Catalytic Upgrading of Biomass through the Hydrodeoxygenation (HDO) of Bio-oil Derived Model Compounds
by Manish Shetty
B.Tech., Indian Institute of Technology, 2011 M.Tech., Indian Institute of Technology, 2011 M.S., Massachusetts Institute of Technology, 2014
SUBMITTED TO THE GRADUATE PROGRAM IN CHEMICAL ENGINEERING IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
AUGUST2017 1_sere
-00. I
0 2017 Massachusetts Institute of Technology. All rights reserved.
Signature redacted
Signature of Author:Manish Shetty Department of Chemical Engineering
4 August 30, 2017
Signature redacted
Certified by: _ _ _ _ _ _RomanYuriy Roman Associate Professor of Chemical Engineering Thesis Supervisor
___________:Signature redacted
Certified by:_________________
William H. Green Prof ssor of Chemical Engineering Thesis Supervisor
Accepted by:
Signature redacted
Patrick Doyle Professor of Chemical Engineering MASSACRUM"SINSTITUTE Graduate Officer of the Department of Chemical Engineering
OF TECHNOLOGY
DEC 13 2017
LIBRARIES
MITLibraries
77 Massachusetts Avenue Cambridge, MA 02139 http://Iibraries.mit.edu/ask
DISCLAIMER NOTICE
Due to the condition of the original material, there are unavoidable flaws in this reproduction. We have made every effort possible to
provide you with the best copy available. Thank you.
The following pages were not included in the original document submitted to the MIT Libraries. This is the most complete copy available.
Catalytic Upgrading of Biomass through the Hydrodeoxygenation (HDO) of
Bio-oil Derived Model Compounds
by Manish Shetty
Submitted to the Department of Chemical Engineering on August 30, 2017 in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy in Chemical Engineering ABSTRACT
Lignocellulosic biomass is an attractive renewable source for fuels and chemicals. Of the many conversion alternatives, catalytic fast pyrolysis has emerged as an attractive technology to convert biomass into fuel additives and value-added chemicals. Current pyrolysis oils or bio-oils are incompatible with refinery streams due to their high acid, water, and water content. The key roadblock in its commercial exploitation is development of catalytic platforms for selective deoxygenation along with minimum hydrogen consumption and carbon loss. Current catalytic solutions including zeolites, and conventional hydrotreating catalysts employ high hydrogen pressures, leading to aromatic ring hydrogenation, and hydrogen consumption. This thesis focusses on developing fundamental catalytic understanding on cheaper and earth-abundant reducible transition metal oxide catalysts for selective hydrodeoxygenation (HDO) of bio-oil derived model compounds using reactivity, computation and characterization studies.
The first section focuses on developing structure-reactivity relationships on bulk and supported MoO3 catalysts for the HDO of lignin-derived model compounds. Characterization reveals that MoO3 undergoes reduction to catalytically inactive MoO2 at a temperature of 673 K, and stabilization of partially reduced MoO3 surface through its partial carburization to oxycarbide phase (MoOxCyHz) at temperatures < 623 K. Thereafter, TiO2 and ZrO2 supports prevent the reduction of dispersed oligomeric MoOx species to catalytically inactive species, enhancing their stability. In addition, the overall catalyst reactivity inversely correlates to the maximum hydrogen consumption temperature during hydrogen temperature programmed reduction (H2-TPR).
Furthermore, a near-monolayer oligomeric MoOx dispersion on ZrO2 support was found to be optimum for HDO reactivity. The second section focuses on developing mechanistic insights into the HDO on bulk and supported MoO3 with the aid of density functional theory (DFT) computations and kinetic studies. DFT computations were carried out on the elementary steps for HDO of acetone-a model compound on pristine U-MoO3 (010) surface to reveal dissociative H2 adsorption on the (010) surface to be the rate-limiting step. Kinetic studies on MoO3 supported on ZrO2 reveal the differences in reaction mechanism and the nature of active sites for HDO on MoO3/ZrO2 as compared to bulk MoO3. The third section focuses on generalizing the low-temperature (< 523 K) selective HDO on other reducible base metal oxides, specifically cobalt oxide and demonstrates oxides to have significantly higher reactivity than base metals for HDO. Finally, lanthanum strontium cobaltite (Lao.8Sro.2CoO3), a perovskite oxide, was demonstrated as a novel HDO catalyst at < 523 K. Overall, this thesis provides a toolkit for developing structure-reactivity relationships on reducible metal oxides for their use as HDO catalysts.
Thesis Supervisors: 1. Yuriy Roman, Associate Professor of Chemical Engineering. 2. William H. Green, Professor of Chemical Engineering
Acknowledgements
I am grateful to many people who have helped me immensely during the past 5 years. It
will not be an exaggeration to say that without these individuals, I would not have finished my PhD work.
Firstly, I am immensely grateful to Yuriy for giving me an opportunity to work in the Roman group. Being in the third batch of PhD students to enter the group, it has been an amazing learning curve and very humbling to see the lab go through the transformation from an early-stage startup to an established lab. Specifically, I thank him for the high standards he set for me and the other lab members to follow from the earliest days. I want to thank him for always aiming high and at times help me see the value in my own work. I also thank him for all the tough discussions and feedback throughout my stay here. As I look back, all those discussions have helped become a better researcher. I believe that I will take with me, an enthusiasm towards solving tough scientific problems from this lab. I am sure Roman group will continue to achieve even greater heights in the coming years.
Secondly, I am immensely fortunate to have Bill as my co-advisor. Technically, Bill is a genius and if there is any person I aspire to emulate, it will be Bill. Bill is immensely humble inspite of being incredibly smart and successful. It is a unique trait that is difficult to find at MIT. Bill has been very supportive professionally and more importantly, in helping me deal with my personal struggles all these years. I will always be grateful to him for his support, especially, in
my final year.
I would also like to thank my committee members Prof. Heather Kulik and Randall Field
(Randy) for their support and useful suggestions and comments during the committee meetings. A special thank you to Randy for being patient in dealing with my requests as he facilitated my
interactions with BP. I would also like to acknowledge all the technical feedback and guidance from Dr. Casey Hetrick and Dr. John Shabaker from BP in all our update meetings and always encouraging and supporting me.
I would like to acknowledge Prof. Claude Lupis for his guidance and mentoring during my
time at Practice School. I would also like to thank Prof. Harry Tuller, Sean Bishop and Prof. Vanessa Mortola for teaching me about Impedance Spectroscopy. I would like to thank Charlie at
CMSE for going out of his way to help me with in-situ PXRD. Joel, Fran, Sydney, Eileen and
Sharece and both Suzannes have helped a lot to make sure admin was not issue that I faced during my stay at MIT. A special shout out to Joel, Gwen and Sharece for their incredible support!
I have to thank Roman Group soldiers, both past and present, for their useful discussion,
collaboration, help, training, mentorship and entertainment. From the past members, Stijn has been a great friend even after MIT. Anthony and Marco made the atmosphere in the lab very lively. Yuran is a bundle of energy and recently been a patient ear and a sounding board as I was exploring career and research options. Sean and Helen set very high standards for us to follow, and it was always great to have long chats with Karthik (the angry one) about life, research, sports or American society. I am grateful for Dr. Teerawit Prasomsri (Air) for his training and mentorship. The initial training helped shape a large part of my thesis. I would also like to thank Tarit, Kenta, Caroline, David, William and Maria for their immense help in my initial years. A special thank you to Chris for helping me negotiate a couple of tricky situations.
