrode surface is subject to change as concentration gradients in reactants and products de-velop, see below, and these experimental variables can further con-volute order data collected at constant η. We believe that this dis-tinction between order data collected at constant E16,18 and η9,20 has been the source of inconsistencies in the literature.
Data collected under irreversible reaction conditions at constant
E reveal that polycrystalline Au electrodes (see SI and Fig. S1 for
electrochemically active surface area measurements) display zero order dependence in bicarbonate for CO production, consistent with previous studies.18 The j
CO was measured at applied potentials of –0.80 V, –0.90 V, and –1.00 V vs SHE (all potentials are refer-enced to the pH-independent Standard Hydrogen Electrode) span-ning bicarbonate concentration values from 30 to 300 mM, Fig. 2a. Rate-Limiting Step of Proposed Mechanism 𝜕𝐸
𝜕log𝑗 ,-#3 #$%&'()at RT 𝜕 𝑙𝑜𝑔 𝑗 ,-𝜕𝑙𝑜𝑔[𝐻𝐶𝑂@–] ,-A,3 𝜕 𝑙𝑜𝑔 𝑗 ,-𝜕𝑙𝑜𝑔𝑃,-1 +,-.C,3
A.1 θM + CO2(aq) + e– ⇌ θMCO2•– 2.3RT/βF 60/β 0 1
A.2 θMCO2•–+ HCO3–⇌ θMCOOH + CO32– 2.3RT/F 60 1 1
A.3 θMCOOH + HCO3– + e– ⇌ CO + H2O + CO32– 2.3RT/(β+1)F 60/(β+1) 0* 2*
B.1 θM + CO2(aq) + e– + HCO3–⇌ θMCOOH + CO32– 2.3RT/βF 60/β 1 1
B.2 θMCOOH + HCO3– + e– ⇌ CO + H2O + CO32– 2.3RT/(β+1)F 60/(β+1) 0* 2* C.1 CO2(aq) + H2O + HCO3–⇌ I– ∞ ∞ 1 1 C.2 θM + I– + e– + HCO3–⇌ θMCOOH + H2O + CO32– + HCO3*** 2.3RT/βF 60/β 2 1 C.3 θMCOOH + e– + HCO3– ⇌ CO + H2O + CO32– 2.3RT/(β+1)F 60/(β+1) 0* 2* D.1 θM + CO2(aq) + e–⇌ θMCO2•–– 2.3RT/βF 60/β 0 1 D.2 θM + HCO3– + e– ⇌ θMH + CO32– 2.3RT/βF 60/β 1 0 D.3 θMCO2•–+ θMH + HCO3– ⇌ CO + H2O + CO32– 2.3RT/2F 30 0* 2*
Table 1. Diagnostic criteria of proposed paths for CO production from CO2 with HCO3– as a proton donor. Rate-limiting step (RLS) of the proposed mechanism (column 1), expected current-potential behavior (column 2), expected current-potential behavior calculated at room temperature (column 3), and expected HCO3– (column 4) and CO2 reaction order (column 5). *PCO2, [HCO3–], and pH are interre-lated, so these reaction orders are calculated in the limit of extremely rapid buffer equilibration and/or transport to the catalyst surface (see SI for details). Observed orders for these sequences are expected to be particularly sensitive to the transport characteristics of the cell and electrode. ***We note that the proton donor invoked in reference 9 is H2O not HCO3–. See Table S1 for derivations with H2O.
The [HCO3–] was varied at fixed ionic strength and constant so-dium concentration by addition of soso-dium perchlorate, and the elec-trolytes were purified25 until voltammetric features associated with the oxidative stripping of Zn impurities were no longer observed following catalysis (Fig. S2). The use of Pt counter electrodes has been hypothesized9 to alter the Au working electrode surface struc-ture over time via deposition of leached Pt2+, leading to the accu-mulation of irreversibly bonded CO species on the Pt/Au surface as a result of CO2RR. CO stripping analyses (see SI text and Figs.
