Assembly and Substrate Recognition Properties of Human CCT
Subunits of the TRiC Chaperonin
by
Oksana A. Sergeeva
B.S. Chemistry/Biology Harvey Mudd College, 2009
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENT
FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
SEPTEMBER 2014
© Massachusetts Institute of Technology. All rights reserved.
Signature of Author: ____________________________________________________________ Department of Biology May 2014
Certified by: __________________________________________________________________ Jonathan A. King Professor of Molecular Biology, Thesis Supervisor
Accepted by: _________________________________________________________________ Amy E. Keating Co-chair, Department Committee on Graduate Students
Assembly and Substrate Recognition Properties of Human CCT
Subunits of the TRiC Chaperonin
by
Oksana A. Sergeeva
Submitted to the Department of Biology at the Massachusetts Institute of Technology on May 30th, 2014 in the Partial Fulfillment of the
Requirements for the Degree of Doctor of Philosophy in Biology
ABSTRACT
Group II chaperonins are large multi-subunit complexes that fold cytosolic proteins to their native structures. They are composed of two back-to-back rings of 7-9 subunits. The eukaryotic cytosolic type II chaperonin Tailless Complex Polypeptide-1 (TCP-1) Ring Complex (TRiC) consists of eight different subunits identified as Chaperonin Containing TCP-1 (CCT) α (1) - θ (8). TRiC is necessary for folding about 10% of newly synthesized proteins and is essential for folding actin and tubulin. Most of the research on TRiC in the last 20 years has focused on yeast and bovine TRiC. However, recently, there has been inquiry into TRiC as a target for disease therapy for Huntington’s disease, cataract, and some cancers. Consequently, to understand human TRiC, we purified endogenous TRiC from HeLa cells for characterization. These complexes contained all eight of the CCT subunits as determined by immunoblot. The structures were well organized as double-rings of eight subunits each, using negative stain electron microscopy (EM). Human TRiC was active in suppressing aggregation and refolding two different substrates: luciferase (a model substrate) and human γD-crystallin (HγD-Crys; a physiological substrate found in the eye lens).
To further understand human TRiC, we expressed all of the human CCT subunits, one at a time in E. coli. This was done so that the subunit specificities of the CCT subunits could be studied and so we could have a system where these proteins could be genetically manipulated. Theoretically, all eight subunits in the mature TRiC-complex are needed to successfully recognize all substrates that need to be folded in the cell. We found that two CCT subunits, CCT4 and CCT5, but not the others, formed TRiC-like homo-oligomeric rings in the absence of the other CCT subunits. Purification of these complexes and subsequent structural assays by negative stain and cryo-EM showed that they formed double rings of eight subunits per ring. Biochemically, we found that CCT4 and CCT5 hydrolyzed ATP at the same rate as human TRiC, could refold luciferase to the same level as human TRiC, and suppressed aggregation of HγD-Crys. The homo-oligomeric complexes also assisted the refolding of HγD-Crys, a property not observed in the lens specific α-crystallin chaperone. On the substrates studied, CCT4 and CCT5 homo-oligomers worked as efficiently as hetero-oligomeric TRiC. More stringent substrates such as actin and tubulin need to be studied to further understand CCT specificity.
4
Two mutations, one in CCT4 (C450Y) and one in CCT5 (H147R), have been implicated in hereditary sensory neuropathy. In order to study the defective mutant proteins, we introduced these mutations into the CCT4 and CCT5 constructs. We found that for CCT4, the newly translated mutant polypeptide chains aggregated much more than wild-type (WT) CCT4. While the mutant formed some rings in the E. coli lysate, as assayed by sucrose ultracentrifugation gradients and negative stain EM, they were not stable throughout the purification and the final purified sample contained few homo-oligomers. The mutant CCT5 polypeptide chains were properly folded and assembled in homo-oligomers. H147R CCT5 was able to hydrolyze ATP at a similar rate as WT CCT5. However, in the HγD-Crys aggregation suppression and refolding assay, mutant huntingtin aggregation suppression assay, and actin refolding assay, mutant CCT5 was not as efficient in suppression or refolding as WT CCT5. Therefore, the H147R mutation in CCT5 led to a chaperoning defect while the C450Y mutation in CCT4 led to a folding/stability defect.
In order to understand features of partially folded intermediates that group II chaperonins recognize in a substrate, we investigated whether the archaeal group II chaperonin from
Methanococcus maripaludis (Mm-Cpn) could recognize a variety of HγD-Crys mutants. These
mutations were in regions of the protein that could act as recognition signals of substrate – unpaired aromatics, domain interface, and buried core residues. We found that Mm-Cpn was able to recognize all of these mutants, better than it recognized WT HγD-Crys. In addition, Mm-Cpn could refold most of the mutants to levels higher than WT HγD-Crys. Therefore, we concluded that Mm-Cpn doesn’t recognize any of the proposed recognition signals but recognizes some β-sheet interface exposed in these mutants.
These studies further our knowledge of group II chaperonins and specifically human TRiC, and open up some new avenues for the investigation of the folding, assembly and function of this eukaryotic protein essential for the reproduction of all cells.
Thesis Supervisor: Jonathan A. King Title: Professor of Molecular Biology
ACKNOWLEDGEMENTS
There are many people who have helped me get to this point of finally writing up this thesis and graduating. First and foremost, I want to thank my advisor Jonathan King. I definitely could not have made it through the last four years without his support, encouragement, and guidance. I have learned a great deal from him as a scientist, mentor, and person. Jon, thank you for believing in me when I did not and supporting me through everything.
I need to thank all of the people in the King lab that I have interacted with and learned from in the last four or five years: Ligia Acosta-Sampson, Jeannie Chew, Dan Goulet, Cammie Haase-Pettingell, Althea Hill, Fan Kong, Kelly Knee, Kate Moreau, Jacqueline Piret, Liliana Quintinar, Dessy Raytcheva, Nathaniel Schafheimer, Meme Tran, Takumi Takata, Cindy Wooley, and Ginger Yang. Cammie: thank you for helping me in every random way you could: running sucrose gradient and gels, making buffers (and pH-ing everything – what will I do without you?), growing cells, and taking care of things when I had to run off to class. Cindy: thank you for always being there to chat about random current events and taking care of all the financial/grant issues. You both have made my life in lab less stressful and more entertaining. Kate, Kelly, and Dessy: I am so glad that I met each one of you in lab and that we’re still friends years later. I learned so much from each one of you in lab and life, and I know you all will go on to great things. Nathaniel: you have kept me sane and grounded for the last few years. I thoroughly enjoyed our lunches, venting sessions, and good times in lab. Thank you for always being there for me and making me feel less alone. Meme: you were the best undergrad anyone could ever ask for. Thank you for being hard working, amazing, and a joy to be around. I think you have a bright future ahead of you! Eugene: I feel like I not only failed you with everything I forgot to teach you, but also because I am leaving you all alone. However, I know you will be just fine and go on to awesome things!
I would like to thank my committee: Amy Keating, Thomas Schwartz, and Susan Lindquist. They have attended many meetings and offered valuable advice on my project, my progress in the program, and my future. I especially want to thank Amy (who I also TAed for) for always being encouraging and looking out for me and my training as a scientist. I also want to acknowledge Frank Solomon for giving me advice and taking an interest in my project.
Next, I need to thank Wah Chiu. I am so lucky to have been part of the Nanomedicine consortium and interacted with Wah and his lab over the last few years. He has always been excited about my projects and about moving research along. Wah: thank you for letting me come to BCM so often and giving me the opportunity to learn about cryo-EM from both the technical and computational sides. At BCM, there are many colleagues I would like to acknoweldge: Steve Lutdke, David Tweardy, Sarah Shahomoradian, Bo Chen, Michele Darrow, Corey Hecksel, Rebecca Dillard, Soung-Hun Roh, Moses Kasembeli, Yao Cong, and Zhao Wang. Thank you for putting up with all of my questions and helping out on all of my many cryo-EM projects. The Nanomedicine consortium has also given me the opportunity to interact with many professors and students in the chaperonin field. I would like to specifically thank Bill Mobley, David Housman, and Chengbiao Wu for stimulating conversations; and, Koning Shen, Tom Lopez, Em Sontag, Ryan McAndrew, and Henrique Pereira for sharing reagents, protocols, and expertise in the chaperonin field. In addition, I want to acknowledge Zach Crook, the only other grad student from MIT in our Nanomedicine group. Zach: thank you for answering all of my huntingtin questions and giving me great feedback on all of my work and concerns. You are one of the most hard-working people I know.
