Determination of ethyl glucuronide and ethyl sulfate from dried blood spots
Texte intégral
Figure

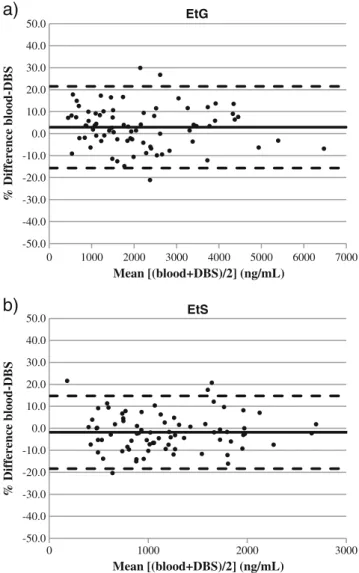
Documents relatifs
Our study showed that for diagnosis, outbreak monitoring and virological surveillance of CHIKV infection and circulation, capillary blood samples taken from the finger and spotted
Table 2 Characteristics of studies included in the systematic review on HCV antibody detection from DBS compared to venous blood sampling (Continued) Author, Country Study design
Cette étude nous montre que la Dimedone et l’ortho-phénylènediamine deux produits commerciaux et bon marché, réagissent pour donner l’intermédiaire
The expression of the ENaC a-subunit (A, upper panel, full-length protein; lower panel, cleaved protein), b-subunit (B, full-length protein), and c-subunit (C, upper panel,
lisseuse et finiss.. Sauter, Fs-Jos., fabr. Meyer frères, fabr. Quartier, Just., fabr. Finck, Fritz, échapp. Jung, Ph., graveur et guill. Meyrat, F., graveur et guill. Vulliet,
Validated method for the determination of the ethanol consumption markers ethyl glucuronide, ethyl phosphate, and ethyl sulfate in human urine by reversed-phase/. weak anion
To increase familiarity of some laboratory staff in the Region to measles IgM EIA testing and the use of dried blood samples (DBS) as an alternative method of sampling, the
To describe the procedure for collecting blood by venipuncture and for preparing thick and thin blood films from venous blood collected in tubes containing anticoagulant..