S3-S5) suggest that long-lived irreversibly-bound CO features persist
on Au surfaces when employing Au counter electrodes, indicating that the presence of a Pt counter is insufficient to account for their existence. Nevertheless, studies discussed below use a Au counter electrode to eliminate the possibility of adventitious Pt deposition. Data shown in Fig. 2a show that over the concentration range probed, jCO is zero order in bicarbonate within error. Data were col-lected at E values where the reaction is known to exist under acti-vation-control due to a lack of rotation-rate dependence on the measured current.18 We note that the pH changes from 6.2 to 7.2 as the bicarbonate concentration is altered from 30 to 300 mM at con-stant 1 atm CO2 partial pressure, however, jCO is independent of pH, see below, suggesting that the zero-order dependence observed is not due to equal and opposite bicarbonate and pH dependences18. In contrast to what we observe in Fig. 2a, previous studies have reported a 0.90 order in bicarbonate using phosphate (Pi) as a co-electrolyte.9 In this study, the sum of the P
i and bicarbonate con-centrations were fixed, meaning that ρ was extracted while simul-taneously lowering the Pi concentration. Therefore, we postulate that this discrepancy may arise from an inhibitory role of Pi, the
attenuation of which would lead to the observed forward order in bicarbonate. To probe the dependence on Pi explicitly, we investi-gated the difference in jCO between CO2RR conducted in CO2 -saturated 50 mM NaPi ([HCO3–] = 45 mM) and CO2-saturated 45 mM NaHCO3 with 50 mM NaClO4 at applied potentials of –0.80 V to –1.00 V, Fig. 2b. The data shown in Fig. 2b highlights that Pi is a potent inhibitor of jCO on polycrystalline Au surfaces, and there-fore convolutes reaction order studies conducted in this medium. Thus, we take the zero order dependence on bicarbonate measured in absence of Pi as the true reaction order.
The diagnostic criteria in Table 1 can only be compared with experimental reaction order data (Fig. 2a) if variations in the bulk concentrations of the relevant species correlate with changes in their concentration near the electrode surface. Steady-state mathe-matical modeling26–30 and experimental observations28,31–36 suggest a rise in the pH local to the electrode surface, which may also trans-late into a variation in bicarbonate concentration near the surface. Indeed, numerical models26,27 demonstrate that the necessary con-sumption of protons local to the catalyst surface results in a spatial and temporal concentration gradient in bicarbonate, however, these studies neglected to examine its effects on the rate of catalysts. To determine if the formation of a bicarbonate concentration gradient significantly convolutes the measured reaction order in bicarbonate we employed a stochastic method for kinetic simulations37–39 (com-putational details and input parameters in the SI and Table S3). We modeled the differences in reaction rate between a bicarbonate-me-diated CPET (B.1. in Table 1) and bicarbonate-independent ET (A.1. in Table 1) mechanism as a function of the bulk [HCO3–]. This represents an advance relative to prior models that assume ET26 or do not explicitly delineate a RLS27–30. This model also in-corporates the following key elements: (1) designation of bicar-bonate as the explicit proton donor in contrast to prior models that use H2O;26–30 and (2) inclusion of the slow hydration kinetics of CO2 (k2 and k–2 in Fig. 1) in contrast to prior models26–30 that as-sume fast equilibration of CO2 and bicarbonate. This model as-sumes a boundary layer thickness of 1 µm. The model excludes any convective or migratory components, and thus, the calculated gra-dients are expected to be greater than those existing at a planar elec-trode in a stirred electrolyte or on a rotating disk elecelec-trode18 and significantly smaller than those existing within a higher surface area, nanostructured, or highly porous electrode31,35. On highly nano/meso structured electrodes, the current may no longer be sub-ject to purely activation control across the entire surface, impeding the accurate determination of intrinsic reaction orders and poten-tially explaining discrepancies16,40 in the literature.
Using this model, we probed the expected bicarbonate concen-tration gradients for CO2RR proceeding via rate-limiting ET (A.1. in Table 1) or bicarbonate-mediated CPET (B.1. in Table 1). Fig.