6
I would like to thank my neighbors across the hall: the Gilbert lab. Boris, MK, Josh, Thomas, Kristen, Pavan, Audra, Maria, Julia, and Wendy: thank you for always letting me bother you and hang out in Starbucks+ with you. I have always been amazed by how well you get along and how fun your lab atmosphere is. To my neighbors in the same hall, the Sinskey lab (JQ, Jingnan, Chris, Tony, and Claudia): thanks for being so welcoming to me, inviting me to Friday wine and cheese, and always having that random chemical I didn’t think actually existed.
I need to acknowledge my biology classmates in the entering class of 2009 for being an exceptional group and somehow still remaining friends so many years later. I am so glad our class data clubs, building 68 lunches, and random Muddy hang-outs are still going on. More specifically, I would like to thank Paul Fields, Glenna Foight, Genny Gould, Heather Hoke, David Kern, Katie Mattaini, and Boris Zinshteyn for all our coffee dates, trivia nights, knitting & game nights, and in general for keeping me sane. You are all amazingly intelligent and dedicated, and have made my last five years fun and engaging. I also want to acknowledge MIT classmates outside of the biology department, in particular: Aimee Gillespie and Bridget Wall. Thanks for all of the road trips, coffee dates, articles clubs, and dinners. I am incredibly grateful we met and have become great friends! To my college friends in Boston/Cambridge (Nadia Abuelezam, Hallie Kuhn, Christina Snyder, Terence Wong, Ken Loh, and Trevor Ashley): thank you for making this transition to grad school easier, always being supportive, and strengthening our friendship over these years. Finally, to my BFFs from middle school (Olga Obraztsova, Sakina Palida, and Mika Wilbur): thanks so much for being there for me all of these years in every way. I have enjoyed all of our random traveling to see each other, and all the letters, postcards, and phone calls. I can’t believe we’ve been friends for more than half our lifetimes, and I can’t wait to see what the next decade or two will bring for us all!
Last, but not least, I would like to thank my family. All four of my grandparents and both my parents received PhDs in the sciences, so they have always encouraged me to pursue my education. I thank them for instilling a love for science and learning in me. A big thank you (even though I’m sure he doesn't want it) goes to my brother, Ivan. He started MIT as an undergrad a year before I got here, and we had a great time both living in Cambridge and hanging out together for the four years we overlapped. He was the one who gave me the courage to move across the country (into the cold) to go to grad school here. Finally, I want to thank my husband, Nate Jones and our son Chase. Nate somehow endured all of my grad school frustrations and still respected me as a scientist and a person. Nate: thank you for always believing in me and always being on my side. I can’t imagine these last five years of my life without you. Chase: thanks for giving me the easiest pregnancy ever. You better be cute! [Edit: You are super cute and the easiest baby! Thanks for your continued cooperation in letting me finish this thesis!]
This research was supported by National Institutes of Health grants (R01EY015834, P41GM103832, and Common Fund Roadmap PN2EY016525). The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-007031) is gratefully
BIOGRAPHICAL NOTE EDUCATION
Ph.D. Massachusetts Institute of Technology, Cambridge, MA Expected 2014 Department of Biology
Chemical Biology Interface Training Program Student
B.S. Harvey Mudd College, Claremont, CA
May 2009 Joint Chemistry/Biology with Psychology minor
Graduated with Distinction and Biology Department Honors
RESEARCH EXPERIENCE
2010-2014 Graduate Research Assistant with Professor Jonathan King Biology Department, MIT, Cambridge, MA
2005-2009 Undergraduate Researcher with Professor David Asai Biology Department, Harvey Mudd College, Claremont, CA
Summer 2008 SURF Student with Professor Seth Darst
Biophysics Department, Rockefeller University, New York, NY
Summer 2007 SURF Student with Professor Paul Insel
Pharmacology Dept., U. of California, San Diego, La Jolla, CA
Summer 2006 REU Student with Professors Bogdan Olenyuk and Katrina Miranda Chemistry Department, University of Arizona, Tucson, AZ
2001-2004 NewBiotics, Inc. and ThioPharma, Inc., San Diego, CA
TEACHING EXPERIENCE
Spring 2013 Teaching Assistant, MIT, Cambridge, MA 7.41: Topics in Chemical Biology
Spring 2011 Teaching Assistant, MIT, Cambridge, MA
7.10/20.111: Physical Chemistry of Biomolecular Systems
Fall 2008 Teaching Assistant, Harvey Mudd College, Claremont, CA Chem 24: Chemistry Laboratory
8 PUBLICATIONS
Sergeeva OA, Tran MT, Haase-Pettingell C, King JA (2014). “Biochemical Characterization of Mutants in Chaperonin Proteins CCT4 and CCT5 Associated with Hereditary Sensory Neuropathy.” J. Biol. Chem. Submitted.
Sergeeva OA, Yang J, King JA, Knee KM (2014). “Group II archaeal chaperonin recognition of partially folded human γD-crystallin mutants.” Protein Sci. 23: 693-702.
Sergeeva OA, Chen B, Haase-Pettingell C, Lutdke SJ, Chiu W, King JA (2013). “Human CCT4 and CCT5 chaperonin subunits expressed in E. coli form biologically active homo-oligomers.” J.
Biol. Chem. 288:17734-17744.
Knee KM, Sergeeva OA, King JA (2013). “Human TRiC complex purified from HeLa cells contains all eight CCT subunits and is active in vitro.” Cell Stress and Chaperones 18:137-144.
Wilkes DE, et al. (2009). “Identification and characterization of dynein genes in Tetrahymena.”
Methods Cell Biol. 92:11-30.
Sergeeva OA, Khambatta HG, Cathers BE, Sergeeva MV (2003). “Kinetic properties of human thymidylate synthase, an anticancer drug target.” Biochem. Biophys. Res. Commun. 307:297-300.