Among the current members, I would also like to thank Dr. Daniela Zanchet for her incredible knowledge, help, mentorship, support, and helping me get out of my comfort zone and think outside-the-box. I would like to thank Dr. Karthick Murugappan for being a partner-in-crime on the HDO project. Together, we achieved quite a lot over our stay here and I wish him the best
for his career and life. I thank Jen for making sure I stayed alive in the lab and running to my rescue when I had a lab accident. Eric Anderson is one of the quirkiest and smartest guys that I have met and I always enjoyed all the research talk that I have had with him. The discussions with him have always been fruitful and valuable. Working with Taka has been fun and a little bit of zeolites that I understand is thanks to him! Sharece and Gwen for being the best administrative assistants at MIT! Aaron, Mike, Daniel, Hoyoung, Kim, Stan and Mickey have all made the lab a great place to work every day. A special shout out to Kebar Geleta for being a great UROP in my final semester!
My friends at MIT have made my time at MIT bearable and made sure I did not lose my
sanity! Thank you to Siva, Shamel and Harshad for all the long chats over Chai and being supportive even after MIT. Wenda, Isaac, Andong, Xiaoxue, Lisa, Kathryn, Leslie and Jared for fun times during Practice School. Yamini, Rohit, Vishnu and Anasuya for all the random coffee, lunch and dinners. A special shout out to Sue and Vibhuti for being there for me during some dark times! Dawn and Larry as Housemasters have been amazing and I will always be thankful to Dawn for being ready with a smile and a word of encouragement. Finally, THRA for being an outlet outside lab!
My friends outside MIT have always been there for me inspite of the distance and
time-zones! Puranik, Darunte, Pandu, Akshay, Rahul Srinivasan, Sallu and all the soccer and undergraduate buddies. The list will be endless if I keep writing all your names!
I could not have been where I am without my family. My parents brought me up with
limited money and they made a lot of sacrifices to make sure me and my brother got the education and support we needed. The grit and determination instilled in me has been due to their perseverance. My brother has always set a high standard for me to follow, and showing me how
to never give up. He has been a constant in my life as things have changed and turned. He will always remain a source of inspiration for me!
Last but not the least, I would like to thank the love of my life Ashwini for agreeing to marry a crazy guy and being my partner-in-crime for the last few years. Without you, I would not have successfully navigated the dark times!
Table of Contents
A b s t ra c t ... ii A c k n o w le d g e m e n ts ... iii
T a b le o f C o n te n ts ... v ii
1 In t ro d u c tio n ... 1 2 Insights into the catalytic activity and surface modification of bulk MoO3 during the
hydrodeoxygenation of lignin-derived model compounds into aromatic compounds under low
h y d ro g e n p re ssu re s ... 2 1 3 Reactivity and stability investigation of supported molybdenum oxide catalysts for the
hydrodeoxygenation (HDO) of m -cresol ... 49 4 Structural properties and reactivity trends of molybdenum oxide catalysts supported on zirconia
for the hydrodeoxygenation of anisole ... 109 5 Computational investigation of hydrodeoxygenation (HDO) of acetone to propylene on a-MoO3
(0 1 0 ) s u rfa c e ... 1 4 5 6 Elucidation of the reaction mechanism for the vapor-phase anisole conversion on bulk and
supported zirconia molybdenum oxide catalysts with kinetic analysis and in-situ titrations ... 173 7 Low temperature hydrodeoxygenation (HDO) of anisole on cobalt oxide ... 233 8 Lanthanum strontium cobaltite as a versatile perovskite material for low temperature
hydrodeoxygenation (HDO) of lignin-derived model compounds ... 253 9 C o n clu sio n s a n d O utlo o k ... 28 1
Chapter
1
Introduction
1.1. Challenges in Mitigating Climate Change
Climate change is arguably one of the greatest challenges facing our generation.[1] The rising surface temperatures and increase in sea-levels can potentially lead to catastrophic con-sequences, including submerging of low-lying countries such as Maldives.[2] Domestically, the effects of the rising sea-levels can be seen by the flooding of coastal areas during high tides in the state of Florida.[3] Anthropogenic greenhouse gas (GHG) emissions contribute to the increasing effects of climate change, with ~75% of the global GHG emissions contributed by C02.[4] In the recent past, CO2 emissions have increased by 90% since 1970, with
contribu-tions from combustion of fossil fuels and industrial use accounting for ~80% of the total in-crease, fueled by the increasing global energy demand.[5] Currently, most energy-consuming sectors, especially the transportation sector, rely heavily on non-renewable fossil-based fuels to meet their energy requirements.[4] In 2010, fossil fuels accounted for ~90% of the total energy consumed worldwide.[5] Not only are fossil fuels undergoing fast depletion, but also their use results in the emission of greenhouse gases, which can increase up to 130% by 2050 at current rates.[6] In order to mitigate the effects of climate change, anthropogenic sources of
C02 emissions need to be controlled. However, the global energy demand is not expected to
slow down in the foreseeable future. By 2035, there will be a 30% increase in the energy de-mand, largely fueled by the demands of rapidly growing developing economies: China and India.[7] While fossil fuels will continue be the main source of energy both domestically and
internationally, increasing scientific, industrial and political consensus on the need to mitigate
effects of climate change has facilitated governments to reach agreements to limit CO2
emis-sions, most notably, the recent Paris accord signed in the COP 21 summit by over 197 parties and entering into force in November 2016.[8] Alternative energy sources must be developed to meet the projected energy demand in the future in a sustainable and environmentally con-scious manner.
1.2. Lignocellulosic biomass as a potential near-neutral source of energy
Lignocellulosic biomass is an attractive source of sustainable renewable carbon source for production of liquid hydrocarbon fuels and chemicals.[9] Lignocellulosic biomass comes from plant sources like wood, grass, forest residues, agricultural wastes, wood processing in-dustries etc. and hence does not compete with food crops. [10, 11] With an emphasis on utilizing renewable energy sources for current energy requirement, it becomes desirable to utilize this source of biomass for transportation fuels and chemicals. Governments across the world in the recent past have introduced regulations to compulsorily blend renewable fuel with fuel derived from conventional fossil sources. It is also a renewable source of aromatics4. The US
Environ-mental Protection Agency (EPA) required 7.5 billion gallons of renewable fuel to be blended with gasoline by 2012. [11] The US Department of Agriculture and Oak Ridge National Labor-atory have estimated that the SU can sustainably produce 1.3 x 109 metric tons of dry bio-mass/year which translates to ~4 x 109 barrels of oil energy equivalent (boe).[1 1] Efficient utilization of biomass can potentially reduce the GHG emission and be a near-neutral source of energy.
