Directed evolution of TurboLD for efficient proximity labeling in living cells and organisms by
Tess C. Branon B.S. Chemistry, B.S. Biology Western Carolina University, 2013 Submitted to the Department of Chemistry in partial fulfillment of the requirements for the
Degree of Doctor of Philosophy at the
Massachusetts Institute of Technology September 2018
2018 Massachusetts Institute of Technology All rights reserved
Signature
Signature of Author:redacted
Department of Chemistry August 14, 2018Signature redacted
Certified by:Professor of Genetics, Biology, and Chemistry
Alice Y. Ting (by courtesy), Stanford University
Thesis supervisor
Signature redacted
Accepted by: MASSACHUSETTS INSTITUTE OFTECHN9 LGY.NOV 14 2018
LIBRARIES
ARCHIVES
1Aobert W. Field Haslam and Dewey Professor of Chemistry Chair, Departmental Committee on Graduate Students--A
This doctoral thesis has been examined by a committee of the Department of Chemistry as
follows:
Signature redacted
JoAnne M. StubbeProfessor of Genetics,
Novartis Professor of Chemistry and Professor of Biology
Signature redacted
ice Y. Ting Biology, and Chemistry (by courtesy), Stanford University Thesis supervisor
-Signature
redacted
Ronald T. Raines Firmenich Profe or of Chemistry
I
Directed evolution of TurbolD for efficient proximity labeling in living cells and organisms
by
Tess C. Branon
Submitted to the Department of Chemistry on August 14, 2018 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
ABSTRACT
Protein interaction networks and protein compartmentalization underlie all signaling and regulatory processes in cells. Traditional approaches to proteomics employ mass spectrometry (MS) coupled to biochemical fractionation or affinity purification but require cell lysis prior to analysis which often results in false-negatives from missed interactions or incomplete purification and false-positives from contaminants. Enzyme-catalyzed proximity labeling (PL) has emerged as a new approach to study the spatial and interaction characteristics of proteins in which a PL enzyme can be genetically targeted to a subcellular region and used to tag surrounding endogenous proteins with a chemical handle that allows their identification by MS. Tagging is carried out in living cells in a distance-dependent manner, allowing data collection from a physiologically relevant environment with preservation of spatial information.
Current PL methods are limited by poor catalytic efficiency or toxic substrates that limit their application in vivo. Therefore, we have developed a new proximity labeling method, called TurboID, that uses non-toxic labeling conditions and has high catalytic efficiency that allows its use in a wide variety of biological contexts. Here, we describe our use of yeast display-based directed evolution to engineer two promiscuous mutants of biotin ligase, TurbolD and miniTurbo. We describe our characterization of the evolved PL enzymes in microbes, cultured cells, in vitro, and in vivo in flies and worms, and show that TurbolD and miniTurbo have much greater catalytic efficiency than any other biotin ligase-based PL method currently available. Lastly, we demonstrate that TurbolD and miniTurbo can be used to obtain proteomes with the same size, specificity, and depth-of-coverage as existing biotin-ligase based PL techniques with over 100-fold shorter labeling times.
In the Appendix, we discuss two separate projects. In Part I, we describe how fusion of the PL enzyme APEX2 to various mitochondrial proteins could be used to map the proteomes of mitochondrial subdomains and be used to visualize the localization of mitochondrial proteins in mitochondrial subdomains using APEX2 to generate contrast for electron microscopy imaging. In Part II, we discuss the development of two platforms that could be used to temporally control genome editing using light.
Thesis Supervisor: Alice Y. Ting
Acknowledgements
I would like to thank Alice, my advisor, for showing me how to be fearless in vision and measured in approach. She has taught me to tolerate no less than the highest scientific rigor, and to always strive to work on the most exciting and impactful questions. I am also thankful to her for teaching me the importance of constructing a compelling narrative so that I can effectively communicate my work and share my passion for science with others. I will always be grateful to her for her support and mentorship during my graduate studies. I would also like to thank my thesis chair, JoAnne. She has helped me understand how important it is to sharpen my chemical intuition and to ask the right questions to get at the heart of the mechanisms behind the biochemistry. She is a constant reminder to me of why I decided to study chemistry in order to understand biology. As a woman in science, I know I wouldn't be where I am today if women like JoAnne did not forge a path into the incredibly male-dominated world of academia, and even more so physical sciences. I am beyond thankful to have had her as my thesis chair. I would like to thank the last member of my thesis committee, Matt, for his unending kindness, support, and mentorship. Matt has inspired me to always keep the biological applications of the tools I develop in mind and showed me the importance of critically considering the approaches I take in developing such tools. I thank him for taking so much of his time and energy to discuss my project with me, and for helping me discover what I want from my future career in science.