TABLE OF CONTENTS PREFATORY MATERIAL Cover Page ... 1
Abstract ... 3
Acknowledgements ... 5
Biographical Note ... 7
Table of Contents ... 9
List of Figures ... 13
List of Tables ... 15
List of Abbreviations ... 17
CHAPTER 1: Introduction Protein Folding and Aggregation ... 20
ATP-Dependent Chaperones ... 22
Hsp70 ... 22
Hsp90 ... 25
Chaperonins ... 27
Group I Chaperonins ... 28
History ... 28
Structure and Function ... 31
Group II Chaperonins ... 34
History ... 34
Structure and Function ... 37
Substrate Recognition by Group II Chaperonins ... 39
Chaperonin Subunits/Domains Involved in Recognition ... 39
Features of the Substrate Recognized ... 42
Chaperonin Complex Evolution ... 43
Homo-oligomeric Chaperonins ... 43
Hetero-oligomeric Chaperonins ... 43
Arrangement of CCT Subunits in TRiC ... 43
Role of Chaperonins in Human Disease ... 46
Mutations in Human Chaperonin Genes ... 46
Using TRiC to Ameliorate Diseases ... 46
Thesis Context ... 49
CHAPTER 2: Human TRiC Complex Purified from HeLa Cells Contains All Eight CCT Subunits and is Active In Vitro Abstract ... 52
10
Materials and Methods ... 55
TRiC Purification from HeLa Cells ... 55
SDS-PAGE and Immunoblots ... 56
Electron Microscopy ... 56
Luciferase Refolding Assay ... 56
Human γD-Crystallin Aggregation Suppression Assay ... 57
Results ... 58
Purification ... 58
Structure ... 58
Activity ... 64
Discussion ... 67
CHAPTER 3: Human CCT4 and CCT5 Chaperonin Subunits Expressed in E. coli Form Biologically Active Homo-oligomers
Abstract ... 70
Introduction ... 71
Materials and Methods ... 73
CCT Subunit Expression ... 73
CCT Subunit Purification ... 73
Human TRiC and Mm-Cpn Purification ... 74
Sucrose Gradient Sedimentation ... 74
SDS-PAGE and Immunoblots ... 74
Electron Microscopy ... 74
Cryo-Electron Microscopy ... 75
Thermal Denaturation by Circular Dichroism ... 76
ATP Hydrolysis Assay ... 76
Luciferase and Human γD-Crystallin Refolding Assays ... 76
Results ... 78
Expression and Purification of CCT Subunits ... 78
Structural Characterization of the CCT4 and CCT5 Homo-oligomers ... 83
Functional Characterization of the CCT4 and CCT5 Homo-oligomers ... 88
Discussion ... 95
CHAPTER 4: Biochemical Characterization of Mutants in Chaperonin Proteins CCT4 and CCT5 Associated with Hereditary Sensory Neuropathy Abstract ... 98
Introduction ... 99
Materials and Methods ... 104
Mutagenesis and Expression ... 104
Long-term Lysate Supernatant/Pellet Separation ... 104
Sucrose Gradient Sedimentation ... 104
SDS-PAGE and Immunoblots ... 104
Electron Microscopy and Circular Dichroism ... 105
Native Gel Electrophoresis ... 106
ATP Hydrolysis and Human γD-Crystallin Refolding Assays ... 106
Mutant Huntingtin Aggregation Suppression Assay ... 106
Actin Refolding Assay ... 106
Results ... 108
Mutant Protein Expression and Stability ... 108
Mutant Protein Sedimentation ... 110
Mutant Protein Purification ... 113
Mutant Protein Structure ... 115
CCT5 Mutant Activity ... 119
Discussion ... 128
CHAPTER 5: Group II Archaeal Chaperonin Recognition of Partially Folded Human γD-Crystallin Mutants Abstract ... 132
Introduction ... 133
Materials & Methods ... 136
Purification of HγD-Crys and Mm-Cpn ... 136
Thermal Denaturation by Circular Dichroism ... 136
Aggregation Suppression of HγD-Crys by Mm-Cpn ... 137
Quantification of Refolded HγD-Crys ... 137
Results ... 138
Buried Aromatic Pairs ... 138
Domain Interface Residues ... 142
Buried Core Hydrophobic Mutants ... 146
Discussion ... 150
CHAPTER 6: Final Discussion and Future Directions Final Discussion ... 154
Future Directions ... 159
CHAPTER 7: References ... 161
CHAPTER 8: APPENDIX A: Co-expression of CCT Subunits to Explore Subunit Assembly Abstract ... 184
Introduction ... 185
Materials and Methods ... 189
Plasmid Construction ... 189
12
Sucrose Gradient Sedimentation ... 189
SDS-PAGE and Immunoblots ... 189
Quantification ... 190
Results ... 192
Summary of Each CCT Profile ... 195
Effect of Homo-oligomers on Full-length CCT Subunits and Their Fragments ... 199
Discussion ... 204
APPENDIX B: Aggregation Suppression of Mutant Huntingtin by Chaperonins Abstract ... 208
Introduction ... 209
Materials and Methods ... 211
Mutant Huntingtin Aggregation Suppression Assay ... 211
LIST OF FIGURES
Figure 1-1: Effect of chaperones on protein folding and aggregation energy landscape ... 21
Figure 1-2: Hsp70 structure and states ... 24
Figure 1-3: Hsp90 structure and states ... 26
Figure 1-4: Group I chaperonin structure and mechanism ... 33
Figure 1-5: Group II chaperonin structure and mechanism ... 38
Figure 1-6: Alignment of apical domains of CCT subunits ... 40
Figure 1-7: TRiC subunit arrangement differs between laboratories and methods ... 45
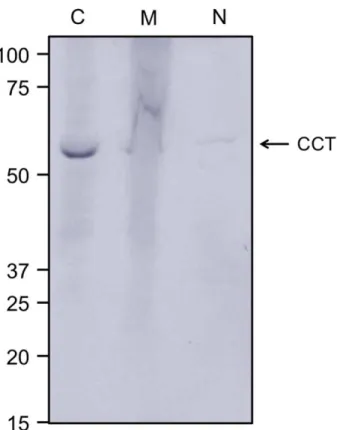
Figure 2-1: Human TRiC primarily limited to the cytoplasmic fraction of HeLa cells ... 59
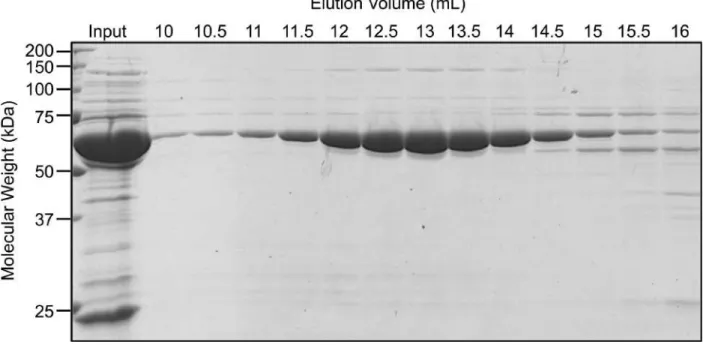
Figure 2-2: Human TRiC purified by size exclusion chromatography ... 60
Figure 2-3: Hsp70 and Hsp90 co-purified with TRiC when heparin affinity chromatography was omitted ... 61
Figure 2-4: All eight subunits present in purified human TRiC ... 62
Figure 2-5: Negative stain TEM of purified human TRiC reveal double rings ... 63
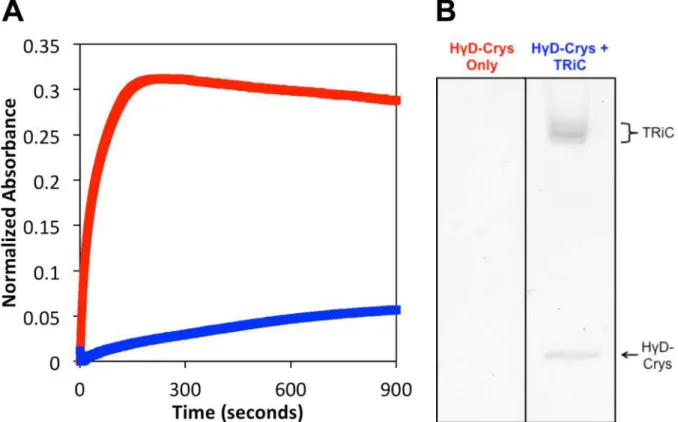
Figure 2-6: Purified human TRiC active in refolding luciferase ... 65
Figure 2-7: Purified human TRiC suppression of HγD-Crys aggregation and HγD-Crys native-like state refolding ... 66
Figure 3-1: Expression of human CCT subunits in BL21 (DE3) RIL E. coli cells ... 79
Figure 3-2: Sucrose ultracentrifugation gradients of CCT subunits ... 81
Figure 3-3: CCT5 purified by size exclusion chromatography as a 1 MDa complex ... 82
Figure 3-4: Negative stain TEM of purified CCT4 and CCT5 homo-oligomers showed morphology similar to human TRiC, and distinct from GroEL/ES ... 84
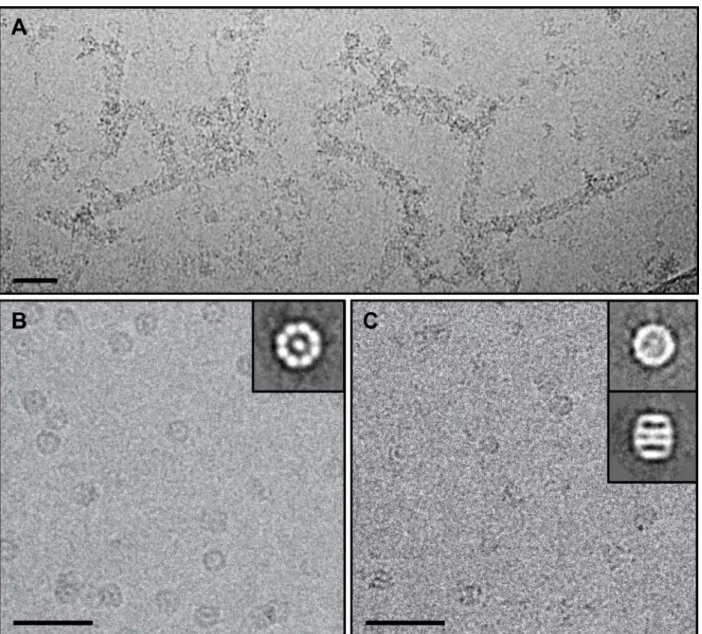
Figure 3-5: Raw cryo-EM images of CCT5 homo-oligomers and 2D class averages indicated two rings of eight subunits per ring ... 85
Figure 3-6: Cryo-EM reconstructions of CCT5 homo-oligomers suggested TRiC-like structures ... 87
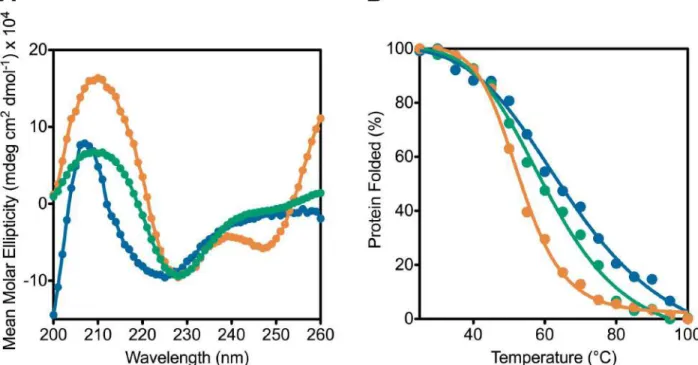
Figure 3-7: Human TRiC is more stable than CCT4 and CCT5 homo-oligomers by thermal denaturation using CD ... 89