1.2.1. Composition of lignocellulosic biomass
Lignocellulosic biomass is a complex mixture made up building blocks of mainly cel-lulose (28-55%), hemicellulose (17-35%) and lignin (17-35%), which are oxygen containing organic polymers.[12] The composition of the building blocks varies depending on the source. The lignocellulosic content of some biomass sources are given in Table 1-1. Cellulose is a linear polymer of P-D-anhydroglucopyranose units (AGUs) with basic unit called a cellobiose unit which is made of 2 AGUs.[12] The degree of polymerization may vary between 10-15,000 monomer units. Hemicellulose contains different sugars including glucose, xylose, mannose, galactose and arabinose. [12] These are highly branched and substituted with acetic acid which is one of the major components of bio-oil.[10, 12] The branched nature of hemicellulose gives it its amorphous structure. Lignin has an irregular structure constituting 'hydroxyl' and 'meth-oxyl' substitutes phenylpropane units with hydrophobic and aromatic nature. Lignin is found in higher proportions in woody biomass.[12]
Table 1-1: Lignocellulose content of some biomass sources[12]
Lignocellulose content (wt %)
Biomass species Cellulose Hemicellulose Lignin
Pinewood 46-50 19-22 21-29
Poplarwood 40-46 17-23 21-28
Switchgrass 28-37 23-29 17-20
1.2.2. Strategies for lignocellulosic biomass conversion
Several methods have been explored for the conversion of lignocellulosic biomass into fuels and chemicals, as shown in Figure 1-1. The first route is the gasification of biomass, involving the oxidation of biomass at high temperatures (1100-1500 K) to produce CO-rich
syngas. This must be further upgraded using the Fischer-Tropsch process to produce liquid hydrocarbons with higher carbon number. [11] In addition, this does not allow us to selectively form fuels or a specific platform/target molecule. The second route is the liquid-phase hydrol-ysis of biomass to form monomeric molecules from cellulose and hemicellulose components of biomass. The lignin-component precipitates out of the biomass.[ 1] The monomers hence produced, can be upgraded by either upgrading of monomers to platform molecules or conver-sion of monomeric sugars through a bio-catalytic processes to second-generation bioetha-nol.[l 1] However, the process does not utilize the total biomass content. The third promising route is the pyrolysis of biomass, where the biomass is thermochemically decomposed in the absence of oxygen to form a liquid bio-oil. The liquid yields can be as high as 65%. [20] Py-rolysis along with subsequent upgrading is considered to be the most cost effective and pro-vides utility in ability to transport the liquid product [11]. It also utilizes all three components of biomass; namely, cellulose, hemicellulose and lignin. [12]
Biomass components
z CO, H2 Fischer-Tropsch Hydrocarbon
OH Cellulose
IOH
OH OH HOH OH HH 0 H H OH OHGlucose Xylose Mannose
HO OH 0 OHPyoyiCaayi Hyrcrn HO I _ OH O4Huprdnfel Galactose Arabinose Hemicellulose HO HO HO
'. .-Aqueous Catalytic Hydrocarbon
p HcoHarH sugar processin fuels
p-COUmaryl Coniferyl Sinapyl
alcohol alcohol alcohol
Lignin
Figure 1-1: Potential routes for the conversion of biomass into fuels and chemicals. 1.3. Challenges in exploitation of pyrolysis of lignocellulosic biomass
The decomposition temperatures of the building blocks of biomass are in the range of 200-5000C.[12] Pyrolysis decomposes the biomass to char, condensable gases (bio-oil) and
gases. Pyrolysis methods differ in heating rate, residence times and temperatures and affect percentages of char, bio-oil and gases. Nearly 50-90% of biomass can be converted to liquid [12]. Pyrolysis can be broadly classified into slow (residence time from minutes to hours, tem-perature of 400-6000C and 0.1 to 10C/min) and fast pyrolysis (residence time of < 2 s, temper-ature of 400-650"C and 10 to 1000C) [11]. Fast pyrolysis has shown good promise for pro-duction of bio-oil. A number of reactions including depolymerization, dehydration, C-C bond cleavage leads to a complex mixture of >200 oxygenated compounds. Advanced fast pyrolysis leads to a higher yield while catalytic fast pyrolysis (pyrolysis integrated with a catalytic pro-cess) gives better quality bio-oil, however is faced with a problem of low yields[13]. Pyrolysis
carried out in the presence of another gas like CO, C02, CH4, H2 and steam have also been
attempted.[15] Some of the main components of bio-oil are acetic acid, acetol, furfural, levoglucosan, guaiacol, syringol.[14, 15] The components in a typical bio-oil are given in Ta-ble 1-2. [15]
Table 1-2: Constituents of bio-oil[15]
Typical Compounds Constituents
Nonaromatic compounds
Carboxylic acids Acetic acid, propionic acid, butyric acid Nonaromatic esters Oxopropanoic acid methylester,
2-Nonaromatic aldehydes Hydroxyacetaaldehyde, cis crotanaldehyde, propanal Nonaromatic ketones Acetol, 1-hydroxy 2-butanone, 2-hydroxy
cyclopenten-1-one
Heterocyclic compounds
Furans Furfuryl alcohol, furaldehyde, furanone
Pyrans Maltol
Carbohydrates
Sugars Levoglucosan
Aromatic compounds
Benzenes Alkyl substituted benzenes, benzofuran, naphthalene
Catechols Catechol, alkyl substituted catechols
Aromatic aldehydes Benzaldehyde, hydroxy benzaldehyde, alkyl hydroxy ben-zaldehyde
Phenols Phenol, alkyl substituted phenols
Guaiacols Guaiacol, coniferyl alcohol, vanillin, alkyl substituted guai-acols
1.3.1. Challenges in upgrading of bio-oil
Bio-oil has poor volatility, high viscosity, coking tendency, corrosiveness, cold flow problems and high oxygen and water content [10, 11]. Further, there exists no standard for bio-oil production. The bio-oil and conventional fuel-oil properties are given in Table 3-3. [11] The bio-oil does not have good combustion properties mainly due to low heating value and high water content. The presence of acids and thermally unstable compounds leads to corrosive properties and high coking.[ 1] The bio-oil is not in thermodynamic equilibrium and compo-nents polymerize and condense with time which change the viscosity and phase separa-tion.[11,12,14] There are a number of reactions which cause degradation of bio-oils like reac-tion of organic acids with alcohols and olefins, aldehydes and alcohols forming hemiacetals or acetals, polymerization of olefins, etc.[ 11] These limitations make bio-oil practically unusable without upgrading. Hence, it must be refined in order to stabilize and upgrade the depolymer-ized products before transporting or blending with any transportation fuel.
Properties of pyrolysis oil, liquefaction oil and heavy fuel oil [10, 11]
Pyrolysis oil Liquefaction oil Heavy fuel oil
Property Moisture content, 15-30 5.1 0.1 wt% pH 2.5 Elemental composi-tion, wt% C 54-78 73 85 H 5.5-7.0 8 11 0 35-40 16 1.0 Higher heating 16-19 34 40 value, HHV, MJ/kg Viscosity (cP and 40-100 15000 180 500C) Distillation residue, Up to 50 1 wt%
Bio-oil can be upgraded to transportation fuels by primarily three different routes which have gained attention; namely, hydrodeoxygenation (HDO), zeolite upgrading and emulsifica-tion with diesel[10]. Some other upgrading techniques include gasificaemulsifica-tion of bio-oil to H2 or syn-gas for further applications, esterification to enhance stability and reduce acidity and aque-ous phase reforming[ 11]. Zeolite upgrading forms a number of products including hydrocar-bons (aliphatic and aromatic), water soluble organics, organic soluble organics, water and gases (CO, C02). However, the poor hydrocarbon yields and high coking (30-40 wt% of
bio-oil) limits the use of zeolites.[10] Bio-oil emulsions with diesel have good ignition character-istics but have higher cost due to surfactants and energy apart from having higher corrosion and lower cetane number.[10, 14] HDO increases the energy density and stability of bio-oil along with reduction in fuel the viscosity, by forming stabilized deoxygenated products.[10]
8
Thus, in the refining step following pyrolysis, bio-oil may be subjected to HDO for removal of oxygen and stabilize the liquid product.
Hydrotreating is a key process in petrochemical refining involving hydrodesulphuriza-tion (HDS), hydrodenitrogenahydrodesulphuriza-tion (HDN) and HDO to remove sulphur, nitrogen and oxygen, respectively from petrochemical feedstocks.[ 16] In petrochemical literature, less attention has been devoted to HDO due to the low oxygen content petrochemical feeds (< 0.3 wt%).[9] Due to the high oxygen content in bio-oil (20-50 wt%), HDO has recently received closer attention in the context of bio-oil upgrading.[10, 17]
For large-scale production of biomass-derived fuels and chemicals, the refining pro-cesses must be low cost. In general, the use of noble metal catalyst and high pressure hydrogen
(> 30 bar) negatively impact the economics of bio-refineries.[18] Bio-oil has a high prevalence
of C=O and unsaturated C=C bonds.[12] It is highly desirable to obtain selective deoxygena-tion of C=O bonds without saturating the highly prevalent C=C bonds, in order to minimize the H2 consumption and maintain a high aromatic content along with a high octane
num-ber. [19] Furthermore, the catalyst must be stable under reaction conditions and regenerative in order to minimize the catalyst cost. In addition, in order to facilitate smoother integration with a pyrolysis unit, the refining process must operate at conditions compatible with a pyrolysis unit (T = 400-6500C, P = 1 atm).[20] One class of materials that show promise are transition
metal oxides, which we will discuss in the later sections.
