USING GEL PERMEATION CHROMATOGRAPHY
by
HARRY ELLIS JOHNSON B.S., Stanford University
(1981)
Submitted to the Department of Chemical Engineering
in Partial Fulfillment of the Requirements of the Degree of
MASTER OF SCIENCE at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY May 1983
@c
Massachusetts Institute of Technology 1983Signature of Author
Certified by
Accepted by
Sig
Signature
redacted---06e r md nt o femical Engineering
1/4 May 1983
Signature redacted
Professor Charlds N. Satterfield Thesis Supervisor
iature redacted
Archives
MASSACHUSETTS INSTiTUTE OF TECHNOLOGYOCT
2
41983
LIBRARIES
Professor Glenn C. Williams Chairman, Department Committee
2
ANALYSIS OF HEAVY PARAFFINIC FISCHER-TROPSCH WAXES USING GEL PERMEATION CHROMATOGRAPHY
by
HARRY ELLIS JOHNSON
Submitted to the Department of Chemical Engineering on May 1, 1983 in partial fulfillment of the requirements for the degree of Master of Science in
Chemical Engineering ABSTRACT
The technique of Gel Permeation Chromatography (GPC) was utilized in analysis of paraffinic wax samples generated via the Fischer-Tropsch synthesis in a slurry reactor. Chromto-grams were obtained by using standard GPC equipment with Waters 100 and 500A Ultrastyragel TM columns. Differential number fraction molecular weight distributions were obtained to C70 and C105 for ambient and elevated temperature injections re-spectively. Corresponding relation of heavy wax hydrocarbon-product distributions to Flory plots show chain growth proba-bility factor a value of 0.92 - 0.94. These values are within experimental accuracy of results found by previous investiga-tors obtained by vapor-phase gas chromatography analysis. The results indicate that GPC may be used successfully in obtaining quantitative data for heavy solid products formed by slurry-reactor operation of the Fischer-Tropsch synthesis.
Thesis Advisor: Charles N. Satterfield
INSTITUTE OF TECHNOLOGY
DEPARTMENT OF
CHEMICAL ENGINEERING
Room number: 6 6-2 5 0 Cambridge, Massachusetts 02139
May 1, 1983
Professor Jack P. Ruina Secretary of the Faculty
Massachusetts Institute of Technology
Dear Professor Ruina:
Telephone: 253-6546
In accordance with the regulations of the faculty, I
submit herewith a thesis entitled "Ai1alysis of Heavy Paraffinic Fischer-Tropsch Waxes Using Gel Permeattion Chromatography," in partial fulfillment of the requirements for the degree of Master of Science in Chemical Engineering at the Massachusetts
Institute of Technology.
Respectfully submitted, ,/
4
ACKNOWLEDGEMENTS
I would like to thank my thesis advisor Professor Charles Satterfield for his helpful support and critism. I hope that I will remember to "stick to the facts". I also wish to thank Professor Jeff Tester and Michel Boudart for their constant encouragement throughout my graduate career.
I wish to thank the GEM program, Shell Development Co., Amoco, and Chevron for their financial support. Thanks are also extended to the Department of Energy for its support of the project.
To my collegues in the department, I have unmeasureable gratitude for all of the assistance offered during my stay at M.I.T.. Special acknowledgements are given to Harvey Stenger, Rich Pekela, Al Horn, Lisa Jungherr, and my officemates. I also wish to thank all of my friends who suffered with me through this very trying ordeal. I am especially grateful for
the friendship and support of Pieter VanderWerf, Brian Smiley, Westley Spruill, and Scott Slate. To Eugenia Brown, I offer the sincerest thanks for her friendship and love. Thanks also to Sally Kreuz for the exceptional assistance in production of this thesis.
And finally to my family, I offer a thunderous chorus of thanks. To my parents I give my love and appreciation for all they've done. Thanks for teaching me to believe in myself and giving me the courage to try and accomplish my goals. And most of all, thanks for instilling in me a devotion to God.
TABLE OF CONTENTS
Page
I. Introduction 11
II. Objectives 15
III. Literature Review 16
IV. Experimental 35
IV.A. Experimental Apparatus 35
IV.A.l. Fischer-Tropsch Reactor 35 IV.A.2. Ambient Temperature Apparatus 35 IV.A.3. Elevated Temperature Apparatus 35
IV.B. Experimental Procedures 49
IV.B.l. Ambient Temperature Procedures 49 1. Sample Preparation 49 2. Chromatographic Analysis 50
2a. Preparation
2b. Analysis 51
2c. Calculation 52
IV.B.2. Elevated Temperature Procedures 53 1. Sample Preparation 53 2. Chromatographic Analysis 53 2a. Preparation 53 2b. Analysis 54 2c. Calculation 55 V. Results 56
V.A. Ambient Temperature GPC Results 56 V.B. Elevated Temperature GPC Results 84
VI. Discussion 103
VII. Conclusion 113
VIII. Appendices 114
6
LIST OF FIGURES
Figure No. Page
3-1 Development and Detection of Size Separation by 21 GPC.
3-2 General Schematic of GPC Equipment.
24 4-1 Slurry Reactor Apparatus.
36 4-2 Waters M6000A Solvent Delivery System, Exploded
View. 38
4-3 Waters M6000A Solvent Delivery System, Circuit
Diagram for Hydraulic Components. 39 4-4 Injectors: (1) Rheodyne Model 7125 Sample
Injector; (2) Waters U6K Sample Injector. 40 4-5 Schematic of Waters R401 Refractive Index
De-tector Optical Unit. 42
4-6 Schematic Waters 150C Main Pump.