I also want to take the time to thank every member of the Ting lab I have had the pleasure to meet and share time with. Dan, Jeff, Phillip, Kurt, Vicky, Stephanie, Ken, Jake, Monica, Kayvon, Oom, Fayqal, Ozan, Cathy, Austin, Chai, Mitch, Wenjing, Mateo, Yisu, Shuo, Robert, Kelvin, Elbeg, Martin, Tina, Boxuan, and Sanjana, I thank you all for your friendship, support, brainstorming, and laughs that we've shared over the years - I would not have made it here without you. The work presented here would have been possible without my wonderful collaborators, Namrata Udeshi, Tanya Svinkina, Steven Carr, Justin Bosch, Norbert Perrimon, Ariana Sanchez, and Jessica Feldman - thank you all for your hard work, critical discussion, and passion for our work together. I would also like to thank the hundreds of other collaborators we have partnered with over the past several years who are making the dream we envisioned for TurbolD a reality.
I would like to thank the hundreds of friends I've made from all corners of the country and all around the world. You have all been there for me through the life changing transitions I've gone through, from leaving my home in North Carolina, to giving me warmth during the cold Boston winters, to welcoming me with the sincerest embrace in California. You show me that there is in fact a world beyond science that is worth living for. I would also like to thank my family for their truly unconditional love and support. I would like to thank my parents, Tony and Jessie, for their generosity and for making me feel like every superlative imaginable applies to me even when I'm feeling nearly worthless. I'd like to thank my sister, Simone, for showing me the importance of taking the time to enjoy my life, and my brother, Joshua, for helping me realize what is truly important to me and why. Last but of course not least, I would like to thank my husband, Paul, who has been my life partner through everything. You are my best friend, my lover, my soul mate, and I would be entirely lost without you. Thank you for holding me through all of the low times, and for enjoying all of the highs by my side.
Table of contents
ABSTRA CT ... 3
Acknow ledm ents ... 4
Table of contents ... 5
List of Figures ... 7
List of Tables ... 10
List of abbreviations ... 11
Chapter 1: Introduction to proximity labeling for spatial proteomics in living cells... 13
Introduction ... 14
Traditional m ethods for spatially resolved proteom ics ... 14
Enzym atic proxim ity labeling m ethods... 15
Conclusion... 20
References ... 21
Chapter 2: Directed evolution of TurbolD and m iniTurbo... 24
Introduction ... 25
Rational m utagenesis of E. coli biotin ligase (BirA ) ... 26
First generation of yeast display-based directed evolution and implementation of signal am plification ... 27
Second generation of yeast display-based directed evolution and implementation of reductive cleavage of ligase ... 29
Third and fourth generations of yeast display-based directed evolution and implementation of negative selections... 31
Conclusion... 34
M aterials and m ethods ... 36
References ... 48
Chapter 3: Characterization of TurbolD and m iniTurbo ... 51
Introduction ... 52
TurbolD and m iniTurbo in m icrobes ... 53
TurbolD and m iniTurbo in m am m alian cells... 54
TurbolD and m iniTurbo in vitro ... 64
TurbolD and miniTurbo in vivo characterization with collaborators... 71
M aterials and m ethods ... 83
References ... 96
C hapter 4: Proteom ic m apping TurbolD and m iniTurbo... 99
Introduction ... 100
Characterizing the specificity and coverage of proteomes obtained with TurbolD and m iniTurbo... 100
Characterizing the labeling radius of TurbolD ... 107
Conclusion... 114
M aterials and m ethods ... 116
References ... 130
A ppendix ... 133
Part I: Investigation of m itochondrial cristae junctions ... 134
Introduction ... 134
G enetic targeting of A PEX to m itochondrial subdom ains ... 135
Electron microscopy imaging of mitochondrial cristae junctions using APEX... 140
