Influence of Na, P and (Na + P) poisoning on a model copper-ferrierite NH3-SCR catalyst
25
0
0
Texte intégral
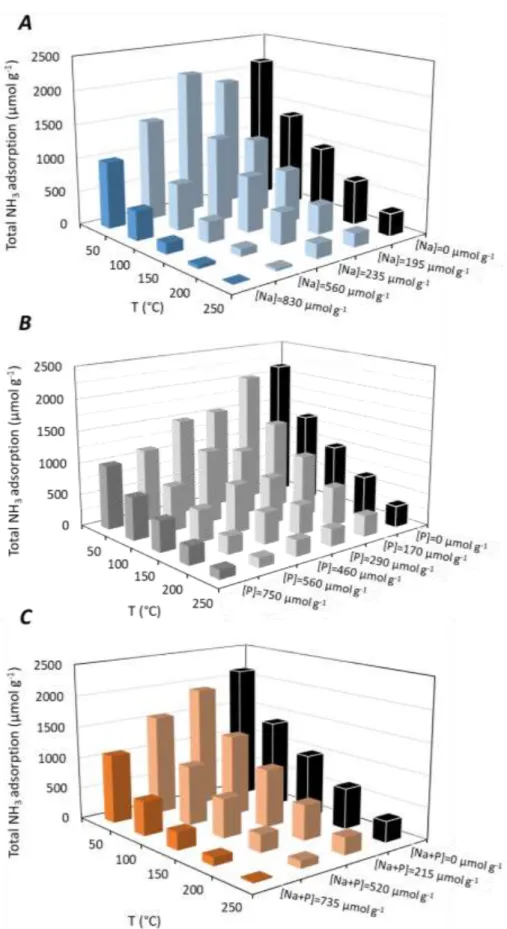
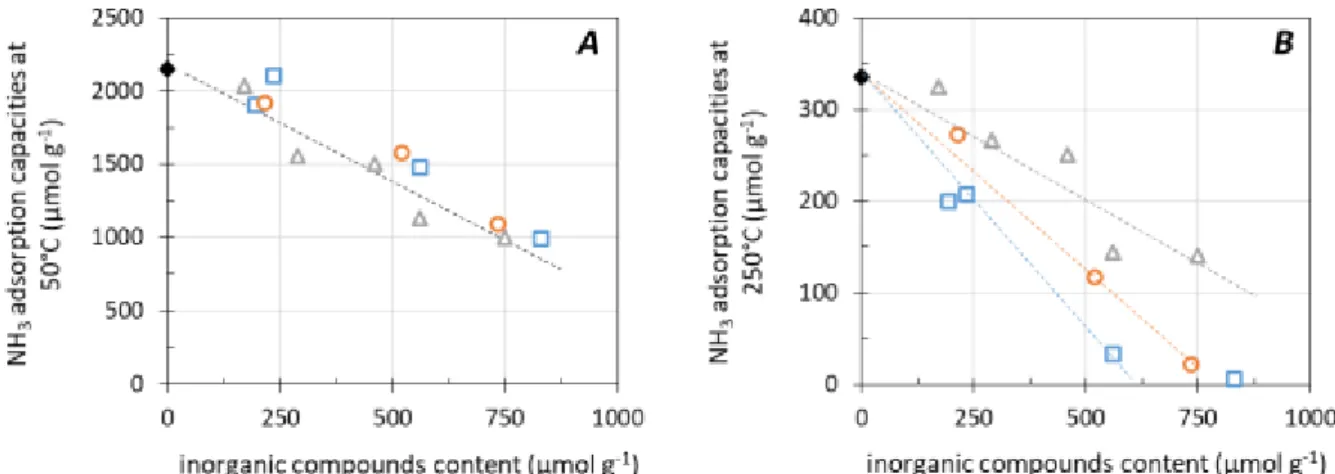
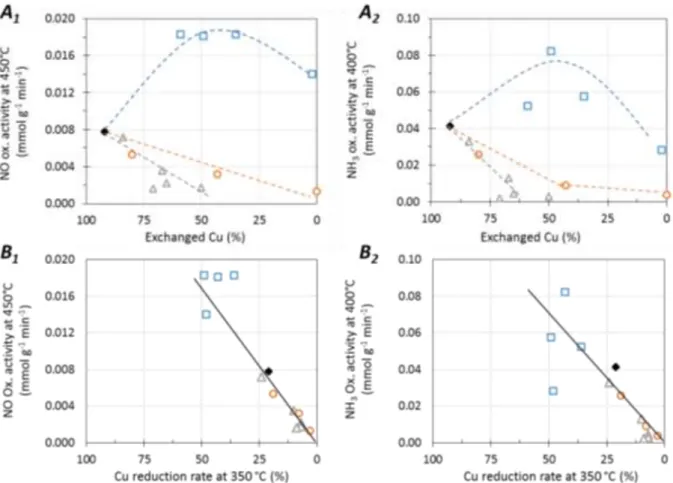
Figure




+7



Documents relatifs