45 4-7 Schematic Waters 150C Injection System. 47 4-8 Schematic Waters 150C Differential
Refractor-meter. 48
5-1 GPC Chromatogram - Injection 1-4: Mixture of n-Hydrocarbon Standards C22, C28, and C38. 59 5-2 GPC Chromatogram - Injection 1-7: Mixture of
n-Hydrocarbon Standards C24, C25, C26, C28, and
C30. 60
5-3 GPC Calibration Curve Run 2: Toluene Mobile Phase, 1 x 100A Ultrastyragel Column, 22C, and n-hydrocarbon standards C19, C20, C21, C22, C23, C24, C25, C26, C28, C30, C32, C36, C38, and C40. 61 5-4 GPC Chromatogram - Injection 2-15: SS-9A 62 5-5 GPC Calibration Curve Run 3: Toluene mobile
phase, 1 x 100A Ultrastyragel column, 24.4C, and n-Hydrocarbon Standards C19, C22, C26, C28, C36,
Figure No. Page 5-6 GPC Chromatogram - Injection 3-11: SS-9E,
Attenuation 2X, Chart Speed changed to 4 inch/
min @ 5.8 minutes. 67
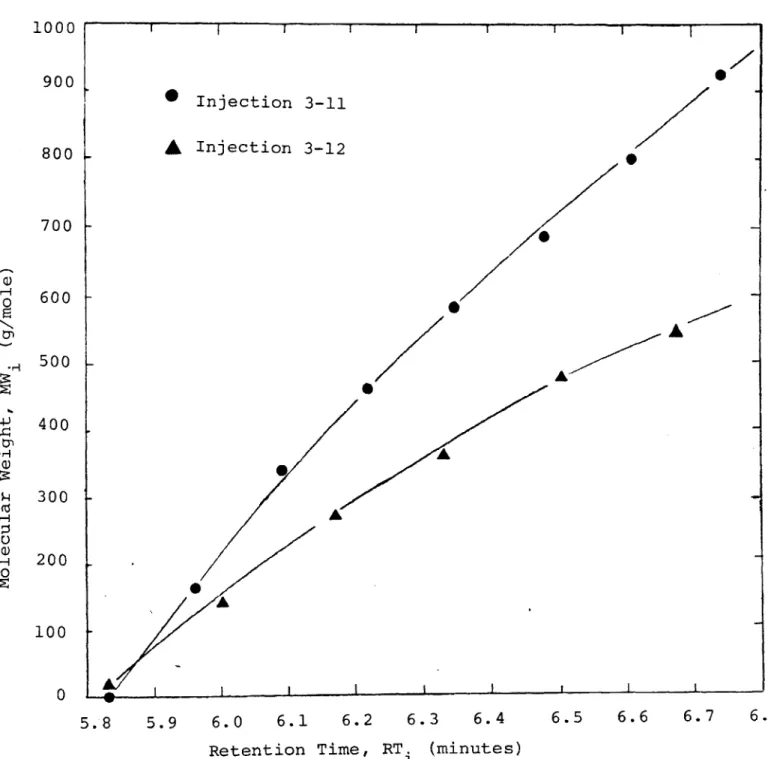
5-7 GPC Chromatogram - Injection 3-12: SS9D, Atten-uation 2X, Chart Speed 4 inch/min. 68 5-8 Peak Height and Retention Time Distribution:
Injections 3-11 and 3-12. 70
5-9 Cumulative Weight Fraction Molecular Weight Dis-tribution: Injections 3-11 and 3-12. 71 5-10 Differential Number Fraction Molecular Weight
Distribution: Injections 3-11 and 3-12. 72 5-11 GPC Calibration Curve Run 4. Toluene Mobile
Phase, 1 x 100A Ultrastyragel Column, 23C, n-hydrocarbon Standards C20, C28, C32, C36, C40
and Polystyrene Standards 800, 1800, and 2000. 74 5-12 Peak Height-Retention Time Distribution:
Injec-tion 4-11. 77
5-13 Cumulative Weight Fraction Molecular Weight
Dis-tribution: Injection 4-11. 78
5-14 Differential Number Fraction Molecular Weight
Distribution: Injection 4-11. 79
5-15 GPC Calibratiop Curve Run 5. Toluene Mobile Phase, 1 x 100A and 1 x 500AO Ultrastyragel
Columns, 25C, n-Hydrocarbon Standards C20, C24,
C28, C36, C40 and Polystyrene Standard 1800. 81 5-16 Area Percent Molecular Weight Distribution:
In-jections 5-12 and 5-17. 83
5-17 Cumulative Weight Fraction Molecular Weight Dis-tribution: Injections 5-12 and 5-17. 85 5-18 Differential Number Fraction Molecular Weight
Distribution: Injections 5-12 and 5-17. 86 5-19 GPC Calibration Curve0Run 6: Trichlorobenzene
Mobile Phase, 1 x 100A and 1 x 50OX Ultrastyragel Columns, 50C, n-Hydrocarbon Standards C20, C24,
8
Figure No. Page
5-20 GPC Calibration Curve Run 7: Trichlorobenzene Mobile Phase, 1 x 100A and 1 x 500l
Ultrastyra-gel Columns, 50C, n-Hydrocarbon Standards Stan-dards C20, C24, C28, C38, C40 and Linear
Poly-ethylene Standard 1800. 91
5-21 Area Percent Molecular Weight Distritubion:
In-jections 7-7, 7-8, 7-9, 7-11, and 7-12. 99 5-22 Cumulative Weight Fraction Molecular Weight
Dis-tribution: Injections 7-7, 7-8, 7-9, 7-11,and 7-12. 100 5-23 Differentioal Number Fraction Molecular Weight
Distribution: Injections 7-7, 7-8, 7-9,7-11, and7-12. 101 5-24 Weight Percent Molecular Weight Distribution:
Injection 7-13. 102
6-1 Form of Flory Plot Postulated for 2-Site Reac-tion and AccumulaReac-tion of Products in Liquid
Carrier 104
6-2 Carbon Number Distribution for Run 9 at 248C and
H2/CO Feed of 1.81. 106
6-3 Carbon Number Distribution of Liquid Carrier
After Run 9. 107
6-4 Theoretical Carbon Number Distribution Based on
Flory Equation. 108
6-5 Carbon Number Distribution of Heavy Paraffinic Fischer-Tropsch Wax at Ambient Temperature Using
GPC. 110
6-6 Carbon Number Distribution of Heavy Paraffinic Fischer-Tropsch Waxes at Elevated Temperature
LIST OF TABLES
Table No. Page
3-1 Gel Permeation Chromatography Operating
Condi-tions - Hillman, 1971. 32
5-1 Summary of Iun 1. Operating Conditions: Sol-vent - Toluene, Temperature - 25C, Columns
-1 x 100A Ultrastyragel, Flowrate - 1 ml/min,
Injection Volume 100 pl, Polarity - Positive. 57
5-2 Summary of Run 2. 58
5-3 Summary of Run 3. 64
5-6 Cumulative and Differential Weight Fraction Molecular Weight Distribution Data: Injections
3-11 and 3-12. 69
5-5 Summary of Run 4. 77
5-6 Cumulative and Differential Weight Fraction Molecular weight Distribution data: Injection
4-11. 76
5-7 Summary of Run 5. 80
5-8 Cumulative and Differential Weight Fraction Mole-cular Weight Distribution Data: Injections 5-12
and 5-17. 82
5-9 Summary of Run 6. 87
5-10 Summary of Run 7. 90
5-11 Fischer-Tropsch Synthesis Operating Conditions. 92 5-12 Cumulative and Differential Weight Fraction
Molecular Weight Distribution Data: Injection
7-7. 93
5-13 Cumulative and Differential Weight Fraction Molecular Weight Distribution Data: Injection
7-8. 94
5-14 Cumulative and Differential Weight Fraction Molecular weight Distribution Data: Injection
7-9. 95
5-15 Cumulative and Differential Weight Fraction Molecular Weight Distribution Data: Injection
10
Table No. Page
5-16 Cumulative and Differential Weight Fraction Molecular Weight Distribution Data: Injection
7-12. 97
5-17 GPC Weight Percent Data: Injection 7-13. 98
I. INTRODUCTION
The production of synthetic fuels to supplement dwin-dling supplies of natural fuels has directed attention towards development of processes which utilize abundant resources of indigenous reserves such as coal. One such process is the Fischer-Tropsch synthsis. In this procedure the indirect liquefaction of coal to hydrocarbons is accomplished via the catalytic reaction of synthesis gas, a mixture of carbon monox-ide and hydrogen produced by gasifaction of coal in the presence of oxygen and steam. The hydrocarbons produced from the
Fischer-Tropsch synthesis are predominantly linear paraffins and olefins with some oxygenates (primarily alcohols). The overall reaction stoichiometry may be represented as:
Paraffins : nCO + (2n + 1)H 2 C H + nH2 0 + 123 kcal
2
n 2n+2
2
(1)
Olefins: nCO + 2nH 2 C H + nH 0 + 93 kcal (2)
2
n 2n
2
Alcohols: nCO + 2nH + CnH 2n+OH + (n - 1)H 20 + 102 kcal (3) where the heat of reaction is based on n=3 at 227C. Two im-portant side reactions may also occur:
Water-Gas-Shift: H20 + CO = CO2 + H2 + 10 kcal (4)
Boudouard: 2CO -+ C(s) + CO2 + 42 kcal (5)
The synthesis of hydrocarbons from carbon monoxide and hydrogen has been known since the classical methane synthesis
12
of Sabatier and Senderens (1902). In 1922 Franz Fischer and Hans Tropsch obtained their first patent on "Synthol", a mix-ture of oxygen-containing derivatives of hydrocarbons. Further developments by Fischer and Tropsch in the 1920's and 1930's directed the synthesis to produce predominately hydrocarbons by using cobalt-based catalysts in fixed-bed, vapor-phase reactors. In 1943, Germany optained a peak production of 16,000 bbl per day. The products consisted of 46% gasoline, 23% diesel fuel, 23% waxes and detergents, and 3% libricating oil. With World War II, Germany's supply of cobalt from the Belgium Congo was severely cut and active search was initiated to develop iron catalysts. In the United States during the 1950's a fluidized-bed process using an iron catalyst to con-vert synthesis gas from then inexpensive natural gas to gaso-line was installed by Hydrocol, but it never operated satis-factorily. And as petroleum supplies became plentiful further investigation of the Fischer-Tropsch synthesis became uneconomi-cal. Until recently, South Africa has been the only country actively pursuing Fischer-Tropsch technology. At South African Synthetic Oil Limited (SASOL), fixed- and fluidized-bed proce-dures have been developed which utilize iron catalysts at inter-mediate pressures (5 - 25 amt), with production capacity of
50,000 bbl per day. Current expansion is afoot which will ultimately increase capacity to over 100,000 bbl per day.