Figure 3-8: CCT4 and CCT5 homo-oligomers hydrolyze ATP at a similar rate to human TRiC 90
Figure 3-9: CCT4 and CCT5 homo-oligomers were active in refolding luciferase ... 92
Figure 3-10: CCT4 and CCT5 homo-oligomers suppressed aggregation of partially folded HγD-Crys and promoted HγD-HγD-Crys native-like state refolding ... 94
Figure 4-1: Location of neuropathy mutations in CCT4 and CCT5 ... 102
Figure 4-2: Expression levels of CCT4, CCT5, and their neuropathy mutants ... 109
14
Figure 4-4: Sucrose ultracentrifugation gradients of CCT4, CCT5, and their neuropathy mutants
... 112
Figure 4-5: CCT4 and CCT5 purification off of the Co-NTA column ... 114
Figure 4-6: Negative stain transmission electron micrographs of CCT4, CCT5, and their neuropathy mutants ... 116
Figure 4-7: Native gel electrophoresis of CCT5 and its neuropathy mutant ... 117
Figure 4-8: Far-UV circular dichroism scans and thermal denaturation of CCT5 and its neuropathy mutant ... 118
Figure 4-9: ATP hydrolysis of CCT5 and its neuropathy mutant ... 120
Figure 4-10: Aggregation suppression of HγD-Crys by CCT5 and its neuropathy mutant ... 121
Figure 4-11: SDS-PAGE and quantification of HγD-Crys refolded by CCT5 and its neuropathy mutant ... 123
Figure 4-12: Mutant huntingtin aggregation suppression by CCT5 and its neuropathy mutant ... 125
Figure 4-13: Quantification of β-actin refolded by CCT5 and its neuropathy mutant ... 126
Figure 4-14: Variations in protein concentration and ionic strength of β-actin refolded by CCT5 and its neuropathy mutant ... 127
Figure 5-1: HγD-Crys mutants chosen fall into three sets ... 135
Figure 5-2: HγD-Crys aromatic pair mutants suppressed by Mm-Cpn ... 140
Figure 5-3: HγD-Crys aromatic pair mutants refolded to native-like state by Mm-Cpn ... 143
Figure 5-4: HγD-Crys mutants refolded by Mm-Cpn have native-like fluorescence ... 144
Figure 5-5: HγD-Crys interface pair mutants suppressed and refolded to native-like state by Mm-Cpn ... 145
Figure 5-6: HγD-Crys hydrophobic core mutants suppressed and refolded to native-like state by Mm-Cpn ... 147
Figure 5-7: Most HγD-Crys mutants refolded to higher levels than WT HγD-Crys ... 148
Figure 8-1: Immunoblot SDS-PAGE of sucrose gradient ultracentrifugation of CCT1-CCT4 .. 193
Figure 8-2: Immunoblot SDS-PAGE of sucrose gradient ultracentrifugation of CCT5-CCT8 .. 194
Figure 8-3: Quantified densities of full-length CCT species for each set of sucrose ultracentrifugation gradients ... 198
Figure 8-4: Quantified densities of fragmented CCT species for each set of sucrose ultracentrifugation gradients ... 201
Figure 8-5: Heat maps of CCT subunit complex formation alone, with Mm-Cpn, CCT4, or CCT5 ... 202
Figure 8-6: Possible models for TRiC formation assuming assembly is started from CCT4 or CCT5 homo-oligomers ... 206
Figure 8-7: CCT5 and human TRiC suppress aggregation of mutant huntingtin while CCT4 and Mm-Cpn do not ... 213
LIST OF TABLES
Table 1-1: CCT subunits implicated in substrate binding varies for different substrates ... 41
Table 1-2: Mutations in chaperonin subunits lead to human disease ... 48
Table 4-1: Mutations in chaperonin genes leading to neuropathy diseases ... 101
Table 5-1: All HγD-Crys mutants are destabilized compared to WT HγD-Crys ... 139
Table 5-2: Kinetics of Mm-Cpn suppression of HγD-Crys aggregation vary by mutant ... 141
Table 8-1: Human CCT subunit expressed from eight different chromosomes ... 186
Table 8-2: Antibodies against the CCT subunits ... 191
LIST OF ABBREVIATIONS (in alphabetical order) ANC: actin non-complementing
AP: alkaline phosphate BBS: Bardet-Biedl syndrome BCA: bicinchoninic acid assay BIN: binucleated BME: β-mercaptoethanol CD: circular dichroism CCT: chaperonin containing TCP-1 DHFR: dihydrofolate reductase DTT: dithiothreitol
E. coli: Escherichia coli
EDTA: ethylenediaminetetraacetic acid FSC: Fourier shell correlation
GdnHCl: guanidine hydrochloride HγD-Crys: human γD-crystallin Hsc: heat shock cognate Hsp: heat shock protein
HSPD1: human mitochondrial Hsp60 HtpG: high temperature protein G Htt: huntingtin
IP: immunoprecipitation
IPTG: isopropyl-β-thiogalactoside mHtt: mutant huntingtin
Mm-Cpn: Methanococcus maripaludis chaperonin NAC: nascent-chain associated complex
NaPi: sodium phosphate
NEF: nucleotide exchange factor OE: overexpression
PDB: protein data bank PEI: polyethelenimine
PVDF: polyvinylidene difluoride
18 RuBisCO: ribulose-biphosphate carboxylase sHSP: small heat shock proteins
SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis SEC: size exclusion chromatography
TAP: TCP-1 associated proteins
TCP-1: Tailless Complex Polypeptide-1 TEM: transmission electron microscopy TLC: thin layer chromatography
TPR: tetratricopeptide repeat TRiC: TCP-1 Ring Complex
Tris: Tris(hydroxymethyl)-amino methane
WD40: 40 amino acid sequences ending in tryptophan and aspartate residues WT: wild-type
CHAPTER 1:
20 Protein Folding and Aggregation
The cell cytoplasm is a crowded space with a high concentration of macromolecules, organelles, and small molecules. In this environment, not all newly synthesized proteins can fold up into their native conformations. Roughly 30% of the proteins in both prokaryotic and eukaryotic cells misfold or are degraded after translation, making protein misfolding a serious obstacle for cell viability and reproduction (Schubert et al. 2000; Hartl and Hayer-Hartl 2009). This failure to assume native state often leads to partially folded states that can result in aggregation or the formation of toxic species (van den Berg et al. 1999; Slavotinek and Biesecker 2001; Ellis 2003). These toxic species can be amyloid in nature, such as in prion disease and other neurodegenerative disease (Berthelot et al. 2013; Olanow and Brundin 2013; van der Putten and Lotz 2013). On the other hand, the toxic species may also be non-amyloid (frequently termed “amorphous”) aggregation as in cataract (Wang and King 2010; Moreau and King 2012). There are also cases where the oligomer species is toxic, as in many of the neurodegenerative diseases such as Alzheimer’s Disease, Huntington’s Disease, and Parkinson’s Disease (Frid et al. 2007; Berthelot et al. 2013; Denny et al. 2013; Margulis et al. 2013). Whatever the toxic conformation, many cell types have systems in place to decrease or eliminate this species.