Conclusion ... 141
M aterials and m ethods... 143
References ... 149
Part II: Development of platforms to control genome engineering with light... 151
Introduction ... 151
A dapting SPA RK for light-gated genom e engineering ... 152
Employing a photocleavable protein for light-gated genome engineering... 157
Conclusion ... 161
M aterials and m ethods... 162
List of Figures
Figure 1-1. Proximity-dependent biotinylation catalyzed by proximity labeling enzymes... 16
Figure 1-2. Proximity-dependent biotinylation catalyzed by APEX peroxidases ... 17
Figure 1-3. Proximity-dependent biotinylation catalyzed by promiscuous biotin ligases... 18
Figure 2-1. Testing mutations in the active site E. coli biotin ligase (BirA). ... 26
Figure 2-2. Yeast display-based selection scheme ... 27
Figure 2-3. Tyramide signal amplification (TSA)23 improves biotin detection sensitivity on the y east su rfa ce ... 2 8 Figure 2-4. Employing TSA to amplify biotinylation signal on the yeast surface. ... 28
Figure 2-5. E volution of G I... 29
Figure 2-6. Employing TCEP treatment to de-enrich self-labeling mutants ... 30
Figure 2-7. E volution of G 2 ... 3 1 Figure 2-8. E volution of G 3 ... 3 1 Figure 2-9. G3 can utilize low concentrations of biotin and N-terminal domain deletion reduces ligase affin ity for b iotin ... 32
Figure 2-10. Employment of negative selections to de-enrich mutants that carry out biotinylation in the absence of exogenous biotin ... 32
Figure 2-11. Evolution of m iniTurbo... 33
Figure 2-12. Evolution of TurbolD ... 33
Figure 2-13. Mutations of TurbolD and miniTurbo. ... 35
Figure 2-14. Progress of directed evolution... 35
Figure 2-15. Examples of various gates drawn for FACS sorting ... 39
Figure 2-16. Summary of yeast display-based directed evolution of TurbolD and miniTurbo ... 41
Figure 3-1. Com parison of ligases in yeast... 53
Figure 3-2. Com parison of ligases in E. coli ... 54
Figure 3-3. Cooling samples to 4'C terminates ligase-catalyzed biotinylation... 55
Figure 3-4. Comparison of different generation ligase activities in HEK cytosol... 56
Figure 3-5. Comparison of BioID, TurbolD, and miniTurbo promiscuous biotinylation activity in H E K cy to so l... 5 7 Figure 3-6. Comparison of TurbolD and miniTurbo to three other promiscuous ligases (BioID 1, BioID2", and BASU13) in the cytosol of HEK 293T cells... 58
Figure 3-7. Comparison of ligase activities in HEK cytosol by fluorescence microscopy ... 60
Figure 3-8. Comparison of promiscuous ligases in multiple HEK organelles. ... 61
Figure 3-9. Comparison of ligase activities in HEK cytosol by fluorescence microscopy. ... 61
Figure 3-10. Long incubations with exogenous biotin cause mislocalization of mitochondrial matrix-targeted TurbolD and BiolD in HEK 293T cells... 62
Figure 3-11. Viability assays in HEK 293T cells expressing promiscuous ligase variants... 63
Figure 3-12. miniTurbo cannot be purified using standard protocols for BirA purification... 64
Figure 3-13. miniTurbo cannot be solubilized using standard BirA purification protocol ... 65
Figure 3-14. Detergent-based lysis does not solubilize miniTurbo for purification... 66
Figure 3-15. Maltose binding protein-fusions to ligases result in degradation... 66
Figure 3-17. in vitro biotinylation of BSA by purified ligases. ... 68
Figure 3-18. in vitro biotinylation of HEK 293T whole-cell lysate by purified ligases... 69
Figure 3-19. Time-course of ligase-catalyzed synthesis of biotin-5'-AMP ... 70
Figure 3-20. Comparison of ligases in the larval wing disc of D. melanogaster... 71
Figure 3-21. Comparison of ligases in adult flies ... 72
Figure 3-22. Further comparison of ligase activities in D. melanogaster with shorter labeling tim e s... 7 3 Figure 3-23. Morphology and viability assays in D. melanogaster expressing promiscuous biotin lig a se s... 7 4 Figure 3-24. Comparison of ligase activity in embryonic C. elegans intestine by fluorescence m icro sco p y ... 7 8 Figure 3-25. Comparison of ligase activity in adult C. elegans intestine by Western blotting. ... 79