The Fischer-Tropsch synthesis is a linear polymerization process. The process begins with an adsorbed single-carbon
another carbon unit or terminate by desorption into the gas (or liquid) phase as product. Debate still exists over the mechanism and nature of this carbon unit. But this does not
affect the mathematical development of an expression to pre-dict carbon number distribution if the probability of chain growth is independant of molecular size.
Flory (1936) statistically derived the basic relation-ship for any polymerization process where the primary step is addition of monomer units one at a time onto the terminus of a growing linear chain. The chain growth proability factor * is defined as:
r
a = (r + (6)
r )
pt
where r and rt are the rates of propagation and termination respectively (a is independant of molecular size). The mole fraction m of molecules in the polymer mixture which contains n structural units is given by the Flory Equation:
n-mn
=(1
- a)(7-)
If the added weight of each carbon unit is proportional to chain length n, the weight fraction w is given by:
n
w
=(l -c
)2n(n-l)(8
A more covenient form for expressing experimental data is the logarithmic form of the Flory Equation:
ln (m ) = n ln (x) + ln (1-) (9) n
14
Therefore a plot of ln (m n) versus carbon number n should be linear with slope ln (a) and ordinate intercept (1 - a)
at n = 1.
The products formed in Fischer-Tropsch synthesis depend on the hydrogen to carbon monoxide ratio in the synthesis gas as well as on the catalyst and reactor conditions selected. These products range from methane to high molecular weight compounds such as heavy paraffinic wax. Gas chromatography has been successfully applied in the analysis of most Fischer-Tropsch products. However utilization of gas chromatography becomes ineffective in examination of high carbon number pro-ducts (C30 +) due to the low volatility of these heavier
hydrocarbons.
For complete analysis, an additional technique is needed that will provide quantitative data of the heavy Fischer-Tropsch fractions. One such technique is Gel Per-meation Chromatography (GPC), which involves the separation of molecules based upon differences in their effective size in solution. The size sorting takes place by repeated transfer of solute molecules between the bulk mobile phase and stagnant liquid phase within the pores of the packing, allowing sample characterization by its molecular weight distribution.
II. OBJECTIVES
The present investigation focused on gel permeation
chromatography analysis of heavy paraffinic waxes formed during the Fischer-Tropsch synthesis in a slurry reactor. This is of fundamental and practical importance. Fundamentally, one would like to know how high in molecular weight the Fischer-Tropsch synthesis proceeds and if the Flory equation is applic-able in this high carbon number product region. Practically, it is needed to know how long the reaction can be allowed to continue before excessive accumulation of heavy hydrocarbons might occur in the reaction apparatus. First, an understanding of GPC was sought. Then application of GPC analysis to Fischer-Tropsch wax samples was undertaken. Finally GPC data were
16
III. LITERATURE REVIEW
Conventional founding of liquid chromatography has been attributed to work performed by Michael Tswett. In 1903
-1906, Tswett recognized chromatography as a general method in description of separation of colored vegetable pigments in petroleum ether on calcium carbonate. From Tswett's early
findings, a large number of workers continued to develop liquid chromatography to its present high performance capa-bilities and it has found application in various forms of scientific disciplines (Synder and Kirkland 1974).
The phenomena of gel chromatography were first observed
with adsorption of different sized ions in 1925 (Ungerer 1968).
The term "molecular sieving" was first used in 1926 by McBain (Porath 1962a). The crystalline crosslinkages of natural and synthetic aluminum silicates, known as molecular sieves,
made possible separation of molecules according to size and
shape (Wiegner 1931, Tiselius 1934, Claesson and Claesson 1944,
1948). Later, Barrer and Brook established and proved correla-tions of adsorption and molecular size in molecular sieves
(1953).
Sieving properties were also found during application of ion exchange resins (Samuelson 1944, Rauen and Felix 1948). A correlation between the number of crosslinkages, the degree of swelling, and the ion exchange capacity of the large ions was found in the structure of ion exchange resins (Wofattite, Amberlite, Kumi and Myers 1949, Mikes 1958). This property
amino acids, peptides, and proteins (Richardson 1949, 1951, Thompson 1952, Partridge 1952). This experience directed attention to larger-pored polysaccharide matrices. Uncharged crosslinked galactomannane gel was used in the desalting of colloids (Deuel and Nenkom 1954). Peptides and proteins were separated on granulated starch particles (Lindquist and
Storgards 1955, Lathe and Ruthuen 1955). Later, it was estab-lished by Lathe and Ruthuen that the penetration of molecules into the gel phase depended upon structure and contration of the gel, and a relationship between molecular size and chroma-tographic behavior was found (1956).
The study of electrophoresis played an important role in the development of gel permeation chromatography. Preparative and analytical methods of gel electrophoresis prompted the examination of macromolecues and biopolymers (Smithies 1955, Raymond and Wintraub 1959, Davis and Ornstein 1959, Porath and Bennich 1962). The electroosmosis of natural substances and proteins in dextran, a characteristic polysaccharide, investi-gated by Tiselius in Sweden led to the production of a new
semi-synthetic gel (1959). Sephadex, dextran gel copolymerized with epichlorhydrin and packed into a column, achieved good
separation even without an electric current. A closer examina-tion of Sephadex by Porath and Flodin marked the beginnings of what may be called an explosive development of gel
chromato-graphy (1959).