1.3.2. Catalysts for the hydrodeoxygenation (HDO) of bio-oil
The various catalysts that have been explored for HDO include different active phases, promoters and supports for HDO of bio-oils and their components. The rational and effective
design of HDO catalysts necessitate the fundamental understanding of the mechanism, kinetics and site requirements for HDO. Hence, the elucidation of the HDO mechanism have been con-ducted on model compounds including phenolics, furans and carboxylic acids which are the major components of bio-oil. [9] Furthermore, other reactions including C-C hydrogenolysis and cracking also need to be avoided. [12] In the context of HDO of bio-oil components, large attention has been given to bulk and supported transition metal sulphides, and base and noble metals catalysts.
Transition metal sulphides
Metal sulphides are ubiquitously used in hydroprocessing technology for HDS and
HDN reactions.[9,12] In general, these catalysts have been largely cobalt and molybdenum or
nickel and molybdenum sulphide catalysts. [21] Due to the high presence of sulphur in petro-chemical feeds, the phase of the sulphide catalysts are maintained. However, for refining of bio-oil, due to the high presence of oxygen, these catalysts undergo oxidation leading to
cata-lyst deactivation, unless a sulphur source such as hydrogen sulphide were added as a co-feed.
[9, 12, 13] Along with unnecessary use of sulphur, it leads to additional safety concerns due to
the likely formation of sulphur dioxide. In addition, these catalysts are active for hydrogenation reactions leading to saturation of C=C bonds and leading to reduction of aromaticity of'bio-oil. [9, 12]
Noble and base metals
Noble metals such as Pt, provide high catalytic activities for HDO reactions. [9] How-ever, they are expensive and rare. At low temperatures and high H2 pressures, they are active
for hydrogenation reactions which leads to high H2 consumption, and undesired products. [9]
At high temperatures, for Pt catalysts, the selectivity towards HDO products is low. For exam-ple, Resasco and coworkers have demonstrated low product selectivity towards deoxygenated product benzene from anisole HDO, forming a side product phenol with a very high selectiv-ity.[22] As a result, many researchers have investigated base metals for HDO, which are less expensive and less reactive than noble metals.[9] Base metals including iron, cobalt, nickel and rhodium exhibit reactivity towards HDO. [9] Among base metals, iron shows high selec-tivity towards selective HDO products as compared to cobalt and nickel which exhibit high
C-C hydrogenolysis activity.[23] For example: Olcese exhibited that although Fe/SiO2 exhibited
lower reactivity towards HDO for guaiacol HDO than supported Cobalt catalyst. [23] However, Cobalt-containing catalysts produced methane from C-C hydrogenolysis with a high selectivity towards methane favored at high temperatures and high conversions.[23]
Base metal oxides
Surprisingly, limited attention has been devoted to transition metal oxides for HDO applications. In general, oxides including transition metal oxides, have been used as supports to disperse active metal species on the support surface. Typically, acidic supports such as
y-A1203 and zeolites, basic supports such as MgO, and reducible supports such as ZrO2 and TiO2 have been studied with transition metal sulphides or metals as the active species.[9]
The reducible metal oxides including MoO3, V205 and W03 are well established to be
effective catalysts for a number of redox reactions, including oxidative dehydrogenation and partial oxidation of alkanes, and oxidation reactions of alcohols and olefins.[24-26] They have been known to go through a Mars van Krevelen mechanism, through the involvement of lattice oxygen (Figure 1-2).[27] It is indeed possible to envisage deoxygenation of bio-oil derived oxygenates through a reverse Mars van Krevelen mechanism occurring on the reducible metal
oxides mediated through an oxygen-vacancy created in the presence of a reducing source such as H2 (Figure 1-2).
"Mars-van Krevelen"
CXHY CXHyOZ
(hydrocarbons) (oxidized products)
* lattice 0
3 0 defect Oxygen r Mn+ Vacancies
Surface oxidation
0
"Reverse Mars-van Krevelen"
CXHY CXHYOZ (hydrocarbons) (oxygenates) 0 lattice 0 1101 J 0 defect oxygen
1555
Mn+ Vacancies 0M(n-l)+ H2 Surface reduction H20Figure 1-2: Schematic of Mars van Krevelen and reverse Mars van Krevelen mechanism on reducible metal oxides.
The efficacy of MoO3 as a selective HDO catalyst of bio-oil derived oxygenates was suggested by Mei et al. through the results of a density functional theory (DFT) study on acet-aldehyde deoxygenation through an oxygen vacancy mechanism on MoO3 (010) surface.[28] Our previous work experimentally demonstrated MoO3 as an efficient catalyst for selective C-O bond cleavage for a number of bio-oil derived oxygenates including acetone, cyclohexanone, furans and anisoles at a temperature 673 K and near atmospheric PH2 (< 1 bar).[29] As such,
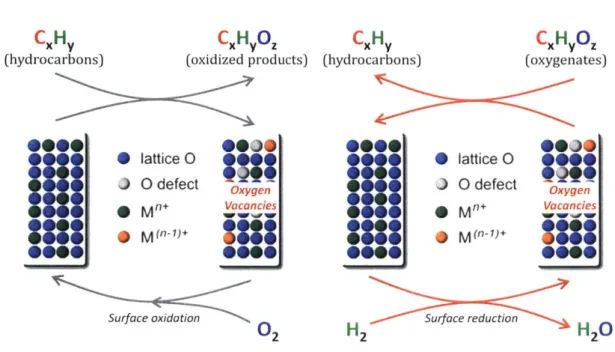
the catalytic deoxygenation was proposed to occur through a reverse Mars van Krevelen mech-anism.[29] Figure 1-3 shows a schematic of a reverse Mars van Krevelen mechanism for ace-tone HDO occurring on an oxygen vacancy.
0 0
0
Mo-O-Mo-O-Mo
0
0
0Mo-o-Mo-o-Mo
Oxygen Vacancy Deoxygenation0
0
0
Mo-O-Mo-O-Mo
I
H
2 Vacancy Site Regeneration 0 0 0 I1 11 I1Mo-oMo-o-Mo
Figure 1-3: Schematic of a reverse Mars van Krevelen mechanism for acetone HDO.[29]
Apart from our previous work, studies on hydrodeoxygenation of bio-oil derived
oxy-genates on reducible metal oxides have been limited to RuO2, CeO2-ZrO2 and Fe2O3
cata-lysts.[30-32] Sievers and coworkers suggested the role of oxygen vacancies towards HDO by corTelating HDO reactivity on CeO2-ZrO2 catalysts to the oxygen uptake obtained by oxygen chemisorption. [31] Vlachos and coworkers suggested a conjugation-driven "Reverse Mars van Krevelen" radical mechanism for low temperature C-O bond activation on RuO2 catalysts.[32] In a number of reports, Yong Wong and coworkers have suggested that stabilization of base metals in their metallic form is critical for maintaining HDO reactivity.[33] While this may be true for base metal catalysts, to the best of our knowledge no other systematic study has been carried out to obtain a fundamental understanding on deoxygenation kinetics on reducible
metal oxides. A detailed study understanding the structure-activity, stability, the role of oxida-tion states and the nature of active sites is required for design of efficient reducible metal oxide
catalysts for HDO of bio-oil and the field of biomass conversion in general.
1.4. Aims of the Thesis
The primary goal of the thesis is to develop a fundamental catalytic understanding for HDO on reducible metal oxides in order to enable design of efficient catalytic process for conversion of biomass-derived feedstocks to fuels and chemicals. Specifically, we develop structure-reactivity relationships for understanding the reactivity on reducible metal oxides for HDO using biooil-derived model compounds. Further, we utilize kinetic and computational tools to develop understanding on the elementary steps and nature of active sites relevant for
HDO reactions.