Molecular chaperones in the cell guide nascent and misfolded proteins to their native states or protect misfolded proteins from aggregation (Figure 1-1) (Feldman and Frydman 2000; Frydman 2001; Slavotinek and Biesecker 2001; Lee and Tsai 2005; McClellan et al. 2005; Ellis 2006; Broadley and Hartl 2009; Chen et al. 2011; Hartl et al. 2011). Direct experiments suggest that up to 50% of newly synthesized proteins in both yeast and E. coli utilize chaperone assistance (Teter et al. 1999; Feldman and Frydman 2000; Hartl and Hayer-Hartl 2009; Hartl et al. 2011). Not only do chaperones act on nascent proteins, but they can also recognize and act on proteins that become unfolded due to cellular stress or inherent destabilization from mutations (Gregersen and Bross 2010). Chaperones can also pass substrates that are unable to fold onto the proteasome system, therefore ridding the cell of these species that have potential to aggregate (Chen et al. 2011; Hartl et al. 2011). Chaperones are divided into two classes: ATP-dependent chaperones (termed foldases) and ATP-independent chaperones (also termed small heat shock proteins or holdases) (Frydman 2001; Hartl et al. 2011). The ATP-dependent chaperones are actually able to bind to partially unfolded or misfolded substrates and help them in folding to more native-like states.
Figure 1-1: Effect of chaperones on protein folding and aggregation energy landscape
Protein conformations from the unfolded state (top) are shown as either having intermolecular (left, orange) or intramolecular contacts (right, blue). Intramolecular contacts are shown as
partially folded states and the native state. Intermolecular contacts are shown as amyloid, oligomers, and non-amyloid aggregation. Chaperones assist (green arrow) in driving proteins
from partially folded states into their native states, and inhibit (red bar-headed arrow) proteins from going from the partially folded state to amyloid, oligomers, and non-amyloid aggregation. Figure based off of Hartl et al. reviews (Hartl and Hayer-Hartl 2009; Hartl et al. 2011).
Unfolded State Native State Partially Folded States Oligomers Amyloid Non-amyloid Aggregation
Intermolecular contacts Intramolecular contacts
En
e
rg
y
22 ATP-Dependent Chaperones
There are three main classes of chaperones that use ATP to drive proteins from partially folded states to their native states. These are heat shock protein (Hsp) 70, Hsp90, and Hsp60 (chaperonins). They act in the cell at different times in the folding pathways, with Hsp70 acting as the first chaperone to encounter a protein off of the ribosome, and the chaperonins and Hsp90 acting downstream of Hsp70 (Hartl et al. 2011).
Hsp70
The first chaperones to bind to newly synthesized proteins and influence their folding in an ATP-dependent manner are part of the Hsp70 family (DnaK in prokaryotes) (Frydman 2001; Hartl et al. 2011; Mayer 2013). In eukaryotes, the nascent-chain associated complex (NAC), a cluster of Hsp70-family chaperones at the ribosome exit site, greets the polypeptide chain (Hartl et al. 2011). On the other hand, in prokaryotes, a protein called trigger factor binds to almost all chains coming out of the ribosome, before these substrates can be transferred to DnaK (Teter et al. 1999; Hartl et al. 2011). Hsp70 that is constitutively expressed is termed Heat shock cognate (Hsc) 70 (Saibil 2013). However, other isoforms of Hsp70 can be induced under stress conditions (Saibil 2013). Many human cancers overexpress Hsp70 family proteins and this overexpression is linked to poor prognosis (Murphy 2013). Therefore, Hsp70 has been a recent therapeutic target (Assimon et al. 2013).
Hsp70 has two domains: the N-terminal nucleotide-binding domain (43 kDa) and the C-terminal substrate-binding domain (27 kDa) (Figure 1-2) (Mayer 2013). Using ATP binding and hydrolysis, it cycles between two conformational states: an ATP-bound low-affinity open state where the substrate has high association and dissociation rates in the substrate-binding domain, and a high-affinity closed state after ATP is hydrolyzed where the substrate has low on and off rates (Mayer 2013). In general, Hsp70s have low ATP hydrolysis and release rates, but with the help of a nucleotide exchange factors (NEFs; specifically GrpE in prokaryotes), the ADP can be released, opening the Hsp70 and releasing the substrate (Mayer 2013). In addition to NEFs, Hsp70s are assisted by other co-chaperones such as Hsp40 (DnaJ in prokaryotes), which help bind substrates, bring them to Hsp70 and increase the rate of ATP hydrolysis (Hartl et al. 2011). Hsp40 has a conserved J-domain, that interacts with the nucleotide-binding domain of Hsp70 (Clare and Saibil 2013).
The substrate-binding domain of Hsp70 specifically binds stretches of five hydrophobic amino acids with positive amino acids on either side, which occur on average every 40 amino acids in many globular proteins (Mayer 2013). These recognition elements are most likely
buried in the hydrophobic core of folded proteins. Therefore, Hsp70 only acts on unfolded or partially folded chains. By binding to these substrates, Hsp70 decreases their potential to aggregate in the cell. When released, the substrate is more competent to fold by burying the bound hydrophobic patches into its core (Hartl et al. 2011). If the substrate cannot fold, it may be bound again by Hsp70, passed on to the other chaperones in the cell, or targeted for degradation (Hartl et al. 2011; Saibil 2013).
24 Figure 1-2: Hsp70 structure and states
Hsp70 has two domains: the substrate binding domain (cyan) and the nucleotide binding domain (magenta). It cycles between a low-affinity ATP-bound state where the substrate binding site is exposed and there are high rates of substrate association and dissociation (A) and a high-affinity state where the substrate binding site is closed and therefore the substrate is tightly bound (B). The substrate and nucleotide binding sites of both states are labeled. Low-affinity state: PDB: 4B9Q; high-affinity state: PDB: 2KHO.
Hsp90
Hsp90 acts on substrates (specifically termed clients) to regulate their conformations and mature them (Hartl et al. 2011). These clients are involved in crucial cell processes such as signal transduction, innate and adaptive immunity, and protein trafficking (Picard 2008; Taipale et al. 2010). Because of its central role in important cell functions, Hsp90 seems to act as a buffer for protein evolution, allowing destabilized proteins to fold and mature (Rutherford and Lindquist 1998; Lindquist 2009). Like Hsp70, Hsp90 is also overexpressed in most human cancers, making it a robust therapeutic target (Whitesell and Lindquist 2005). Interestingly, while Hsp90 is essential in eukaryotic cells, the bacterial homolog high temperature protein G (HtpG) is not essential and Archaea do not possess any Hsp90 homologs (Taipale et al. 2010).
Hsp90 is made up of three domains: a N-terminal ATP binding domain, a middle domain, and a C-terminal dimerization domain (Figure 1-3) (Taipale et al. 2010; Clare and Saibil 2013). It functions as a homo-dimer and cycles through two conformations: an open conformation where only the terminal domains interact and a closed ATP-bound conformation in which both the C-terminal and N-C-terminal domains are interacting and the N-C-terminal domains are twisted relative to each other (Clare and Saibil 2013). Clients on their own seem to bind to any of the three domains, but co-chaperones preferentially bind to the C-terminal domain (Clare and Saibil 2013). These co-chaperones have tetratricopeptide repeat (TPR) domains that bind to the MEEDV sequence in Hsp90 C-terminus (Taipale et al. 2010; Hartl et al. 2011). The co-chaperones are specific for their clients, increasing the diversity of interactions by Hsp90 (Taipale et al. 2010). These co-chaperones also regulate the ATP hydrolysis cycle of Hsp90, allowing clients to undergo various conformational transitions that lead to their maturation (Hartl et al. 2011).
26 Figure 1-3: Hsp90 structure and states
Hsp90 has three domains: the N-terminal ATP-binding domain (orange), the middle domain (blue), and the C-terminal dimerization domain (green). It cycles between an open state (A) where substrates can easily associate and dissociate and an ATP-bound closed state where substrates are tightly bound (B). Open state: PDB: 2IOQ; closed state: PDB: 2CG9.