Since the properties of natural polysaccarides, such as dextran, were difficult to reproduce, application of such materials were not found wholly suitable for chromatographic
18
uses. In the early 1960's semi-synthetic and synthetic poly-mers replaced the natural gel forpoly-mers (Polson 1961, Lea and
Sehon 1962, Hjerten and Mosback 1962). Initially hydrophilic polyacrylamide gels produced by copolymerization of acrylamide and methylene bisacrylamide found widest application. The examination of gels which swelled in organic solvents began
simultaneously with that of hydrophilic gels, particularly with attention to polystryene matrices copolymerized with divinylbenzene.
In 1964 J.C. Moore disclosed the use of cross-linked polystrene gels for separating synthetic polymers soluble in organic solvents (Moore 1964). It was recognized that with proper calibration, gel permation chromatography was capable of providing molecular weight and molecular weight distribution information for synthetic polymers.
Gel permeation chromatography is used as an analytical technique for separating small molecules according to size difference and to obtain molecular weight distribution inform-ation of polymers. (The raw-data GPC curve is a molecular weight distribution curve.) With a concentration sensitive
differential refractometer detector, the GPC curve can become a size distribution curve in weight concentration. And with proper calibrating, molecular weight averages can be calculated.
A convenient quantity which measures the average chain length in a polymer sample is the number-average molecular weight, Mn, defined as:
EN Mw W M = = n EN W '
E()
(10)
Mw.
1where W and N are the weight and number of molecules with molecular weight Mw , respectively. From GPC:
M Ehi (11)
S (h /Mw )
where h is the GPC curve height at the ith volume increment and Mw is the molecular weight of the species eluted at the ith retention volume, and N is the number of chains present. Another convenient quantity obtainable from GPC data is the weight-average molecular weight, M w, given by:
2w
ZN
Mw
2VW.
Mw
M E N __MW 2L~d
1W1(12) w N. Mw. EW. l l 1 and from GPC:Z(h. Mw.)
M = (13) wEhi
The value of M is always greater than Mn except when the
w
n
values are identical in monodisperse systems. The dispersity, M /M , is a measure of the broadness of the molecular weight distribution. The Z-average molecular weight, Mz, is related to a higher moment of the distribution and is defined as:
Mw
3M =
ZN
1 (14)20 (Dallas and Abbott 1979).
In the (theoretical) model to describe GPC, the gel is composed of a porous matrix whose pores are closely controlled in size. The separation mechanism involves differences in ability of molecules to penetrate the pores. Very large mole-cules cannot penetrate into the gel pores and thus migrate down the column through the interstitial volume between parti-cles and emerge first from the column. Smaller molecules can to some degree penetrate into the gel pores, and thus they are slowed in their migration through the column. Very small molecules that are able to completely penetrate into the gel pores will be retained the most, and elute from the column last (Lawrence 1981). This is represented by the illustration shown in Figure 3-1.
The retention time, RT, is the time required for a peak to elute from the column following sample injection. Its value is sensitive to changes in experimental conditions such as flow rate and the specific column used. The retention volume, RV, accounts for flow rate differences and is defined as:
RV = F(RT) (15)
where F is the mobile flow rate. The peak capacity factor, k', is a more basic retention parameter. Physically, k'
represents the ratio of the weight of solute in the stationary phase to that in the mobile phase. This is defined as,
RT - T RV- V
k T 0 _ m (16)
TIME SEQUENCE: Refractive Index Detect (A) Sample Injected 49~K
00C>,
(B) Size SeparationC-Large (C) Solutes Eluted . . Small (D) Solutes Eluted Chromatogram (Concentration Elution Curve) Injection (A) RT = RV/F
Figure 3-1: Development and Detection of Size Separation by
GPC (Yau, Kirkland, and Bly 1979).
22
where T = RT for an untetained peak and Vm = F(T
)
for the retention volume of unretained solute.To account for differences in stationary-phase loading, the solute distribution coefficient, KLC, is defined as:
k' V
K =
m
(17)LC
V
S
where Vs is the equivalent liquid volume for a stationary phase. Physically, KLC is the ratio of solute concentration in the stationary phase to that in the mobile phase.
Transfer between mobile and stationary phases occurs as solute molecules migrate through the column to continually redistribute themselves between the phases to satisfy thermo-dynamic equilibrium. This implies equivalance of chemical potential of each solute component in the two phases. For dilute solutions at equilibrium, solute distribution can be related to the standard free energy difference, AG*, given by:
AG* = - RTlnK (18)
with
AG* = AH* - TAS* (19)
where K is the solute distribution coefficient, R is the uni-versal gas constant, T the absolute temperature, and AH0 and AS* are standard enthalpy and entropy differences between
phases, respectively. In GPC, solute distribution is governed primarily by entropy changes between phases (Dawkins 1976).
KGPC eAS/R (20) Temperature changes have only a small indirect effect on GPC retention since they only affect polymer solute molecule size, which in turn affects AS*. Large fluctuations of temperature of GPC experiments should be avoided.
The results of temperature, flow rate, and steric mixing experiments show that GPC retention is an equilibrium, entropy-controlled, size-exclusion process. The diffusion in and out of the pores of the solute is fast enough with respect to flow rate to maintain equilibrium solute distribution.
Extensive developments in liquid chromatography have made available a variety of equipment which can be used in gel permeation chromatography. A general schematic for GPC
equipment is shown in Figure 3-2. The instrumentation used in
GPC is needed to function rapidily and reliably. In the solvent delivery system a constant, reproducible supply of solvent
to the column is required. The sample injector must introduce sharp volumes of sample without disturbing the flow of the solvent. High efficiency columns are needed to give maximum separation capability and provide reproduciable information over extended periods of time. Specific detectors with wide sensi-tivity ranges are used to monitor the separation. A detailed description of apparatus used in this study will be presented in Section IV.A.
Gel permeation chromatography was primarily developed for measuring molecular size distribution of polymer products.
24
8 7 6
5
3
2
Figure 3-2: General schematic of GPC equipment: (1) Solvent Supply; (2) Solvent Delivery System; (3) Injector;
(4) Sample Syringe; (5) Columns; (6) Differential Refractometer Detector; (7) Computerized Data Handling Device; (8) Recorder.
A series of narrow molecular weight range fractions of anionic polystyrene samples were efficiently separated in polystrene columns with aromatic and chlorinated solvents (Moore 1964). Further investigation by Hendrickson and Moore (1966) found that by calibrating columns with carefully characterized polymers, it was possible to prepare a curve relating the
elution volume to the logarithm of the average molecular chain length (or effective angstrom chain length) given by:
Angstrom chain length (A) = 2.5 + 1.25 (C.A.U.) (21)
where C.A.U. is the size in carbon atom units. These calibra-tion curves were often linear over a large range of chain
length, and molecular weight distributions of unknown polymer samples could then be calculated. Analysis of polystyrene, polyvinylchloride, and polypropyleneoxide dissolved in THF showed some deviation from the molecular chain length correla-tion. Hendrickson and Moore suggested five basic forces that might be expected to modify elution volume of compounds in
GPC: (1) changes in width of the network openings in the column packing beads, (2) solvent - solute association, (3) dimeriza-tion of solute molecules, (4) intramolecular bonding, and
(5) adsorption of solute onto or into the gel surface. There-fore, simple chain length was an inadequate size parameter in correlation of elution volumes of some non-normal samples.