The first part of the thesis is focused on developing insights on the reactivity and sta-bility for bulk and supported molybdenum oxide catalysts and identifying the role of support towards HDO reactivity. The second part of the thesis is focused on utilizing density functional theory (DFT) and kinetic studies to develop mechanistic insights on the reaction mechanism and the nature of active sites on bulk and supported MoO3 catalysts. In the final part, we gen-eralize our understanding of reactivity on reducible transition metal oxide (specifically, bulk Co304) and novel perovskite material, lanthanum strontium cobaltite (Lao.8Sro.2CoO3) for HDO, and identify the role of oxidation states for HDO reactivity and stability. In summary, we aim to develop a fundamental catalytic understanding on the efficacy of reducible metal oxide catalysts for HDO of model compounds. The fundamental understanding thus obtained,
will play an important role in designing efficient catalysts for biomass conversion in specific, and deoxygenation of bio-oil in particular.
1.5. Outline of the Thesis
The results of my PhD research is summarized in the following 7 chapters (Chapters
2-8), followed by Conclusions and Outlook (Chapter 9).
Chapter 2 demonstrates that bulk MoO3 is an effective catalyst for the hydrodeoxygen-ation (HDO) of lignin-derived oxygenates (contained in bio-oil) to generate high yields of ar-omatic hydrocarbons without ring-saturation, at low H2 pressures ( < 1 bar) and temperatures
593 - 623 K. With the aid of characterization tools, the role of carbon-H2 is investigated to
understand the stabilization and enhanced reactivity of partially reduced MoO3 surface, and its role in preventing its deactivation.
Supported oxides, often inert materials, are used to disperse active species on the sup-port surface to enhance accessibility of active sites for the reaction. Chapter 3 studies the reac-tivity and stability of supported molybdenum oxide catalysts for the hydrodeoxygenation (HDO) of m-cresol, a model bio-oil compound. The genesis of the observed reactivity differ-ences at temperature 593 K and H2 pressures (< 1 bar) across different supported catalysts and
enhanced stability of supported oxides were investigated with detailed characterization tech-niques.
Chapter 4 studies the influence of the loading of active molybdenum on zirconia sup-port on the structural properties and reactivity trends towards HDO reactivity. The findings in the previous chapter were valid for oligomeric MoOx moieties on the support surface. In
Chap-ter 4, MoO3 loadings on supported catalysts were varied to form controlled amounts of iso-lated, oligomeric species, and crystalline MoO3 and Zr(MoO4)2 crystallites. The origin of the observed differences in HDO rates as a function of MoO3 loading was investigated by com-bining reactivity studies with characterization techniques to identity that a near-monolayer dis-persion of MoOx species are the optimum configuration for HDO reactivity.
Chapter 5 details a computational study on the reaction mechanism for acetone HDO on a pristine a-MoO3 (010) surface following an oxygen vacancy-driven pathway with the aid of density functional theory (DFT) calculations. The dissociative H2 adsorption on the surface was found to be the rate-limiting step for the catalytic cycle. The lower reactivity on bulk MoO3 in the initial phase of the reaction (Chapter 2) can be potentially explained to be due to the high reaction barriers for H2 dissociation.
There are few detailed kinetic studies for HDO on bulk and supported MoO3 catalysts, as the focus on HDO using reducible metal oxides have gathered interest only in the recent past. Chapter 6 presents a detailed kinetic study for HDO reactivity on bulk and supported MoO3 catalysts. The kinetic studies is complemented by a titration and co-feed studies using titrants such as water (H20), methanol, pyridine and di-tertbutyl pyridine (DTBP), which re-veal the nature of the active sites involved in HDO, on bulk and supported MoO3 catalysts. Finally, reaction mechanisms for both bulk and supported MoO3 are proposed consistent with kinetics and titration studies.
Chapter 7 demonstrates that the base metal oxides including bulk Co304 exhibit HDO reactivity with atleast an order of magnitude greater their corresponding bulk metallic forms, Co metal. Consequently, a major mechanism of deactivation on these base metal oxides is their
reduction to a metallic form. In contrast, the metals deactivate by surface passivation caused
by oxygenates. Furthermore, with Co304 as a starting material, the catalyst exhibits reactivity
towards C-C bond hydrogenolysis along with HDO at temperatures > 523 K. The selectivity towards C-C bond hydrogenolysis increases at low PH2 and high temperatures in the presence
of Co metal, suggesting the role played by dehydrogenation and Co metal in C-C bond cleav-age reactions.
Chapter 8 presents lanthanum strontium cobaltite (Lao.8Sro.2CoO3) as the first demon-stration of a novel low-temperature perovskite HDO catalyst for the vapor-phase deoxygena-tion of anisole. The perovskite reactivity mimics the reactivity similar to the corresponding bulk reducible metal oxide catalysts. Interestingly, the perovskite phase is resistant to reduction and hence, enhances subsequently the stability of the catalyst.
Finally, chapter 9 summarizes the main results of the thesis and identifies the major challenges and opportunities in the field in order to obtain not only a better fundamental cata-lytic understanding on these oxides, but also their applicability to biomass conversion pro-cesses.
1.6. List of publications
Each chapter of the thesis was written based on a separate manuscript; either in print, or in preparation and can be read independently. Accordingly, some of the introduction and experimental methods may be common across the chapters. Chapters 2-5 are extracted from published journal articles, while chapters 6-8 are based on manuscripts under preparation. Ac-cordingly, the specific articles included in the thesis are listed below.
Chapter 2: T. Prasomsri, M. Shetty, K. Murugappan, & Y. Romin-Leshkov (2014). Insights
into the catalytic activity and surface modification of MoO3 during the hydrodeoxygenation of ligninderived model compounds into aromatic hydrocarbons under low hydrogen pres-sures. Energy and Environmental Science, 7, 2660-2669.
Chapter 3: M. Shetty, K. Murugappan, T. Prasomsri, W.H. Green, & Y. Roman-Leshkov
(2015). Reactivity and stability investigation of supported molybdenum oxide catalysts for the hydrodeoxygenation of m-cresol. Journal of Catalysis, 331, 86-97.
Chapter 4: M Shetty, K Murugappan, W.H. Green, & Y. Romain-Leshkov (2017). Structural
properties and reactivity trends of molybdenum oxide catalysts supported on zirconia for the hydrodeoxygenation of anisole. ACS Sustainable Chem. Engg. 5(6), 5293-5301.
Chapter 5: M Shetty, B Buesser, Y. Roman-Leshkov, & W.H. Green (2017). Computational
Investigation on Hydrodeoxygenation (HDO) of Acetone to Propylene on a-MoO3 (010) Surface. The Journal of Physical Chemistry C 121(33), 17848-17855.
References
[1] R. Prestele, A. Arneth, A. Bondeau, N. de Noblet-Ducoudre, T.A.M. Pugh, S. Sitch, E.
Stehfest, P.H. Verburg, Current challenges of implementing anthropogenic land-use and land-cover change in models contributing to climate change assessments, Earth Syst Dynam,
8 (2017) 369-386.
[2] E. Hirsch, "It won't be any good to have democracy if we don't have a country": Climate change and the politics of synecdoche in the Maldives, Global Environ Chang, 35 (2015)
190-198.
[3] S.W. Shen, X. Feng, Z.R. Peng, A framework to analyze vulnerability of critical
infrastructure to climate change: the case of a coastal community in Florida, Nat Hazards, 84
(2016) 589-609.