Chaperonins
Like Hsp70, chaperonins bind partially folded substrates and through conformational changes, induced by ATP-binding and hydrolysis, release a more native-like substrate (Frydman 2001). However, unlike Hsp70, substrate folding by chaperonins occurs in a cavity within the chaperonin where the substrate is sequestered, in whole or in part, away from the environment of the cell (Tang et al. 2006). Since substrates up to 60 kDa can fold in this cavity, whole proteins and domains can be recognized and encapsulated (Xu et al. 1997). Little is understood about how exactly the substrate achieves a more native-like conformation within this cavity (Gershenson and Gierasch 2011).
Chaperonins are composed of two back-to-back rings with seven to nine subunits each (Horwich et al. 2007). Each subunit is divided into three domains: equatorial, intermediate, and apical (Braig et al. 1994). ATP hydrolysis and inter-ring negative allostery occur at the equatorial domain (Spiess et al. 2004). The hinge-like intermediate domain connects the equatorial and apical domains (Braig et al. 1994). Substrates are recognized via the apical domains, taken into the cavity, folded, and then released through the same opening (Horwich et al. 2007). The chaperonins are divided into two groups: group I (found in prokaryotes and in the chloroplasts and mitochondria of eukaryotes) and group II (found in archaeal and eukaryotic cytosols) (Frydman 2001). Group I chaperonins bind approximately 12% and group II chaperonins bind 9-15% of newly synthesized proteins in their respective cytosols (Ewalt et al. 1997; Thulasiraman et al. 1999; Frydman 2001). Many features of the structure and mechanism of group I chaperonins have been elucidated; however, studies of group II chaperonins are more limited due to their complexity.
28 Group I Chaperonins
History
In the late 1960s and early 1970s, many laboratories were investigating phage mutations that led to defects in lysogenic or lytic propagation or recombination (Georgopoulos 2006). Costa Georgopoulos and Ira Herskowitz decided to look at mutations in E. coli that would hinder the development of λ phage (Georgopoulos 1971). They used C600 E. coli strain containing a
supE amber nonsense suppressor gene (Georgopoulos 1971). After mutagenesis, the bacteria
were spread on LB agar plates with λcI- and 434cI- phage (Georgopoulos 1971). These phage were chosen because they do not lysogenize so their infection would absolutely result in death of the host, and they have distinct host-surface receptor attachment so host surface receptor mutations would only be observed if both surface receptor genes contained mutants. The concentration of phage to apply to the bacteria was very carefully chosen as to get enough bacterial colonies lysed by the phage (rather than just “nibbled”), but not so many that all of the bacterial colonies are killed (Georgopoulos 1971; Georgopoulos 2006). At the correct conditions, most of the bacteria were killed by the phage, but every thousandth colony was large and unaffected by the phage. The bacteria in these colonies had mutations that blocked phage growth and were therefore named gro mutants (Georgopoulos 1971). Two of these mutants,
gro15 and groC3, were studied further and eventually identified as part of two distinct
chaperone systems (Georgopoulos 1971).
Georgopoulos observed that most λcI- phage did not form plaques on the Gro15 E. coli mutant, but with a frequency of about 10-7, plaques did form (1971). Therefore, the phage that formed these plaques were able to compensate for the Gro15 mutation. The phage mutants were purified and tested on a variety of other amber-suppressing and non-suppressing E. coli strains (Georgopoulos 1971). About 20% of these λ phage mutants could not kill the non-suppressing wild-type (WT) E. coli strains but did form plaques on the amber-non-suppressing bacterial strains. This lead to the conclusion that the compensatory mutants found in the λ phage had a suppressible amber mutation in an essential phage gene (Georgopoulos 1971). Herskowitz tested a collection of essential λ phage amber mutants for complementation with the λ phage Gro15 compensatory mutations, and found that the λ phage Gro15 compensatory mutations mapped to gene P of λ phage (Georgopoulos 1971; Georgopoulos 2006). This gene was known to be responsible for λDNA replication. Two other λ phage mutants that possessed mutations in gene P, λPam3 and λPam80, were shown to also form plaques on C600 supE
gro15 mutant E. coli (Georgopoulos 1971). However, many of the isolated λ phage Gro15
phage compensatory mutants were mapped, the mutations in E. coli could further be studied. Herskowitz showed that most of the Gro15 E. coli mutant (and a few other mutants with the same phenotypes named groP mutants) mapped to the dnaB gene (Georgopoulos 1971). Georgopoulos and Herskowitz reasoned that for λDNA replication to occur, the λP protein must interact with DnaB protein of E. coli (Georgopoulos 1971). As DnaB is a helicase, it is not surprising that they found this replication-associated protein in their genetic screen (Zylicz et al. 1984).
There was one groP mutant (groPAB756) that did not map to DnaB, but instead mapped near the thr locus of E. coli and was renamed groPC756 (Georgopoulos 1977). Georgopoulos went on to show that this mutant was responsible for both phage growth defects and host temperature sensitivity, therefore concluding that this GroPC protein forms a complex with the λP protein (1977). Concurrently, Michael Feiss’s laboratory isolated an E. coli mutant defective in λ phage growth and bacterial growth temperature sensitivity (Sunshine et al. 1977). They showed that this mutant (groPC259) was closely linked to groPAB756 (Sunshine et al. 1977). Around the same time, Saito and Uchida were isolating E. coli mutants (named grp for groP-like), which interfere with λDNA replication and are temperature sensitive (1977). As with Georgopoulos and Herskowitz, some of their mutants mapped to DnaB, but one class of mutants (grpC) fell near groPC756 and groPC259 (Saito and Uchida 1977). In collaboration, it was discovered that the groPC and grpC mutants fell into two distinct complementation classes:
groPC756 and groPC259, which Saito and Uchida renamed dnaK and dnaJ, respectively
(Yochem et al. 1978). Saito and Uchida went on to isolate the essential nucleotide exchange factor grpE from another class of their mutants, therefore rounding out the DnaJ/DnaK/GrpE chaperone system (1978).
The other original gro mutant identified by Georgopoulos and Herskowitz was groC3. Georgopoulos et al. isolated compensatory λ phage mutants, about 30% of which had amber suppressing mutations or temperature-sensitive mutations in gene E of λ phage (1973). The gene encoded the capsid of the phage and was referred to as λε, therefore the mutant was renamed GroEAC3 (and similar mutants were designated GroE mutants) (Georgopoulos et al. 1973). The GroE mutants were split into two classes: GroEA on which λεA plaques but not λεB plaques formed, and GroEB on which λεB plaques but not λεA plaques formed (Georgopoulos et al. 1973). Both λεA and λεB phage were isolated as compensatory mutants of GroE bacterial mutants. Both bacterial mutant classes had problems growing at high temperatures. While all of λεA phage mutants mapped to the E gene, the λεB phage mutants fell in both the E and B genes (Georgopoulos et al. 1973). Transmission electron micrographs (TEMs) of groE mutant
30
bacteria infected with WT λ phage showed that the λ capsid (E gene) was incorrectly assembled, showing that groE bacteria affected the λB protein function (Georgopoulos et al. 1973).
One groE bacterial mutant, GroEA44, not only resisted λ phage growth, but also T4 phage growth. As before, T4 plaque forming phage mutations were present at a frequency of 10-7, and referred to as T4ε. One of these mutants, T4ε1, could no longer grow on groE mutants (groEB515), which propagated WT T4 phage (Georgopoulos et al. 1972). Using complementation tests with known T4 amber suppressing mutants, this phage mutation was mapped to gene 31 of T4 (which makes Gp31) (Georgopoulos et al. 1972). Therefore, it was concluded that Gp31 of T4 phage interacted with the groEA44 gene product (Georgopoulos 1971). Concurrently, Ulrich Laemmli et al. showed that Gp31-defective T4 phage had no heads and that without Gp31, the T4 capsid protein, p23, aggregated into insoluble clumps (1970). Employing λgroE+-infected cells, which could overexpress the GroE protein, Georgopoulos and colleagues found that GroE was a protein of approximately 60 kDa, had a tetradecameric structure and could hydrolyze ATP (Georgopoulos and Hohn 1978; Hendrix 1979).