Smith and Kollmansberger (1965) reported on the separa-tion of a number of alkanes and halogenated aromatic compounds
26
the apparant molecular volume is the important parameter
in-fluencing the degree of separation, (where molar volume (ml/nole) is defined as molecular weight divided by density at 25C.).
Later, Edwards and Ng (1968) investigated the use of bulk molar volume for various compounds (including normal hydro-carbons) and showed that there was a linear relationship be-tween logarithm bulk molar volume and elution volume for normal hydrocarbons. The elution behavior of about 100 model com-pounds was studied to show the effectiveness of GPC as an
analytical tool. It was found that organic functional groups exert a systematic influence on GPC elution behavior, re-inforcing the theory that such behavior is the result of solute's association with the solvent, the gel packing, or itself. By measuring these effects with model compounds, gen-eralized observations for characterization of nonfunctional minor components of lower molecular weight epoxy resins was accomplished.
Rather than using standards such as polystyrene and polypropylene glycols to calibrate GPC columns, Sweeney, Thompson, and Ford (1970) used normal paraffins and cali-brated by carbon number. This calibration was used to deter-mine relationships between boiling point, carbon number, and elution volume. A limited number of branched hydrocarbons were also investigated as well as a brief quantitative study to determine response factors for the normal hydrocarbons.
Lambert (1970) recommeded that a set of molar volume values for n-alkanes be used as standards to calibrate GPC
columns. Molar volume is given by:
Molar volume (ml/mole @ 20C) = 33.02 + 16.18 (C.A.U.)
+ 0.004 (C.A.U.) (22)
where C.A.U. is as previously defined.
Debate still exists over the appropriate parameter to use in calibration of GPC columns. Mori and Yamakawa (1980) obtained relationships among oligostyrene, n-hydrocarbons, and oligoethylene glycol in chloroform and tetrahydrofuran. Different elution behaviors of oligomers in different eluents made it difficult to use molar volumes or effective chain
lengths as the calibration parameter. The n-hydrocarbons are non-polar compounds and were assumed to elute without solute-solvent association or the adsorption on the gel. Molecular weight conversion equation for several oligomers based on molecular weight of oligostryene and n-hydrocarbons were de-derived, making it possible to use these as reference stan-dards when molecular weights of oligomers are measured.
A wax is a complete mixture of high molecular weight organic components. Hatt and Lamberton (1956) defined a wax
as "a thermoplastic of low mean molecular weight and low mech-anical strength." They classified waxes into three categories: hydrocarbon, natural (ester), and synthetic waxes. Hydrocar-bon waxes, derived from petroleum, can be further sub-divided into two groups, the paraffinic and the microcrystalline
waxes. Paraffinic waxes consist primarily of long chain nor-mal paraffins, with snor-mall amounts of branched chain and
28
cycloparaffin in the approximate range of C18 - C42. The microcrystalline waxes consist mainly of branched chain par-affins of high molecular weight. The natural (ester) waxes, excreted by most animals and plants, are more complex in composition and consist mostly of mixtures of esters of long chain (C18+) fatty acids and alcohols. Appreciable amounts of free acids, alcohols, paraffins, resins, dihydride alcohols, hydroxy acids, ketones, and sterols are also present. Syn-thetic waxes may be entirely synSyn-thetic, such as polyethylene types, be prepared from petroleum waxes, or be prepared from other natural materials.
Many elaborate procedures have been utilized in complete analysis of waxes (Robinson and Johnson 1966). Prior to
widespread use of chromatographic techniques, lengthy pro-cesses of fractional distillation and fractional crystalliza-tion were used. Infra-red spectroscopy has been applied in the fingerprinting of single wax compounds by comparison with infra-red spectrums of known waxes. Investigation of wax
properties (melting point, inflexions on cooling curves, speci-fic gravity, penetration hardness, and refective index) have been used for the quality control of all types of waxes, how-ever, the information given by these methods is not completely reliable. Robinson and Johnson noted two problems involved in complete analysis:
1. Separation of wax constituents according to their chemical nature, i.e. ester, alcohols, paraffins, etc.
2. Separation of the fractions into individual single chemical species must then be identified.
Chromatographic techniques are superior due to, i) efficiency of separation, ii) simplicity of operation, and iii) direct indication of the identity of components from their rention data.
Paper chromatography was used to separate the mercuric acetate-methanol adducts of the allyl esters of the fatty acids by Kaufman of Pollenberg (1957). Reversed phase paper chromatography was used to separate even-numbered acids up the C36 in Beeswax, Carnuba wax, and Montan wax by Kaufmann and Das (1963). The major problem in paper chromatography
is the low solubility of wax acids in solvents at room tempera-ture, and elevation of temperature increases the complexity of the method.
Gas chromatography of waxes has been largely investigated.
Adlard and Whitham (1958) separated paraffin hydrocarbons up to C36 using a silicone coated column at 270C. Normal paraffins were removed from kerosine by means of Linde 5A molecular
sieve by Whitham (1958). O'Connor and Norris (1960 and 1962) extended this procedure to the higher molecular weight par-affins waxes. Gas chromatography of separated fractions gave the ratio of normal to non-normal paraffins, the relative amounts of each normal paraffin, and the relative amounts of total non-normal paraffin of each carbon number in a paraffin wax. Unfortunately this separation procedure required long
30
for routine analysis. Scheneck and Esima (1963) used Linde 5A sieves in conjuction with a gas chromatography column using silicon stationary phase for paraffins in crude oils and rock extracts.
Gas-solid chromatography was used by Scott and Rowell (1960) to separate C15 and C36 paraffins using an alumina column deactivated with sodium hydroxide at 390C. Levy and co-workers (1961) separated and identified 67 components pre-sent in a refined paraffin using mass spectrometry in conjuc-tion with gas chromatography (8-foot columns packed with 10% microwax distillation residue on Chromosorb W. at temperatures of 300C).
Levy and Paul (1963) used a dual column, dual flame
ionization, temperature programmed gas chromatograph to obtain carbon number distributions of C19 to C36 paraffinic waxes using 12-foot 1/8-inch copper tubing packed with 0.82% micro-crystalline wax distillation residue on 80-100 mesh Diatoport S over the temperature range 140-330C at 2.3C per minute.
Resolution of a homologous series of the even-numbered alkanes from tetradecane through dopentacontane was accomplished on 6-foot aluminum columns packed with 0.1% Apiezon L on 60-80 mesh glass microbeads over the temperature range 40 to 350C at 5.6C per minute by Perkins, Laramy, and Lively (1963). Hydrocarbon components in the range C25 to C68 of four
micro-srystalline waxes were resolved on dual 2-foot columns packed with SE 52 on Chromosorb G using a temperature programmed dual flame ionization gas chromatograph by Ludwig (1965).
in total characterization of waxes. Cole and Brown (1960) used a specially prepared alumina column for separation of complex waxes into fractions according to their chemical na-ture, however incomplete separation often occurred. Wiedenhof
(1959) used column chromatography prior to X-ray diffraction measurements on wax fractions. Ion exchange column chromato-graphy was used in determination of free wax acids, wax soaps, and total hydrocarbon content in waxes by Presting and Janicke (1960). Robinson and Johnson recommended use of ion exchange column chromatography only as a precursor to gas or thin
layer chromatography (1966).