3 displays the calculated spacio-temporal bicarbonate
concentra-tion gradient for CO2RR occurring in 0.1 M NaHCO3 via CPET (a) or ET (b) pathways. Importantly, we observe maximum HCO3– de-pletion of ~2 mM within 1 µm of the electrode surface for both the ET and CPET pathways. In 1.0 M NaHCO3 (Fig. S6), we find the same ~2 mM depletion for the ET pathway, but a ~20 mM deple-tion for the CPET case, reflective of higher jCO (see below). These trends indicate that the percent HCO3– depletion remains constant at ~2% for CPET, whereas the absolute depletion, ~2 mM, remains constant for ET. Thus, irrespective of the mechanism, local deple-tion effects negligibly impact the reacdeple-tion order data collected be-tween 30 and 300 mM, Fig. 2a, and provide additional confidence in assigning a ρ value of 0 in bicarbonate for Au-catalyzed CO2RR.
While [HCO3–] is changing minimally at the electrode surface, the consumption of protons in the overall reaction, combined with the sluggish CO2 hydration kinetics, leads to a substantial buildup in CO32– for CPET and ET pathways (Fig. S7). Using the Hender-son-Hasselbalch equation, we calculate that this buildup in [CO32– ] serves to raise the pH of the medium near the electrode surface to
Figure 2: Electrokinetic data for CO evolution catalysis.
Bicar-bonate concentration dependence (a) of CO partial current density at –0.80 V (black), –0.90 V (red), and –1.00 V (blue) vs SHE rec-orded in CO2-saturated bicarbonate electrolyte at constant 0.3 M ionic strength. Comparison of CO2RR conducted (b) 45 mM bicar-bonate with (black) and without (red) 50 mM sodium phosphate.
Figure 3: Simulated bicarbonate concentrations gradients (a and b)
as a function of the distance away from the electrode surface (x) and CO evolution current densities (c and d) for a reaction sequence in-volving rate-limiting bicarbonate-mediated CPET (a and c) or ET (b and d) in 0.1 M (a, b, or black) or 1 M (red) bicarbonate buffer.
~8.7 and 8.1-8.6 for 0.1 and 1.0 M bicarbonate electrolytes, respec-tively. Thus, although a higher [HCO3–] leads to an elevated bulk pH at constant PCO2, the local pH trends in the opposite direction due to the higher buffering capacity regardless of the mechanism of catalysis. Thus, the zero order [HCO3–] dependence observed in
Fig. 2a also implies a zero order dependence in pH for CO2RR. The stochastic model also provides a direct estimate of jCO for both CPET and ET pathways. Modeling of the bicarbonate-medi-ated CPET sequence shows an increase in initial jCO from ~1.3 to ~13 mA cm–2 as the bulk concentration of bicarbonate increases from 0.1 to 1.0 M. The ~30% decay in current density for the CPET sequence in 1.0 M bicarbonate is attributed to ~30% depletion CO2 at the electrode surface at higher jCO (Fig. S8). In contrast, the ET sequence shows no dependence on bicarbonate concentration, in line with the experimental data (Fig. 2a). Together, the simulations highlight how the rate of CO production catalysis is insensitive to changes in the bicarbonate concentration – bulk or interfacial – if the RLS of the catalysis only involves ET.