Figure 3-26. Effect of promiscuous ligase variants on C. elegans viability and development... 81
Figure 4-1. Characterization of nuclear and mitochondrial matrix-targeted ligases by fluorescence m icroscopy... 10 1 Figure 4-2. Mass spectrometry-based proteomic experiment comparing BiolD, TurbolD, and m iniTurbo in the nucleus or m itochondrial m atrix... 102
Figure 4-3. Western blot and gel characterization of proteomic samples from Figure 4-2 for (a) nucleus and (b) mitochondrial matrix proteomic experiments ... 103
Figure 4-4. Determination of proteins preferentially labeled by ligases in proteomic experiments ... 1 0 5 Figure 4-5. The majority of proteins in the final proteomes are detected by all three ligases.... 106
Figure 4-6. Specificities of nuclear and mitochondrial matrix proteomes obtained via BiolD (18 hr), TurbolD (10 min), and miniTurbo (10 min)-catalyzed labeling ... 106
Figure 4-7. Coverage analysis for the nuclear and mitochondrial matrix proteomic datasets.... 107
Figure 4-8. Proximity labeling with BiolD, TurbolD, and miniTurbo at the ER and mitochondrial m em b ran es. ... 10 8 Figure 4-9. Mass spectrometry-based proteomic experiment comparing TurbolD and BiolD on the ER m em brane (ERM ), facing cytosol... 109
Figure 4-10. Quality control analysis of ERM proteomic samples. ... 110
Figure 4-11. ERM proteins are enriched in the ERM proteomic datasets ... 112
Figure 4-12. Specificity analysis of ERM proteom e. ... 113
Figure 4-13. Coverage analysis for each proteomic dataset ... 114
Figure 4-14. Proteins biotinylated by TurbolD prior to addition of exogenous biotin... 115
Figure 4-15. ROC (receiver operating characteristic) analysis to determine TMT ratio cut-offs for filter 1 to generate final proteom ic datasets... 125
Figure 4-16. ROC (receiver operating characteristic) analysis to determine TMT ratio cut-offs for filter 1 to generate final proteom ic datasets... 127
Figure 4-17. ROC (receiver operating characteristic) analysis to determine TMT ratio cut-offs for filter 1 to generate final proteom ic datasets... 128
Figure 5-1. Characterization of APEX2 fusion proteins targeting the mitochondrial intracristal space by fluorescence m icroscopy... 135
Figure 5-2. Characterization of APEX2 fusion proteins targeting the mitochondrial intracristal space by light m icroscopy ... 136 Figure 5-3. Characterization of APEX2 fusion proteins targeting the mitochondrial peripheral IM S by fluorescence m icroscopy... 137 Figure 5-4. Characterization of APEX2 fusion proteins targeting the mitochondrial peripheral IM S by light m icroscopy ... 137 Figure 5-5. Characterization of APEX2 fusion proteins targeting the mitochondrial cristae
junction by fluorescence m icroscopy... 138 Figure 5-6. Characterization of APEX2 fusion proteins targeting the mitochondrial cristae
junction by light m icroscopy. ... 139 Figure 5-7. Comparison of cristae junction targeted-APEX2 fusion protein activities in HEK c y to so l ... 13 9 Figure 5-8. Transmission electron micrograph of mitochondria containing CHCHD3-APEX2 fu sio n p ro tein ... 14 0 Figure 5-9. Transmission electron micrograph of mitochondria containing mitofilin-APEX2 fu sio n p ro tein . ... 14 1 Figure 6-1. Strategy to control Cas9 genome editing activity using SPARK-like platform... 153 Figure 6-2. Assaying SPARK-like strategy to light-gate dCas9-VPR by fluorescence microscopy ... 1 5 4 Figure 6-3. Assaying reporter gene turn-on by free dCas9-VPR by fluorescence microscopy.. 154 Figure 6-4. Assaying negative controls for SPARK-like strategy to light-gate dCas9-VPR by fluorescence m icroscopy ... 155 Figure 6-5. Assaying SPARK-like strategy to light-gate stably expressed dCas9-VPR by
fluorescence m icroscopy... 156 Figure 6-6. Strategy to control Cas9 genome editing activity using PhoCl... 157 Figure 6-7. Assaying PhoCl strategy to light-gate dCas9-VPR by fluorescence microscopy.... 158 Figure 6-8. Assaying PhoCl strategy to light-gate dCas9-VPR by fluorescence microscopy with m od ified p roto co l... 15 8 Figure 6-9. Intracellular yeast-based directed evolution platform for PhoCl.. ... 159 Figure 6-10. Fluorescence assisted cell sorting (FACS) analysis of yeast with a fluorescent