[4] 0. Edenhofer, R. Pichs-Madruga, Y. Sokona, Climate Change 2014 Mitigation of Climate Change Working Group III Contribution to the Fifth Assessment Report of the
Intergovernmental Panel on Climate Change Preface, Climate Change 2014: Mitigation of Climate Change, (2014) Ix-Xi.
[5] T. Boden, Global, regional, and national C02 emissions, in.
[6] I.E.A.O.o.E. Technology, D., G.o. Eight, Energy technology perspectives, International
Energy Agency, 2008.
[7] I. Sim~es-Filho, BP Energy Outlook: 2017 Edition, in: New Energy Landscape: Impacts
for Latin America, 6th ELAEE/IAEE Latin American Conference, April 2-5, 2017, International Association for Energy Economics, 2017.
[8] C.J. Rhodes, The 2015 Paris climate change conference: COP21, Science progress, 99 (2016) 97-104.
[9] D.A. Ruddy, J.A. Schaidle, J.R. Ferrell III, J. Wang, L. Moens, J.E. Hensley, Recent
advances in heterogeneous catalysts for bio-oil upgrading via "ex situ catalytic fast
pyrolysis": catalyst development through the study of model compounds, Green Chem., 16 (2014) 454.
[10] S. Czernik, A.V. Bridgwater, Overview of applications of biomass fast pyrolysis oil,
Energy Fuels, 18 (2004) 590-598.
[11] G.W. Huber, S. Iborra, A. Corma, Synthesis of transportation fuels from biomass:
Chemistry, catalysts, and engineering, Chem. Rev., 106 (2006) 4044-4098.
[12] H. Wang, J. Male, Y. Wang, Recent Advances in Hydrotreating of Pyrolysis Bio-Oil and Its Oxygen-Containing Model Compounds, ACS Catal., 3 (2013) 1047-1070.
[13] X.Y. Wang, R. Rinaldi, A Route for Lignin and Bio-Oil Conversion: Dehydroxylation
of Phenols into Arenes by Catalytic Tandem Reactions, Angew Chem Int Edit, 52 (2013)
11499-11503.
[14]
Q.
Zhang, J. Chang, T. Wang, Y. Xu, Review of biomass pyrolysis oil properties and upgrading research, Energy Convers. Manage., 48 (2007) 87-92.[15] E. Kantarelis, W.H. Yang, W. Blasiak, Production of Liquid Feedstock from Biomass via Steam Pyrolysis in a Fluidized Bed Reactor, Energy & Fuels, 27 (2013) 4748-4759.
[16] E. Furimsky, F.E. Massoth, Hydrodenitrogenation of petroleum, Catalysis Reviews, 47 (2005) 297-489.
[17] A.V. Bridgwater, D. Meier, D. Radlein, An overview of fast pyrolysis of biomass, Org.
[18] W. Mu, H. Ben, A. Ragauskas, Y. Deng, Lignin Pyrolysis Components and
Upgrading-Technology Review, BioEnerg. Res., 6 (2013) 1183-1204.
[19] T. Prasomsri, M. Shetty, K. Murugappan, Y. Romain-Leshkov, Insights into the catalytic
activity and surface modification of MoO3 during the hydrodeoxygenation of lignin-derived model compounds into aromatic hydrocarbons under low hydrogen pressures, Energy Environ. Sci., 7 (2014) 2660.
[20] K. Murugappan, C. Mukarakate, S. Budhi, M. Shetty, M.R. Nimlos, Y.
Roman-Leshkov, Supported molybdenum oxides as effective catalysts for the catalytic fast pyrolysis of lignocellulosic biomass, Green Chem., (2016).
[21] E. Furimsky, Hydroprocessing challenges in biofuels production, Catal. Today, 217 (2013) 13-56.
[22] Q.H. Tan, G.H. Wang, A. Long, A. Dinse, C. Buda, J. Shabaker, D.E. Resasco,
Mechanistic analysis of the role of metal oxophilicity in the hydrodeoxygenation of anisole, J Catal, 347 (2017) 102-115.
[23] R.N. Olcese, M. Bettahar, D. Petitjean, B. Malaman, F. Giovanella, A. Dufour,
Gas-phase hydrodeoxygenation of guaiacol over Fe/Si02 catalyst, Appl Catal B-Environ, 115 (2012) 63-73.
[24] I. Rodriguez-Ramos, A. Guerrero-Ruiz, N. Homs, P.R. de la Piscina, J.L.G. Fierro, Reactions of propene on supported molybdenum and tungsten oxides, J. Mol. Catal. A: Chem., 95 (1995) 147-154.
[25] K. Chen, S. Xie, A.T. Bell, E. Iglesia, Structure and properties of oxidative
dehydrogenation catalysts based on MoO3/A1203, J Catal, 198 (2001) 232-242.
[26] C.A. Carrero, R. Schloegl, I.E. Wachs, R. Schomaecker, Critical Literature Review of
the Kinetics for the Oxidative Dehydrogenation of Propane over Well-Defined Supported Vanadium Oxide Catalysts, ACS Catal., 4 (2014) 3357-3380.
[27] K.D. Chen, S.B. Xie, E. Iglesia, A.T. Bell, Structure and properties of
zirconia-supported molybdenum oxide catalysts for oxidative dehydrogenation of propane, J. Catal.,
189 (2000) 421-430.
[28] D. Mei, A.M. Karim, Y. Wang, Density Functional Theory Study of Acetaldehyde
Hydrodeoxygenation on MoO3, J. Phys. Chem. C, 115 (2011) 8155-8164.
[29] T. Prasomsri, T. Nimmanwudipong, Y. Roman-Leshkov, Effective hydrodeoxygenation
of biomass-derived oxygenates into unsaturated hydrocarbons by MoO3 using low H2 pressures, Energy Environ. Sci., 6 (2013) 1732-1738.
[30] A.J.R. Hensley, Y.C. Hong, R.Q. Zhang, H. Zhang, J.M. Sun, Y. Wang, J.S. McEwen,
Enhanced Fe2O3 Reducibility via Surface Modification with Pd: Characterizing the Synergy within Pd/Fe Catalysts for Hydrodeoxygenation Reactions, Acs Catal, 4 (2014) 3381-3392.
[31] S.M. Schimming, O.D. LaMont, M. Konig, A.K. Rogers, A.D. D'Amico, M.M. Yung, C. Sievers, Hydrodeoxygenation of Guaiacol over Ceria-Zirconia Catalysts, Chemsuschem, 8 (2015) 2073-2083.
[32] A.V. Mironenko, D.G. Vlachos, Conjugation-Driven "Reverse Mars-van
Krevelen"-Type Radical Mechanism for Low-Temperature C-O Bond Activation, J Am Chem Soc, 138
(2016) 8104-8113.
[33] Y.C. Hong, S.R. Zhang, F.F. Tao, Y. Wang, Stabilization of Iron-Based Catalysts
against Oxidation: An In Situ Ambient-Pressure X-ray Photoelectron Spectroscopy (AP-XPS) Study, Acs Catal, 7 (2017) 3639-3643.