With the introduction of GPC by Moore (1964), column chromatography became a more useful technique in wax analysis. Hillman (1971) found that by selecting the appropriate porosity range of Styragel column packing, waxes in carbon range C15
to C100 could be closely examined. By using a series of
Styragel columns it was possible to characterize hydrocarbon and ester waxes dissolved in organic solvents. Operating conditions for study are shown in Table 3-1.
The GPC unit was calibrated using 0.1% solutions of n-hydrocarbons (C16, C20, C28, C32, C36, C48) and narrow
molecular weight range polystrenes (No. 25168, 26169, 25171 from Waters Associates). Retention times were plotted versus carbon number, the carbon number for polystrenes being cal-culated by effective carbon number (equation 21). Calibration plots were essentially linear over the range interest but
TABLE 3-1: Gel permeation chromatography operating conditions-Hillman, 1971.
I Column Porosities (A)
Column Temperature (*C) 100, 500, 10, 3 x 104 70 II 100, 100, 100, 500 30 III 100, 400, 500, 103 80 Solvent Toluene Inhibitor
Flow rate (ml/min)
Sample Concentration (%) Sample size (ml) Sensitivity Inlet heater (*C) Syphon heater (*C) Detector heater (*C) Nil 1.0 1.0 Tetrahydrofurane Quinol 1.0 1.0 2 x2 70 70 2 x2 30 30 70 30 o-Dichlorobenzene Stavox CP (5g/gall) 0.5-1.0 1.0 2 x2 for fingerprinting x4 for polyethylene 80 80 80 L&J
molecular exclusion of the column system was approached. The extrapolation of the linear region for the n-hydrocarbons
passed through the point for the lowest molecular weight poly-styrene standard. In the actual hydrocarbon wax analysis, carbon numbers corresponding to the retention times of the maxima of the gel permeation chromatograms were read off the applicable calibration curve. A correction factor of twice the standard deviation of the gaussian peak obtained for in-jection of pure C36 was introduced to account for peak broading during elution.
Improvements in the efficiency of small pore packing materials and column preparation advanced the speed and
con-venience of GPC to that of gas chromatography. Lack of vola-tility, or absence of significant differences in polarity, solubility, or ionic characteristics are not significant pro-blems in GPC analysis. Krishen and Tucker found that the
high efficiency of the GPC column affords a separation of com-ponents as distinct separate peaks, in the short time, and provided a useful technique for extending the molecular weight range beyond that covered by gas chromatography (1977).
Harmon (1978) used GPC in characterization of hydrocar-bon waxes, reporting molecular size distribution profiles,
coupled with melting point profiles from differential scanning calorimetry, as the basis for comparison and selection of
replacement waxes for use by the B.F. Goodrich Company.
Styragel columns were used by Sosa, Lombana, and Petit in analysis of crude paraffinic waxes (C16 - C50) waxes (1978).
34
Molecular weights (Mn and Mw) and molecular weight distribu-tions of waxes containing various amounts of oil were deter-mined. The method of GPC was choosen because temperatures re-quired to analyze n-paraffins having chain lengths above C40
are almost at the upper useful limit of routine gas chromato-graphy. Gas and gel permeation chromatograms were presented and were found to complement each other well.
IV. EXPERIMENTAL
A. EXPERIMENTAL APPARATUS
A. 1. Fischer-Tropsch Reactor
The Fischer-Tropsch synthesis reactions were carried out in a semi-continuous, slurry-bed catalytic reactor system. A schematic of the slurry reactor unit is shown in Figure 4-1. The stainless-steel, 1-liter autoclave was operated isothermally
in semi-batch mode. Pre-mixed synthesis gas was feed into the autoclave by a pneumatically-controlled valve while volatile products were removed overhead. The catalyst and inert liquid carrier remained in the reaction vessel. Condensable products were collected in a wax trap (held at 70C) and a cold trap
(held at 2C). A detailed discussion of the reactor system and procedures followed during operation are found in the Ph.D. thesis of G.A. Huff and related publications by Satterfield and Huff.
A. 2. Ambient Temperature Apparatus
Analysis of Fischer-Tropsch wax samples at ambient temper-ature was performed on two gel permeation chromatographic
apparatuses. This equipment may be categorized into five divi-sions: solvent delivery system, injector, columns, detector, and data compiling system.
1. Solvent Delivery System
In both sets of GPC components used at ambient temperature, Waters Associates Model M6000A pumps were utilized to deliver
Vant
Ab
P
-6
Corriar
Som
pla
I-PI
9
10
-8-
_
-111
Wox
Oil
Sompla
Sompla
Figure 4-1: Slurry Reactor Apparatus: (1) Gas Cylinder with Premixed CO/H2 Mixture;
(2) Pressure regulator; (3) Automated Flow controller; (4) Back-Pressure Regulator; (5) 1-Liter, Mechanically Stirred Autoclave with Thermocouple at (a) and Turbine Impeller at (b); (6) Pressure Gauge; (7) Wax Receiver;
(8) Ice-Cooled Receiver; (9) Back-Pressure Regulator; (10) Gas Sample Valve; (11) Soap-Film Flowmeter (Huff 1982).
P P
3*
4LA~)
Figures 4-2 and 4-3. The M6000A is a dual head reciprocating piston positive displacement pump, in which solvent enters through an inlet check valve during the first half of the piston stroke and exits through an outlet check valve during the second half of the stroke. Each pump head delivers 100 microliters of solvent per stroke. A steady flow of solvent was provided by precise timing of the pistons. The flow rate of the pump was adjustable in 0.1 ml/minute increments between 0 and 9.9 ml/minute.
2. Injector
Two types of injectors were used in these experiments, a Rheodyne Model 7125 (Runs 1 - 4) and Waters Associates U6K (Run 5). Schematics of these injectors are shown in Figure 4-4. The Model 7125 is a six port sample injection valve. The sample loop, whose volume corresponds to the injection volume, is loaded by syringe through a needle port built into the valve shaft. While in the load position, solvent from the pump flows directly to the columns and sample from the syringe may be loaded into the sample loop. When flipped to the inject position, solvent from the pump pushes the samples from the sample loop into the column.
The U6K has a 2 ml last-in-first-out sample loop into which any sample amount under 2 ml can be injected. While on
load, the solvent flows through the restrictor loop. To in-ject a sample into the sample loop, the needle port valve is locked by flipping the injector valve to inject, allowing solvent flow through the sample loop to carry the sample into the columns.
I
~Ii I-M~4
- -
-Figure 4-2: Waters M6000A Solvent Delivery System, Equipment Manuals).
Exploded View (Waters GPC
k
~r
1
AC j.L3QA V
-E --OT 2
IREFERENCE TRANSEEREA
HIGM CI4OLA ASSEMBLY PUMP MEA OUTLET
ACESSORY E C. EE NOT &I
OUTAsETOLO VALV
IU I 04CT IS 3 G ME FITFILTER
-414ALs D*asAD UfA CONNECTION
MITLET -XLT LTE- OUTLET
CHIECK CECK
VALVI VALVE
ASSEMBLY ASSEMASLY
LEFT syaPyN PUMP'NG 141GO4
Pump 14EAO CMAVS v.4 CI a PUW 04EAO
-FHLI- 6--FLAVTA wOLET OLET
CHECK CHICK
VALVE VALVI
sEMBLY ASSEMBLY
fRACitOft INLE T PvRG1 iNft TL cC T . MsANIFOLD 14 7O
OUTLET PO0-4 ASSErSIBy O WCTM
ctsE NoE Is) 7-r3 COLL ECT LINE)
"AVTE VALVE FLT WASTEPORT .... m. SOLVENT IETO SOLVE.T DEsTECT.R Pi SOVE4 EOLV ST63) L VAV I L T SOLVENT PIES 0tvC.11 ASLENTlm PLaSIO IR 0 ME
Figure 4-3: Waters M6000A Solvent Delivery System, Circuit Diagram for Hydraulic Components (Waters GPC Equipment Manual).