protein (FP) reporter stably integrated in their genome and transiently expressing the
List of Tables
Table 2-1. Mutations in TurboID, miniTurbo, BiolD and key intermediate clones... 34
Table 2-2. Table of plasmids used in this chapter. ... 36
Table 2-3. List of antibodies used in this chapter. ... 40
Table 3-1. Table of plasmids used in this chapter. ... 83
Table 3-2. Table of antibodies used in this chapter. ... 88
Table 4-1. List of plasmids used in this chapter. ... 116
Table 4-2. List of antibodies used in this chapter. ... 119
Table 5-1. List of plasmids used in this chapter. ... 143
Table 5-2. List of antibodies used in this chapter... 147
Table 6-1. List of plasmids used in this chapter. ... 162
List of abbreviations 8-oxo-dGTP ADP AMP AP APEX APX ATP biotin-5'-AMP BirA BRB BSA CD4 CFP CIBI CIBN CoA CreER CRISPR CRY2 DAB DNA dPTP ER ER ERM FACS FP GFP GOCC GPCR HEK His6 HRP IP IP LOV miniSOG MS NEB Ni-NTA OMM PL 8-oxo-2'-deoxyguanosine-5'-triphosphate adenosine diphosphate adenosine monophosphate acceptor peptide enhanced APX ascorbate peroxidase adenosine triphosphate biotin adenosine monophosphate E. coli biotin ligase BirA reaction buffer bovine serum albumin cluster of differentiation 4 cyan fluorescent protein cryptochrome-interacting basic-helix-loop-helix
truncated CIBI aa 1-170 coenzyme A Cre recombinase estrogen receptor fusion clustered regularly interspaced short palindromic repeats cryptochrome 2 3,3'-diaminobenzidine deoxyribonucleic acid 2'-deoxy-P-nucleoside-5'-triphosphate endoplasmic reticulum estrogen receptor endoplasmic reticulum membrane fluorescence activated cell sorting fluorescent protein green fluorescent protein gene ontology cellular compartment G-protein coupled receptor human embryonic kidney cells histidine hexamer protein tag horseradish peroxidase immunoprecipitation intraperitoneal injection light-oxygen-voltage mini singlet oxygen generator
mass spectrometry New England BioLabs nickel nitrilotriacetic acid outer mitochondrial membrane proximity labeling
PMSF ROC SD/GCAA SDCAA SDS SDS-PAGE SPARK Ste2 TCEP TEV TLC TRE3G VPR phenylmethylsulfonylfluoride receiver operating characteristic synthetic dextrose/galactose plus casein amino acid synthetic dextrose plus casein amino acid sodium dodecyl sulfate sodium dodoceyl sulfate polyacrylamide gel electrophoresis Specific Protein Association tool giving transcriptional Readout with rapid Kinetics pheromone alpha factor receptor tris(2-carboxyethyl)phosphine hydrochloride tobacco etch virus thin-layer chromatography Retro-X Tet-on 3G
Chapter 1: Introduction to proximity labeling for spatial proteomics in living cells
The text and figures in this chapter were adapted from Branon, T. et al. Efficient proximity labeling in living cells and organisms with TurbolD, Nature Biotechnology (2018) and Branon, T. et al. Beyond immunoprecipitation: exploring new interaction spaces with proximity biotinylation,
Introduction
Compartmentalization within cells creates unique biochemical environments that allow for a wide variety of biological processes. For example, the oxidizing chemical environment of the endoplasmic reticulum (ER) facilitates efficient folding of newly synthesized proteins', while differences in pH between compartments on either side of the mitochondrial inner membrane drive the synthesis of adenosine triphosphate (ATP)2. Cellular compartmentalization also allows specific molecules to be concentrated together or to be segregated from others. For example, enzymes involved in a biosynthetic pathway may be concentrated together in a cellular compartment to increase the rate of synthesis; or signaling proteins may be segregated from their downstream targets to control the timing of signal transduction.