Chapter 2
Insights into the catalytic activity and surface
modification of bulk
MoO3
during the
hydrodeoxygenation of lignin-derived model
compounds into aromatic compounds under low
hydrogen pressures
2.1. Introduction
The utilisation of lignocellulosic biomass as a source of renewable carbon for the production of fuels and chemicals has garnered much attention in the last decade. Among the various technologies investigated for lignocellulosic biomass processing, fast pyrolysis and catalytic fast pyrolysis have emerged as attractive and economically viable options.[1-3] Although the liquid products from these pyrolysis processes, (i.e., bio-oil) exhibit moderate energy densities, they cannot be directly utilised as transportation fuels without a prior upgrading step. Further upgrading is required because oxygen and water content of bio-oil is too high, resulting in several drawbacks that include low energy density, immiscibility with hydrocarbons, and wide variation in boiling point temperatures[4, 5]. Moreover, bio-oil has high acid content, making it very reactive and unstable during transportation and storage. Bio-oil is considered a hydrogen-deficient feedstock, requiring the use of hydrogen for all upgrading processes. Hydrogen can be obtained from different sources, including steam reforming of
methane abundant shale gas reserves, catalytic reforming of biomass-derived feedstock, or, ideally, solar-based water splitting. [6]
Lignin-derived phenolic compounds represent a significant fraction of bio-oil. Lignin comprises up to 30 wt% of lignocellulosic biomass and it is composed of a heterogeneous, amorphous matrix of polyaromatic units featuring numerous ether linkages (C-O-C), as well as hydroxyl (-OH), and methoxyl (-OMe) side groups. Unfortunately, these phenolic molecules are much more refractory than sugar-derived oxygenates and their effective processing currently represents one of the grand-challenges in bio-oil upgrading. [7-9]
Hydrodeoxygenation (HDO) of biomass components involves direct removal of oxygen from bio-oil via C-0 bond cleavage. HDO processes for phenolic compounds are challenging since high yields of aromatic hydrocarbons can only be achieved by selectively cleaving the strong C-O bond without hydrogenation the aromatic ring. Excellent reviews are available on HDO catalysts for lignin-derived molecules that highlight the main bottlenecks faced during processing. [10, 11] Supported noble metals such as Ru, Rh, Pd, Pt, Re, as well as base metals, such as Cu, Ni, Fe and their heterometallic alloys are active for hydrogenation/hydrogenolysis reactions, but demand high H2 pressures and high temperatures, which typically promote saturation
of all double bonds.[12, 13] Molybdenum-based sulphides (such as MoS2, NiMoS2, and
CoMoS2), the typical industrial hydrotreating catalysts in refining, are active HDO catalysts under high operating H2 pressures (15-80 bar), but experience a rapid
deactivation in the absence of a sulphur source and are sensitive to coke formation and water poisoning.[14-16] Metal phosphides, such as Ni2P, have shown higher HDO
activity than commercial sulphide catalysts for the conversion of guaiacol into phenol and benzene.[17, 18] Recently, Hicks and co-workers showed that a bimetallic FeMoP
catalyst is active and selective for C-O bond cleavage of phenolics and aryl ethers in the liquid phase, producing benzene with selectivities exceeding 90% at 673 K, but require H2 pressures in the range of 21-42 bar.[19] Bifunctional (metal/acid) catalysts
consisting of noble metals supported on acid supports, such as Pt/H-Beta and Ga/H-Beta, generate aromatics (benzene, toluene and xylene) during vapour-phase processing of anisole and m-cresol feeds under atmospheric H2 pressure at 673-823 K and in
aqueous phase conditions under high H2 pressures (>50 bar) but only in moderate yields.[20-3 1]
Many challenges remain for the development of HDO catalysts for bio-oil upgrading that do not use expensive noble metals, that utilise minimum amounts of H2
(i.e., low H2 pressures), and feature high stability. Recently, we demonstrated that
MoO3 is an attractive, earth-abundant catalyst active for the HDO of various biomass-derived oxygenates, including aliphatic and cyclic ketones, furanics, and phenolic feeds. Importantly, it was shown that MoO3 selectively cleaves C-O bonds to produce olefinic and aromatic hydrocarbons, with high activity and selectivity using low H2 pressures (<1 bar).[32] We inferred that oxygen vacancies play an important role in the reaction
mechanism, but the nature of the active site(s) was not identified. Chen and co-workers presented a similar catalytic behaviour on transition-metal carbide catalysts, for the conversion of C3 oxygenates (e.g., propanal, propanol and acetone) into propylene under low H2 pressures and temperatures between 573 - 653K.[33, 34]
In this contribution, we report the high selectivity, stability, and hydrogen efficiency of MoO3 catalysts to produce aromatic hydrocarbons from a set of lignin-derived model compounds that represent the spectrum of C-O bonds present in real
lignin fractions. Bond-dissociation energies (BDEs) of relevant C-O bonds involved in the HDO of these model compounds were estimated by density functional theory (DFT) computational methods. Next, catalyst performance, temperature effects, stability, and regenerability were investigated and compared with predicted reactivity trends from the BDE analysis. Finally, catalyst characterisation methods during the course of the HDO reaction were used to reveal the transition of the catalyst surface from an oxide phase to oxycarbide and oxycarbohydride phases. Systematic catalyst pretreatments were performed to gain understanding of the nature and genesis of surface active sites.
2.2. Experimental section
Chemicals and materials
Phenol (>99 wt%), m-cresol (99 wt%), anisole (>99 wt%), guaiacol (>98 wt%) and diphenyl ether (>99 wt%) obtained from Sigma-Aldrich were used as feeds with no further purification. H2 (99.999%, Airgas) and He (99.998%, Airgas) were used as a
reactant in the reaction experiments, and as an inert carrier gas, respectively. 02
(99.999%, Airgas) and air (zero grade, Airgas) were used for catalyst regeneration.
Iso-butane (Airgas) and zinc granules (-20 mesh, 99.8%, Alfa Aesar) were used during the synthesis of molybdenum oxycarbohydride. Commercial molybdenum(VI) oxide, MoO3 (>99.5 wt%), molybdenum(IV) oxide, MoO2 (>99 wt%), and molybdenum
carbide, Mo2C (>99.5 wt%) were purchased from Sigma-Aldrich. Prior to reaction, MoO3 was calcined at 873 K (with a ramp rate of 10 K min-') for 3 h under air flow
(100 mL min-').
A phase pure molybdenum oxycarbohydride (MoOxCyHz) sample was synthesised
MoO3 was added into a 4N HCl aqueous solution containing an excess amount of Zn granules. Evolved hydrogen generated in-situ intercalates between the layers of MoO3 to form a hydrogen bronze. The resulting slurry was washed with a generous amount of deionised water to remove ZnCl2 and remaining HCl. The absence of chlorine ion was detected by AgNO3 test, and the excess Zn granules were sieved out. The hydrogen bronze of molybdenum oxide was treated with a flow of H2/i-butane (200 mL min-1, 3:1 v/v
H2:i-butane mixture) at 623 K for 12 h to produce the oxycarbohydride.[36] The final product was dried under vacuum and stored under moisture- and oxygen-free conditions.
Catalyst activity measurement
The catalytic testing experiments were conducted in a vapour-phase packed-bed flow reactor system. A stainless steel tube of 0.95 cm OD (wall thickness 0.089 cm) was used as the reactor, which was mounted in an insulated single-zone furnace (850W/I 15V, Applied Test Systems Series 3210). Temperature was controlled using a thermocouple mounted slightly downstream of the catalyst bed (Omega, model TJ36-CAXL- 116u) connected to a temperature controller (Digi-Sense, model 68900-10). The MoO3 catalyst bed (20-300 mg) was mixed with an inert (a-alumina, Sigma-Aldrich) sieved through a 100-200 mesh. The bed volume was typically 2 mL and situated in the middle of the furnace. Liquid reactants were introduced into the reactor via a syringe pump (Harvard Apparatus, model 703005). In the case of phenol feed, the solid reactant was mixed with an unreactive solvent, (e.g., 20 wt% in mesitylene), prior to loading into the syringe. Carrier He gas or reactant H2 gas was mixed with vapourised reactants
at the inlet of the reactor. Each experiment was carried out at 573-673 K and atmospheric pressure with a constant total gas flow rate of 70 mL min-'. All experiments
were carried out under conditions free of mass transfer limitations. The space-time (W/F), expressed in gcat (mmOlFeed h-')-, is defined as the ratio between the mass of the catalyst and the molar feed rate of the reactants. For catalyst regeneration experiments, spent catalysts were calcined in situ under pure 02 or air at 593-673 K for 3 h.