Further investigation of groE mutants showed that they fell into two complementation groups (Tilly et al. 1981). These were renamed GroEL (large 60 kDa product) and GroES (small 15 kDa product) (Tilly et al. 1981). The GroES product was found to be a co-chaperone of GroEL, and their interaction was ATP-dependent (Chandrasekhar et al. 1986). The T4 phage protein Gp31 is another co-chaperone essential for Gp23 capsid formation and takes over the function of GroES (Ang et al. 2000). The Gp31 co-chaperone is approximately the same size as GroES, but its interaction with GroEL creates a larger cage in which Gp23 can fit and fold (Bakkes et al. 2005; Clare et al. 2006). Interestingly, while λ phage, and obviously E. coli, require the GroES co-chaperone, T4 phage does not (Ang et al. 2000). Therefore, isolation of host mutants by T4 infection would not have identified the crucial GroES co-chaperone. As the temperature sensitive properties of GroEL/GroES were part of the criteria of their discovery, it was not until almost ten years later that their requirement for E. coli cell growth at all temperatures was verified (Fayet et al. 1989).
While Georgopoulos was investigating the effect of bacterial mutation on phage assembly, the laboratory of R. John Ellis was researching a protein that bound and helped assemble Ribulose-biphosphate carboxylase (RuBisCO), a protein complex involved in carbon fixation. RuBisCO subunit binding protein was isolated in a much more biochemical way than GroEL. RuBisCO subunit binding protein was found to bind to RuBisCO in isolated pea chloroplasts, but not be part of RuBisCO itself (Hemmingsen et al. 1988). Biochemical studies
showed that it was made up of 60 kDa and 61 kDa subunits making an approximately 700 kDa complex with ATPase activity (Hemmingsen and Ellis 1986). With the help of protein sequences, Ellis and Georgopoulos assembled their findings to conclude that GroEL and RuBisCO subunit binding protein were members of the same family (Hemmingsen et al. 1988). Around the same time, McMullin and Hallberg had concluded that the protein they found in the mitochondria of Tetrahymena thermophila was homologous to GroEL (1987; 1988). Ellis, Georgopoulos, and colleagues coined the term “chaperonin” for this family of proteins, derived from the term “molecular chaperone” that Laskey used for nucleoplasmin, a protein that bound histones and helped assemble them onto DNA (Laskey et al. 1978). The term “chaperonin” was quickly accepted as work from the Hartl and Horwich groups on the mitochondrial Hsp60 in yeast employed that term in publication shortly afterwards (Cheng et al. 1989).
Structure and Function
The most studied of the bacterial group I chaperonins, GroEL from E. coli, consists of two back-to-back rings of seven 57 kDa identical subunits each with an inner cavity volume of 85,000 Å3 (Figure 1-4A) (Braig et al. 1994; Fenton and Horwich 1997). GroEL is a stress-induced chaperone like Hsp70 and Hsp90 (Horwich et al. 2007). GroEL requires the cofactor GroES, or Hsp10, which acts as a lid to close the cavity when a substrate protein is bound and encapsulated (Hunt et al. 1996). GroES is a dome-shaped structure composed of seven individual subunits (Chen et al. 1994; Fenton and Horwich 1997). When GroES binds to GroEL, the volume of the cavity expands to about 175,000 Å3 (Figure 1-4B), therefore allowing for larger proteins to fit in the cavity (Xu et al. 1997). Substrate binding occurs when exposed hydrophobic residues in the substrate folding intermediate make contacts with the hydrophobic residues in the apical domain of the subunits of one ring of GroEL (cis ring; Figure 1-4C, 2) (Lin and Rye 2006). When ATP binds to the cis ring, the substrate experiences global stretching and local segmental tightening (Figure 1-4C, 3) (Sharma et al. 2008; Kim et al. 2010). GroES can bind to the hydrophobic residues of the apical domains where the substrate is bound, pushing the substrate into the cavity of the ring (Figure 1-4C, 4) (Frydman 2001). The substrate is encapsulated in the cavity and refolded for approximately 15 seconds as the ATP is hydrolyzed (Figure 1-4C, 5) (Tang et al. 2006). ATP and substrate binding to the trans ring cause GroES dissociation and substrate release (Figure 1-4C, 6) (Lin and Rye 2006).
The released, more native-like, substrate may spontaneously assume its native state or may need another round of encapsulation (Weissman et al. 1994). Due to its proximity to the apical domain when released, the substrate can easily rebind to the cis ring of the chaperonin
32
when the trans ring completes its cycle (Weissman et al. 1994). The actual substrate folding mechanism inside the chaperonin is unknown, but there is some evidence that sequestration of the substrate from the environment produces an Anfinsen cage, where the substrate can freely fold (Ellis 2003; Apetri and Horwich 2008; Horwich et al. 2009). Other studies have shown that positively charged residues lining the cavity of GroEL play an active role in substrate folding, especially for larger proteins that directly contact these residues (Xu et al. 1997; Tang et al. 2006). Studies of GroEL substrates show that GroEL prefers substrates with multiple α/β domains with buried hydrophobic β-sheets (Houry et al. 1999; Kerner et al. 2005; Hirtreiter et al. 2009). These architectures are slow to fold and are prone to aggregation (Kerner et al. 2005; Fujiwara et al. 2010; Raineri et al. 2010).
Figure 1-4: Group I chaperonin structure and mechanism
Each subunit has equatorial (blue), intermediate (green), and apical domains (purple). The cavity of the group I chaperonin GroEL expands from the open state (A; PDB: 1OEL) to the closed state (B; PDB: 1AON) in complex with the GroES lid (red). The mechanism of a group I chaperonin involves an ATP hydrolysis transition state in which the substrate is folded (C, see text for details).
34 Group II Chaperonins
History
Genetically unique tailless mice mutants have been studied since the early 1930s (Chesley and Dunn 1936). Two mutations have been isolated from the work: the dominant T-locus mutation and the recessive lethal t allele mutants (Bennett 1975). WT mice have normal length tails, whereas the double heterozygous T/t mice have the tailless phenotype. Further, T/+ mice have short tail but t/+ mice have normal tails. Additionally, tx/ty males were sterile and the sperm transmission ratios of tx/+ males resulted in as many as 99% of the progeny received the t haplotype (Bennett 1975). The double heterozygous T/t mice bred true since both T/T and t/t mice died as embryos (Bennett 1975). This was the first balanced lethal mammalian system (Waelsch 1989). Therefore, the tailless phenotype seems to arise from the interaction of the T-locus and the t allele (Bennett 1975).
It was not until the 1970s that the mapping of the t complex could be achieved. The t chromosome 17 is different than the WT chromosome 17 in a region of inversions (named the t complex) spanning 30 Mbs (Bennett 1975). This region contains around 100 genes (Bennett 1975). As defects were seen in spermatogenesis of male mice, Silver et al. isolated spermatogenic cells from WT and t haplotype mice (1979). Cells were labeled with 35 S-methionine in vitro, the protein fraction was isolated, and the proteins were separated using two-dimensional isoelectric focusing and gel electrophoresis (Silver et al. 1979). They found that a single abundant protein spot, p63/6.9, differed between WT and t haplotype mice. In WT mice, p63 was a bit more acidic, therefore labeled p63/6.9a, whereas in t haplotype mice, the protein was labeled p63/6.9b (Silver et al. 1979). They extended their work to other cell types (splenocytes, thymocytes, and two carcinoma cell lines) to find that these cell types only contained the WT p63/6.9b (Silver et al. 1979). This protein was coined Tailless Complex Polypeptide 1 (TCP-1) (Silver 1985). Although research continued on the t complex in terms of its implication in development (Waelsch 1989) and transmission ratio (Willison 1986), how exactly it caused the tailless phenotype and whether TCP-1 actively played a role is still unclear.
To better understand t haplotype chromosome evolution, Keith Willison and colleagues cloned TCP-1b (WT) and part of TCP-1a (t haplotype) genes. They found no sequence similarity to known proteins and at least six nucleotide differences between the two genes (Willison et al. 1986). Willison’s laboratory went on to study TCP-1 in the cell and found (due to nonspecific antibodies (Lewis et al. 1992)) that it associated with the cytoplasmic part of the Golgi membrane (Willison et al. 1989). Therefore, they concluded that TCP-1 plays a role in transport of proteins through the exocytic pathway (Willison et al. 1989). Working on microtubules in
yeast, Ursic and Culbertson found that an essential gene in yeast shared sequence identity with mouse TCP-1 (Ursic and Culbertson). They isolated and characterized a cold sensitive yeast TCP-1 mutant and concluded that TCP-1 affected the microtubules responsible for spindle pole body positioning (Ursic and Culbertson 1991). Despite their logical conclusions based on their data, both groups were misled. At this time, the publication by Georgopoulos and Ellis outlining the chaperonins gave both groups the direction they needed to continue studying the actual role of TCP-1 in the cell (Hemmingsen et al. 1988). Comparing the protein sequence of TCP-1 to those of the rest of the chaperonins, Ellis speculated that TCP-1 was the eukaryotic cytosolic chaperonin (1990). Unpublished data mentioned in his 1990 review bolsters this theory, because the TCP-1 antibody recognized proteins in the crude extracts of pea leaves but not chloroplasts or mitochondria (Ellis 1990).