40 -,e e'
;Kt
LIFJ
At\/pop
Poll Vets IL .2~ INJFCT (1) Rheodyne Model 7125 Sample InjectorU.I
_J_
ri~~~j;C~
PLU fllene toLJ
C(2) Waters U6K Sample
Figure 4-4: Injectors: (1) Rheodyne Model
(Rheodyne Injector Manual); (2) Injector (Waters GPC Equipment
7125 Sample Injector
Waters U6K Sample Manual).
3. Columns
The columns used were Waters Associates Ultrastyrageltm Gel Permeation Columns. As discussed in Section III, Styragel is a cross-linked styrene/divinylbenzene copolymer. The Ultra-styragel columns are available in a variety of pore sizes
50
ranging from 100 to 10 A. Ultrastyagel columns have better resolution that their precursor, Waters p Stryageltm columns. Ultrastyagel columns provide the highest resolution per column of any GPC column currently used for separation of hydrocarbons, low to intermediate MW polymers, synthesis reaction products, and polymer additives as well as other samples. Each column produces 10,000 - 15,000 plates per cm column. Two sizes of
Q
columns were used in this investigation, 1 x 100A (Runs 1-4)
0 0
and 1 x 100A in series with 1 x 500A (Run 5), each 30 cm in length.
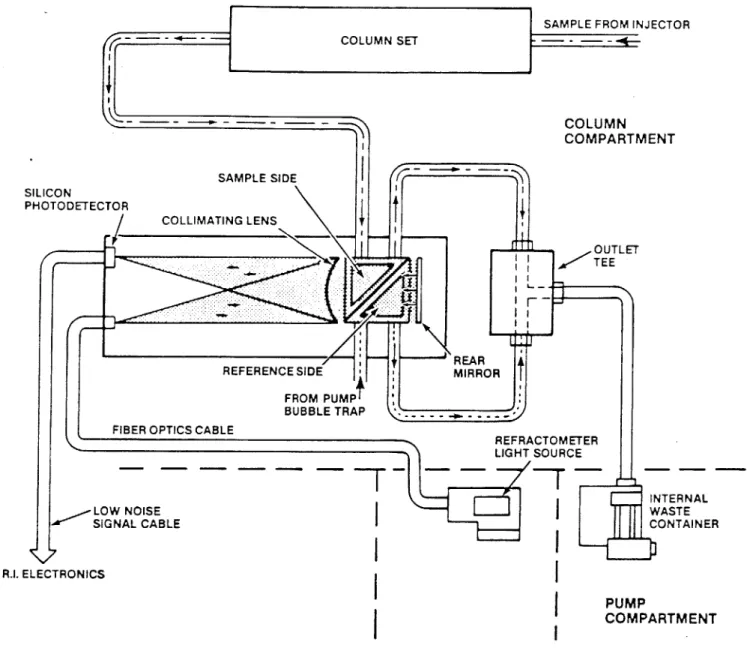
4. Detector
A Waters Associates R401 refractive index detector was used in all GPC analyses. The detector consists of optical and electronic units. The optical unit, a schematic shown in Figure 4-5, consists of a light source shone through a slit, a lens, and the sample cell to a mirror. It reflects off the mir-ror, back through the sample cell and the lens, to a rotable piece of glass used as the zero adjust, and finally to a cadmium sulfide detector which measures the position of the beam. The beam's deflection is proportional to the refractive index dif-ference between the contents of the sample cell, which contains flow from the column and flow directly from the pump.
SAMPLE
MIRROR - LENS MASK LIGHT
SOURCE
- - - DETECTOR
0-TTETL AMPLIFIER & RECORDER
REFERENCE 0PTICAL ZERO PWRSPL
ZERO ADJUST
Figure 4-5: Schematic of Waters R401 Refractive Index Detector Optical Unit (Waters GPC Equipment Manual).
The cadmium sulfide detector outputs a voltage propor-tional to the beam deflection. This signal is sent to the electronics unit, which amplifies the signal and outputs it as a voltage between -100 and +100 millivolts. The amplification of the signal from the optics unit is controlled with an attenu-ator switch with settings from 128X (least sensitive)to 1/4X
(most sensitive). A zero test setting on the attenuator and a potentiometer labeled RECORDER ZERO allow the zero setting of the detector to be matched with the zero setting of the recorder. A polarity switch allows the polarity of the signal to be reversed. a chart marker switch sends a small output to the recorder to
allow events to be marked. 5. Data Compiling
A Houston Instruments 0 - 10 millivolt 2 channel recorder with friction driven chart speeds variable over a wide range was used in Runs 1 - 4. One channel was used to record the out-put of the differential refractometer, the other channel was not used. The output from the detector was passed through a
two resistor voltage divider, which transformed the 100 millivolt output to a 10 millivolt output.
A Waters Associates M730 Data Module was employed in Run 5 to compile chromtographic data. The M730 is a versatile printer-plotter-integrator offering a choice of calculation
methods and baseline corrections, and individual sample calcula-tions with ten calibration files. Parameters for set-up, peak integration, and calculation are entered prior to injection of samples. Integration can be controlled by user selection of timed events. A typical plot is marked with an injection mark,
44
retention times, and start and stop integration marks. The calculated results, retention time, area, molecular weight, and area/molecular weight, are printed after plotting of the chroma-togram. For detailed discussion of operation of the M730, con-sult the Waters Data Module Instruction Manual.
A. 3. Elevated Temperature Apparatus
The waters Associates 150C ALC/GPC was used in all elevated temperature runs. The Model 150C is a fully automated, micro-processor-based system consisting of all the fundamental GPC components plus it has capability for precise temperature con-trol. High temperature control is extremely important when:
1. The sample is difficult to dissolve at room temperature 2. The sample is difficult to keep in solution at room
temperature
3. The viscosity of the sample in solution is high, or 4. The viscosity of the solvent is high.
Each component of the self-contained Model 150C is designed for superior system operation. The 150C operates automatically under microprocessor control and functions in the same manner for single or multiple samples. operating commands are entered via the front panel keyboard. The microprocessor stores the
instructions and organizes them into the required sequence of mechanical operations.
The solvent delivery system, shown in Figure 4-6, con-sists of a noncircular, computer-designed gear drive which
over-lapes piston strokes to provide constant flow for smooth base-line. A small pre-pump, in-line filter and debubbler ensure
TO INJECTOR COMPARTMENT PRESSURE TRANSDUCER OUTLET 1 CHECK VALVES INLET CHECK VALVES BUBBLE I TRAP
LOW PRESSURE 0NLN FLE
DETECTOR INE *
PURGE VALVE MANIFOLD
TO REFERENCE SOLVENT
SIDE OF SUPPLIED
REFRACTOMETER BY PRE-PUMP
Figure 4-6: Schematic of Waters 150C Main Pump (Waters GPC Equipment Manual).