By comparing the experimental and simulated data with Tables
1, S1, and S2, we are able to elucidate the role of bicarbonate during
Au-catalyzed CO2RR. We measure a ρ value of 0 in bicarbonate and also infer a ρ value of 0 in pH. These results, combined with the first order dependence in CO28,9,16–18 and the absence of a measureable H/D KIE,18 argue against mechanisms involving hy-dronium, water, or bicarbonate as a proton donor in the RLS
(Ta-bles 1, S1-S2). This analysis leaves step A.1/D.1, electrosorption
of CO2, as the most viable RLS. Literature reports8,16,17 and our pre-vious studies18 suggest a Tafel slope of >120 mV/dec over the po-tential range examined here, lending further support to mechanisms A and D as the most probable, with step A.1/D.1 as rate-limiting with a β ~ 0.5. Sequences A and D only differ by the pathway for addition of H equivalents to adsorbed CO2 beyond the RLS and are therefore kinetically indistinguishable. Together, the data suggest that bicarbonate is not explicitly involved in the RLS of CO evolu-tion on polycrystalline Au surfaces, and instead, acts as a viable proton donor past the RLS and a sluggish buffer that maintains the bulk but not local pH. While the explicit mechanistic role of bicar-bonate for other catalyst surfaces remains an open question, this insight helps explain why inhibited diffusion in mesoporous elec-trodes, which would further deplete bicarbonate and raise pH, does not lead to a significant suppression in the rate of CO produc-tion.31,35 The inability of bicarbonate to act as general acid moti-vates the development of designer electrolytes capable of promot-ing CO2 activation at electrode surfaces via low-barrier CPET path-ways.
ASSOCIATED CONTENT
Supporting Information
Experimental and computational details, derivations, cyclic volt-ammograms, additional simulation results. The Supporting Infor-mation is available free of charge on the ACS Publications website.
AUTHOR INFORMATION
Corresponding Author
ACKNOWLEDGMENT
We thank Dr. Sahr Khan, Dr. William Hinsberg, Prof. Masatoshi Osawa, Dr. Momo Yaguchi, and Travis Marshall-Roth for helpful discussions. This research was supported by the Air Force Office of Scientific Research under AFOSR Award No. FA9550-15-1-0135.
REFERENCES
(1) Kondratenko, E. V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G. O.; Pérez-Ramírez, J. Energy Environ. Sci. 2013, 6, 3112.
(2) Benson, E. E.; Kubiak, C. P.; Sathrum, A. J.; Smieja, J. M. Chem.
Soc. Rev. 2009, 38, 89.
(3) Koper, M. T. M. Chem. Sci. 2013, 4, 2710.
(4) Hori, Y. In Modern Aspects of Electrochemistry; Vayenas, C., White, R., Gamboa-Aldeco, M., Eds.; Springer: New York, 2008; Vol. 42, pp 89–189.
(5) Pocker, Y.; Bjorkquist, D. W. J. Am. Chem. Soc. 1977, 99, 6537. (6) Adamczyk, K.; Prémont-Schwarz, M.; Pines, D.; Pines, E.;
Nibbering, E. T. J. Science 2009, 326, 1690.
(7) Palmer, D. A.; Van Eldik, R. Chem. Rev. 1983, 83, 651. (8) Hori, Y.; Murata, A.; Kikuchi, K.; Suzuki, S. J. Chem. Soc. Chem.
Commun. 1987, 728.
(9) Dunwell, M.; Lu, Q.; Heyes, J. M.; Rosen, J.; Chen, J. G.; Yan, Y.; Jiao, F.; Xu, B. J. Am. Chem. Soc. 2017, 139, 3774. (10) Hori, Y.; Suzuki, S. J. Electrochem. Soc. 1983, 130, 2387. (11) Zhu, W.; Michalsky, R.; Metin, O.; Lv, H.; Guo, S.; Wright, C.
J.; Sun, X.; Peterson, A. A.; Sun, S. J. Am. Chem. Soc. 2013, 135, 16833.
(12) Zhu, W.; Zhang, Y.-J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A. A.; Sun, S. J. Am. Chem. Soc. 2014, 136, 16132. (13) Peterson, A. A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.;
Norskov, J. K. Energy Environ. Sci. 2010, 3, 1311.
(14) Hansen, H. A.; Varley, J. B.; Peterson, A. A.; Nørskov, J. K. J.
Phys. Chem. Lett. 2013, 4, 388.
(15) Hatsukade, T.; Kuhl, K. P.; Cave, E. R.; Abram, D. N.; Jaramillo, T. F. Phys. Chem. Chem. Phys. 2014, 16, 13814.