While neither Georgopoulos nor Ellis crossed over to the group II chaperonin field, the team of Ulrich Hartl and Arthur Horwich who got their start studying mitochondrial Hsp60 in yeast, stumbled upon the connection between the archaeal thermosome and TCP-1 (Trent et al. 1991). They reasoned that thermophilic factor 55 (TF55) was part of the chaperonin family by showing structural similarity to GroEL via electron microscopy and functional similarity to GroEL as both chaperonins form a complex with a substrate, 35S-methionine labeled Su9-DHFR (part of subunit 9 of F0-ATPase fused to dihydrofolate reductase), diluted out of denaturant (Trent et al. 1991). Additionally, protein sequence similarity was shown to be strongest between TF55 and TCP-1 than any other chaperonins, suggesting that they form a subclass of chaperonins, possibly specialized in cytoskeletal assembly (Trent et al. 1991). This paper propelled the study of TCP-1 as a chaperonin, although its chaperoning function was only speculative at this time.
In the summer of 1992, two papers published back-to-back in Nature, verified the chaperoning function of TCP-1. The first paper, from the tubulin-focused Sternlicht laboratory in collaboration with Horwich, reported that in rabbit reticulocytes, newly made tubulin subunits enter a 900K complex before being competent to assemble into microtubules (Yaffe et al. 1992). This complex consisted of a set of polypeptides between 55-60K and one of which reacted with the TCP-1 antibody (Yaffe et al. 1992). Therefore, the authors concluded that tubulin interacted with the TCP-1 complex to acquire a more assembly-competent form (Yaffe et al. 1992). This was concurrently shown for actin assembly in rabbit reticulocytes by Nicholas Cowan’s laboratory (Gao et al. 1992). The second article in the same issue was a report from the Willison lab preliminarily characterizing human and mouse TCP-1 (Lewis et al. 1992). They found that in both species, TCP-1 made a complex of approximately 900K (Lewis et al. 1992). Additionally, TCP-1 associated with four to six unidentified polypeptides and two Hsp70 homologs, coined
36
TCP-1 associated proteins (TAPs) (Lewis et al. 1992). They speculated that the unidentified polypeptides may be TCP-1 related proteins which were just being found in a variety of organisms by sequence similarity (Lewis et al. 1992). Due to this heterogeneity of the TCP-1 complex, and the observation that TCP-1 levels were not increased in response to stress, the Willison lab concluded that TCP-1 was unique from GroEL (Lewis et al. 1992).
At this time, there was only indirect evidence of the chaperoning properties of TCP-1 complex. Judith Frydman in the Hartl Lab reported that TCP-1, renamed TCP-1 Ring Complex (TRiC), refolded unfolded substrates in vitro and definitively showed that TRiC did not need the Hsp10 co-chaperonin required for GroEL-assisted protein folding (1992). Frydman et al. demonstrated that purified bovine TRiC associated with at least five other polypeptides which were sequenced to show at least 40% identity with TCP-1, suggesting that the TRiC is made up of a number of homologous proteins (Frydman et al. 1992). Additionally, Frydman et al. showed that TRiC can bind to and refold firefly luciferase from denaturant whereas GroEL can bind denatured luciferase but not refold it (1992). This was the first direct evidence verifying that TRiC functions as a chaperonin (Frydman et al. 1992). While this showed that TRiC had the ability to fold substrates other than actin and tubulin, there was still only evidence of TRiC-assisted actin and tubulin folding in the cell (Sternlicht et al. 1993).
The eight subunits of TRiC were first identified by Rommelaere, et al. and subsequently sequenced and mapped by the Hartl and Willison laboratories (Rommelaere et al. 1993; Kubota et al. 1994; Li et al. 1994). Rommelaere et al. also showed that bovine TRiC was structurally consistent and functionally identical with rabbit reticulocyte TRiC (1993). All eight human subunits were sequenced by the end of 1994 (Kubota et al. 1995). The Willison lab renamed TRiC to CCT (Chaperonin Containing TCP-1) (Kubota et al. 1994). Although the term TRiC is more widely used for the complex, CCTx is commonly used to designate subunit x of the complex.
While TCP-1 (now CCT1) was identified from its difference between t haplotype and WT mice, other subunits were also found concurrently through genetic screens. The group of Huffaker was searching for mutants of tubulin that lead to binucleated cells (cells with two nuclei produced by defective spindles) (Chen et al. 1994). Two of their mutants, BIN2 and BIN3 (binucleated), were further characterized, and mapped to CCT3 and CCT2, respectively (Chen et al. 1994). In the meantime, while searching for temperature sensitive mutations of actin in S.
cerevisiae, the Drubin laboratory found non-complementing extragenic mutants of
actin-interacting proteins (Vinh et al. 1993). One of their mutants, ANC2 (actin non-complementing 2), was mapped to CCT4 (Vinh and Drubin 1994). Recently, the actual ANC2 mutant was identified
to be CCT4 G345 and characterized to abolish ATP-induced allostery of TRiC (Shimon et al. 2008).
Structure and Function
The group II chaperonin mechanism is different from the group I chaperonin mechanism due to the structural differences of the two groups. The archaeal group II chaperonins consist of two 7-9 subunit rings that have 1-3 different subunits, while the eukaryotic group II chaperonin Tailless Complex Polypeptide-1 (TCP-1) Ring Complex (TRiC) consists of two identical rings, each with eight different subunits (Bigotti and Clarke 2008). While the archaeal chaperonins can be stress induced like the group I chaperonins, the eukaryotic TRiC is not a stress-inducible chaperone (Horwich et al. 2007). Overall, the subunits have the same domain organization as those of GroEL, but since group II chaperonins do not require a cofactor like GroES, the apical domain has a helical protrusion which acts as a built-in lid (Figure 1-5A) (Reissmann et al. 2007). Unlike expanding in the group I chaperonins, the volume of the cavity of the group II chaperonins contracts from about 350,000 Å3 in the open state to 130,000 Å3 in the closed state (Figure 1-5B) (Huo et al. 2010; Pereira et al. 2010). Although group II chaperonins do not have a lid-like co-chaperone, they do interact with prefoldin, a co-chaperone that binds some substrates and brings them to the apical domain (Gutsche et al. 1999; Martín-Benito et al. 2002; Sahlan et al. 2010). Most of the research on prefoldin structure and function has employed the archaeal group II chaperonins, so little is known about this co-chaperone in eukaryotic cells.
The homo-oligomeric archaeal Methanococcus maripaludis chaperonin (Mm-Cpn) has provided a useful group II chaperonin model because it allows for recombinant expression of site-directed mutations (Spiess et al. 2004). Recent studies using a variety of Mm-Cpn mutants have revealed the mechanism of substrate folding differs from that of the group I chaperonin (Douglas et al. 2011). The substrate folding intermediate binds to the cis ring of the chaperonin at the apical domains (Figure 1-5C, 2). ATP binds to the cis ring and the lid begins to close (Figure 1-5C, 3) (Douglas et al. 2011). The ATP hydrolysis transition state (Figure 1-5C, 4) precedes substrate release into the cavity, which is caused by scraping the substrate from the apical domain via lid closing (Figure 1-5C, 5) (Douglas et al. 2011). Once the substrate is released into the cavity of the chaperonin, it folds into a more native-like conformation (Figure 1-5C, 6) (Douglas et al. 2011). The cause of lid opening is unknown but it may be substrate binding at the trans ring (Figure 1-5C, 7).
38
Figure 1-5: Group II chaperonin structure and mechanism
Each subunit has equatorial (blue), intermediate (green), and apical domains (purple) along with a built-in lid (red). The cavity of the group II chaperonin narrows from the open state (A; PDB: 3KO1) to the closed state (B; PDB: 3KFB). The mechanism of a group II chaperonin involves an ATP hydrolysis transition state which precedes substrate release into the cavity for folding (C, see text for details).