46
highly accurate, resetable solvent delivery to the inlet of the high pressure pump. Compressibility compensation provides
accurate flow rates fo 0 to 9.9 ml/minute in 0.1 ml/minute in-crements.
The programmable injection system, shown in Figure 4-7, provides automatic spin, filtration, and injection volumes from 10 to 500 -pl for each of sixteen sample vials. The 150C per-forms injection of samples just as an operator would manually with a syringe. It cleans the syringe, places the needle into the sample vial, withdraws a specified amount of sample, injects, and marks the chromatogram.
The column oven accepts up to 10 columns. This oven was designed to operate from ambient to 150C. It maintains samples and solvent temperatures constant for consistent and reliable results. The programmable heating rate reduces thermal shock to columns. The columns used were 1 x 100A and 1 x 500A Ultra-styragel columns.
A sensitive, linear universal refractive index detector, shown in Figure 4-8, is utilized in the 150C. Light passes through the flow cell and is reflected by a mirror behind the flow cell. The reflected light then passes through the flow cell again and is focused on a photodetector sending an output volt-age to the recording device. When a difference between refrac-tive indices of the two fluids in the cell chambers is detected, the refracted light beam falls upon a different part of the
photodetector, causing a change in the photodector voltage out-put which is indicated by deflection on the chart recorder. A
SYRINGE
SYRINGE
VALVE
(V2)
HOLDING LOOP (OPENED)
RES TSYRINGE YRI SE VENT A TUATED MOTORl)E PISTON TO COLUMN INJECT fl CLOSED) BYPASS T7 7771RESTRICTOR V'11 SOLVENT FROM
FILTERED SAM PLE PUMP
SA!.MPLE VIAL
Figure 4-7: Schematic of Waters 150 Injection System (Waters GPC Equipment Manual).
48
SAMPLE FROM INJECTOR
CL IMIN COMPART SAMPLE SIDE TOR TO COLLIMATING LENS
_
OUTLET TEE -REARREFERENCE SIDE MIRROR
FROM PUMP+ BUBBLE TRAP FIBER OPTICS CABLE
MENT REFRACTOMETER LIGHT SOURCE INTERNAL W WASTE CONTAINER LOW NOISE SIGNAL CABLE TO R.I. ELECTRONICS PUMP COMPARTMENT
Figure 4-8: Schematic of Waters 150C Differential Refracto-meter (Waters GPC Equipment Manuals).
SILICON PHOTODETEC
COLL
a
counter-current heat exchanger ensures thermal stability to
0.0005C. Fiber optics light transmission provides cooler opera-tion of the light source for sensitive detecopera-tion and longer
life. Usage of a quartz halogen light source provides extra sensitivity by increasing signal to noise ratio.
Rapid and reproducible data recording with automatic calculation of GPC data was obtained with the Data Module M730 (as described in Section 4.A.2.5). For a more complete description of the 150C and its components, consult the Waters Model 150C ALC/GPC Instruction Manual.
B. EXPERIMENTAL PROCEDURES
B. 1. Ambient Temperature Procedures
The procedure of obtaining a molecular weight distribu-tion using GPC is broken into three parts: sample preparation and solvent selection, actual chromatographic analysis to gener-ate chromatograms, and calculation of molecular weight informa-tion from the resultant chromatograms.
1. Sample Preparation
Samples from 0.02 - 0.2 weight% were prepared by dissolving a known weight of sample in a given volume of solvent. Larger concentration of samples could cause "viscous fingering," a phenomenon in which the sample components being separated reach a high enough concentration in the column to begin to interact with each other, causing poor separation. The solvent used in all ambient experiments was HPLC grade Toluene supplied by VWR Scientific. Toluene was chosen because of its capatibility with
50
the column system, refractive index difference from samples, and easy accessibility. Samples were filtered through Millipore Corporation Millex-GV .22 pm filters.
2. Chromatographic Analysis 2a. Preparation
The solvent is vacuum filtered through 1/2 micron Milli-pore filters to prevent particulate contamination to the columns. The pump is then primed by drawing solvent through the pump in-let into a syringe. The pump is turned to a high setting to ensure that the pump outputs to a low pressure. Solvent is
pushed from the syringe into the pump until the pump can operate on its own. The M6000A pump will pump the volume to which it
is set immediately once primed.
The reference valve on the pump is positioned at REFERENCE and the pump is set at 3 ml/minute to purge the reference cell. A few minutes purging is sufficient. Flow is then returned to 0 ml/minute and the reference valve is turned to the column position.
If the Rheodyne injector is used a syringe full of solvent is used to flush the sample loop. If the Waters injector is used, the valve is turned to INJECT and the sample port valve to UNLOCK, the pump turned back to 3 ml/minute, and the loop purged for three sample loop volumes. After purging, the flow
should again be retuned to zero, the sample port valve locked, and the injection valve to LOAD.
After the injector is flushed, the flow rate can be brought slowly up to the operating flow rate, 1 ml/minute in
all ambient experiments. Care must be taken to ensure that the
pressure does not oscillate considerably, which could be due to failure of the pump inlet check valve. If this occurs, the pump must be reprimed with degassed solvent.
The chart recorder is zeroed relative to the refractive index detector by turning the detector to ZERO TEST and adjusting the recording zero knob until flipping the polarity switch
produces little or no deflection of the chart recorder pen.
The optics are zeroed by turning the detector to its lowest sen-sitivity and adjusting the optical zero to the chart recorder zero, and repeating this process in the next higher sensitivity until the desired sensitivity is reached. Sensitivities of 4X and 2X were used in all ambient experiments.
2b. Analysis
Once the baseline has steadied and the appropriate sen-sitivity level reached, samples are ready for injection. The desired amount of sample is injected into the sample loop with a clean syringe. Injection volumes of 100 pl were used in all ambient experiments. When using the Rheodyne injector, the injector must be on LOAD. Several hundred microliters of air-free solvent are injected into the loop and around the injection port to clean the syringe loop. Injection of the appropriate amount of air-free sample then follows. The syringe remains in the injector. When the Waters injector is used, the injector also must be on LOAD and the sample port unlocked. After the sample is unlocked in the injector, it is withdrawn and the sample port locked. If the sample port is not locked injection will cause depressurization of the columns.
52
To inject the sample, flip either injector to INJECT. The Rheodyne injector must be flipped quickly as there is no flow when the injector is between LOAD and INJECT. After one sample loop volume has flowed through the injector, it can be flipped back to LOAD, and in the case of the Rheodyne injector flushed with solvent. In any case, the syringe should be rinsed with clean solvent.
The output of the detector, the chromatogram, is recorded on the chart recorder (Runs 1-4) or the Data Module (Run 5). Sufficient time should be allowed for complete elution of sample before injection of another sample. This time can be approxi-mated from flow rate values and column lengths.
After all samples have been run, the flow rate is slowly returned to zero, and all instrumentation is turned off except the detector. The detector is designed to remain on continuously, and turning it off will decrease its usefullness.
2c. Calculation
In order to obtain molecular weight data from a chroma-togram, calibration of the column is first required. If nar-rowly dispersed standards are available (as is the case in this investigation) calibration is quite simple. Injecting samples of the standards and determing their retention time is all that is required to formulate a calibration curve. A plot of log molecular weight versus peak retention time yields a linear calibration curve which conforms to the equation,