Product analysis and data evaluation
The reactor effluent lines were heated at 523 K to prevent any condensation of products, which were directly analysed and quantified by an online gas chromatographer equipped with a flame ionisation detector (GC-FID, Agilent Technologies, model 7890A) equipped with a DB-5 column (Agilent, 30 m x 0.25 mm id, 0.25 pm). The GC was operated with ultra-high purity helium as the carrier gas at a constant flow rate of 1.0 mL min-. The GC parameters used for the analysis were as follows: injector temperature 523 K; detector temperature 573 K; split ratio 1:100. The temperature program started at 343 K and held for 5 minutes; then the temperature was increased at 20 K min-' to 533 K, followed by a 7-min hold. Product identification was performed using a mass selective detector (MSD, Agilent Technologies, model 5975C). The nature and quantity of carbon residues on spent catalysts were analysed by a combination of thermogravimetric analysis (TGA, TA Instruments TGA-Q500) and mass spectrometry (MS, Hiden Analytical HPR-20/QIC). The TGA-MS experiments were performed by using 5 vol% 02/He with a total flow rate of 100 mL min. The temperature was ramped from 323 K to 923 K with a heating rate of 7.5 K min' and held at the final temperature for 30 min. The evolution of CO2 (m/z = 44) and H20 (m/z
18) were continuously monitored by the MS.
Conversion% = moles of carbon in reactant consumed x moles of carbon in reactant fed 100 (1)
Selectivity to hydrocarbons%
sum of moles in aromatic products 100 (2)
moles of carbon in reactant consumed
Yield% = sum of carbon in product x100 (3)
moles of carbon in reactant fed
The catalyst deactivation profiles were obtained using a first-order decay kinetic model.[37] The activity of the catalyst is given by:
-rA rate of reaction A at any time t -rA(fresh) rate of reaction A with fresh catalyst
For first-order decay:
a(t) = exp(-kDeact t), where a(t) = C In (5) Combining gives:
ln( In(*) )= -kDeact t C (6)
where x = fractional conversion, kDeact = first-order deactivation rate constant, and t
time.
Characterisation
The powder X-ray diffraction (PXRD) patterns of fresh and spent catalyst samples were recorded on a Bruker D8 Discover diffractometer, equipped with a Nickel-filtered Cu-Ka radiation (k = 1.5418
A).
Diffraction data recorded on a 2D image plate were integrated between 200 and 90' 20. The surface area was determinedby nitrogen adsorption/desorption experiment using a Quantachrome Autosorb iQ
automated gas sorption system. X-Ray photoelectron spectroscopy (XPS) data were collected using a PHI Versaprobe II equipped with a multichannel hemispherical analyser and aluminium anode X-ray source operating at 100 W with a 1 00-tm beam scanned over a 1.4-mm line across the sample surface. A dual-beam charge
neutralisation system was used with an electron neutraliser bias of 1.2 eV and Argon ion beam energy of 10 eV.
2.3. Results and discussion
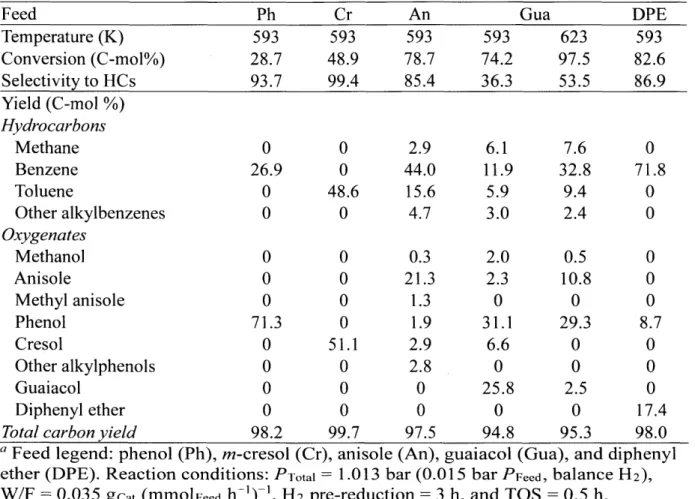
Conversion of lignin-derived model compounds
Table 2-1 shows the total conversion and the corresponding product distribution from reacting lignin-derived model compounds with MoO3 under atmospheric pressure at 593 K and identical contact time (W/F, defined as gcat (mmolFeed h-')-1). Prior to each
run, the catalyst was treated under flowing H2 at 593 K for 3 h. Phenol was converted
to benzene with, a selectivity of 94% at 29% conversion. Neither cyclohexane nor cyclohexene was observed in the product mixture, thus indicating that deoxygenation via ring saturation followed by dehydration is unlikely and instead suggests a pathway involving direct C-O bond cleavage. Similarly m-cresol was converted to toluene with
99% selectivity at 49% conversion. At 79% conversion, anisole yielded 44% benzene
and 20% alkylbenzenes. Minor phenolic products included phenol, methyl anisole, cresol, and alkylphenols with yields of 2%, 1%, 3%, and 3%, respectively. In a similar manner, benzene, alkylbenzenes, phenol, and alkylated phenols were mainly produced
from guaiacol with yields of 12%, 9%, 31% and 9%, respectively, while methane (6% yield) and methanol (2% yield) were also detected as minor products. Diphenyl ether was selectively converted to benzene with 87% selectivity at 83% conversion. Overall carbon balances typically exceeded 94%, with the remaining carbon being assigned to soft coke and carbon intercalation into the oxide lattice, as analysed by TGA-MS and XRD of the spent catalysts (vide infra).
Table 2-1 Conversion and product distribution of lignin-derived model compounds on MoO3
catalystsa
Feed Ph Cr An Gua DPE
Temperature (K) 593 593 593 593 623 593 Conversion (C-mol%) 28.7 48.9 78.7 74.2 97.5 82.6 Selectivity to HCs 93.7 99.4 85.4 36.3 53.5 86.9 Yield (C-mol %) Hydrocarbons Methane 0 0 2.9 6.1 7.6 0 Benzene 26.9 0 44.0 11.9 32.8 71.8 Toluene 0 48.6 15.6 5.9 9.4 0 Other alkylbenzenes 0 0 4.7 3.0 2.4 0 Oxygenates Methanol 0 0 0.3 2.0 0.5 0 Anisole 0 0 21.3 2.3 10.8 0 Methyl anisole 0 0 1.3 0 0 0 Phenol 71.3 0 1.9 31.1 29.3 8.7 Cresol 0 51.1 2.9 6.6 0 0 Other alkylphenols 0 0 2.8 0 0 0 Guaiacol 0 0 0 25.8 2.5 0 Diphenyl ether 0 0 0 0 0 17.4
Total carbon yield 98.2 99.7 97.5 94.8 95.3 98.0
a Feed legend: phenol (Ph), m-cresol (Cr), anisole (An), guaiacol (Gua), and diphenyl ether (DPE). Reaction conditions: PTotal = 1.013 bar (0.015 bar PFeed, balance H2),
W/F = 0.035 gcat (mmolFeed h')-1, H2 pre-reduction = 3 h, and TOS = 0.5 h.
These reactivity data suggest that MoO3 promotes both deoxygenation and demethylation/transalkylation reactions in the presence of methoxy substituents in the ring. To identify primary products, the HDO of anisole was investigated under differential conditions. As shown in Table 2-2, the ratio of total aromatic hydrocarbons to total phenolic intermediates is approximately 1:1 when extrapolated to 0% conversion. Consequently, the selectivity to aromatic hydrocarbons continues to increase with increasing conversion once the phenolic intermediates also undergo HDO

![Figure 1-3: Schematic of a reverse Mars van Krevelen mechanism for acetone HDO.[29]](https://thumb-eu.123doks.com/thumbv2/123doknet/14205385.480823/21.917.115.779.108.543/figure-schematic-reverse-mars-krevelen-mechanism-acetone-hdo.webp)