(16) Chen, Y.; Li, C. W.; Kanan, M. W. J. Am. Chem. Soc. 2012, 134, 19969.
(17) Noda, H.; Ikeda, S.; Yamamoto, A.; Einaga, H.; Ito, K. Bull.
Chem. Soc. Jpn. 1995, 68, 1889.
(18) Wuttig, A.; Yaguchi, M.; Motobayashi, K.; Osawa, M.; Surendranath, Y. Proc. Natl. Acad. Sci. 2016, 113, E4585. (19) Bockris, J. O.; Otagawa, T. J. Phys. Chem. 1983, 87, 2960. (20) Rosen, J.; Hutchings, G. S.; Lu, Q.; Rivera, S.; Zhou, Y.; Vlachos,
D. G.; Jiao, F. ACS Catal. 2015, 5, 4293.
(21) Schouten, K. J. P.; Kwon, Y.; van der Ham, C. J. M.; Qin, Z.; Koper, M. T. M. Chem. Sci. 2011, 2, 1902.
(22) Gileadi, E. Physical Electrochemistry, Fundamentals, Techniques and Applications; Wiley-VCH: Weinheim, 2011; pp
77–92 and 151–164.
(23) Conway, B. E.; Salomon, M. Electrochim. Acta 1964, 9, 1599. (24) Tilak, B. V.; Conway, B. E. Electrochim. Acta 1992, 37, 51. (25) Wuttig, A.; Surendranath, Y. ACS Catal. 2015, 5, 4479. (26) Delacourt, C.; Ridgway, P. L.; Newman, J. J. Electrochem. Soc.
2010, 157, B1902.
(27) Gupta, N.; Gattrell, M.; MacDougall, B. J. Appl. Electrochem.
2005, 36, 161.
(28) Kas, R.; Kortlever, R.; Yilmaz, H.; Koper, M. T. M.; Mul, G.
ChemElectroChem 2015, 2, 354.
(29) Lim, C. F. C.; Harrington, D. A.; Marshall, A. T. Electrochim.
Acta 2017, 238, 56.
(30) Singh, M. R.; Kwon, Y.; Lum, Y.; Ager, J. W.; Bell, A. T. J. Am.
Chem. Soc. 2016, 138, 13006.
(31) Hall, A. S.; Yoon, Y.; Wuttig, A.; Surendranath, Y. J. Am. Chem.
Soc. 2015, 137, 14834.
(32) Varela, A. S.; Kroschel, M.; Reier, T.; Strasser, P. Catal. Today
2016, 260, 8.
(33) Hori, Y.; Murata, A.; Takahashi, R. J. Chem. Soc. Faraday Trans.
1 Phys. Chem. Condens. Phases 1989, 85, 2309.
(34) Yang, K. D.; Ko, W. R.; Lee, J. H.; Kim, S. J.; Lee, H.; Lee, M. H.; Nam, K. T. Angew. Chem. Int. Ed. 2017, 56, 796.
(35) Yoon, Y.; Hall, A. S.; Surendranath, Y. Angew. Chem. Int. Ed.
2016, 128, 15508.
(36) Ma, M.; Djanashvili, K.; Smith, W. A. Angew. Chem. Int. Ed.
2016, 55, 6680.
(37) Bunker, D. L.; Garrett, B.; Kleindienst, T.; Long, G. S. Combust.
Flame 1974, 23, 373.
(38) Gillespie, D. T. J. Phys. Chem. 1977, 81, 2340.
(39) Hinsberg, W.; Houle, F. Kinetiscope: a stochastic kinetics simulator http://hinsberg.net/kinetiscope/ (accessed Apr 1, 2017). (40) Zhao, C.; Dai, X.; Yao, T.; Chen, W.; Wang, X.; Wang, J.; Yang, J.; Wei, S.; Wu, Y.; Li, Y. J. Am. Chem. Soc. 2017, 139, 8078.