To understand the function of a subcellular compartment and its role in the cell, we need to know the identity of the molecules that compose or reside in it, as well as how this composition can change under different cellular conditions. A key class of macromolecules that occupy these subcellular compartments and are essential to our understanding of any biological process is proteins. Proteins play a significant role in nearly every cellular process, from enzymes catalyzing chemical reactions, to structural proteins shaping the cell's internal and external structure, to signaling proteins that facilitate intra- and intercellular communication and sensing of the cell's environment. Therefore, the ability to characterize endogenous proteins - their localization, trafficking, and interactions - within the native context of the living cell would greatly advance our understanding of a multitude of cellular processes and pathologies.
There are several approaches to study proteins in the context of cells, but few that allow for their identification and cataloging in a high-throughput manner. One such technique is mass spectrometry (MS)-based proteomic analysis, which allows for the identification of thousands of endogenous proteins from complex samples, such as those obtained from cells and tissues3. This technique requires cell lysis to extract proteins for identification, which disrupts membranes and macromolecular structures, and results in the loss of spatial information associated with the proteins identified. However, biochemical fractionation or enrichment protocols can be employed to isolate subcellular organelles or macromolecular complexes prior to MS analysis to recover some spatial information. These methods and their limitations are discussed below.
Traditional methods for spatially resolved proteomics
A widely used approach to preserve spatial information in MS-based proteomic analysis is to fractionate lysed cells prior to analysis to yield subcellular fractions that contain different organelles or cellular structures4. To keep organelles and other cellular structures as intact as possible, subcellular fractionation typically employs gentle cell lysis protocols aimed to disrupt only the plasma membrane. This includes mechanical methods such as sonication, or non-physical methods such as hypo-osmotic shock. To separate the lysate into subcellular fractions, centrifugation is often employed. Centrifugation of cell lysates can separate organelles and subcellular structures based on their size and density, with larger and denser organelles and structures sedimenting at lower centrifugal forces. Density gradients can be employed to obtain higher resolution separation of subcellular structures. Lysates loaded on top of gradients of various solutions with different osmolarities, viscosities, or densities, then upon centrifugation, organelles accumulates in the gradient where its density equals the density of the surrounding media. Because
density is determined by properties such as lipid/protein content and shape, organelles that are of a similar size can be resolved from each other and separated into different fractions4.
While subcellular fractionation can allow isolation of organelles or subcellular structures for MS-proteomic analysis, fractionation is imperfect, and it is impossible to obtain pure samples using this technique. This results in high negative rates from loss of material, and high false-positive rates from contaminants. In addition, this technique is inherently limited to subcellular fractions that can be isolated. While organelles such as the nucleus and mitochondria can be isolated intact, organelles such as the endoplasmic reticulum fragment during lysis and centrifugation and cannot be recovered whole. Sub-organellar compartments, such as the mitochondrial matrix or intermembrane space, are also difficult to access by biochemical fractionation techniques. There are also some subcellular structures, such as the ER-mitochondrial contact sites, which cannot be isolated through biochemical fractionation at all.
Spatial information can also be recovered using more focused techniques, such as immunoprecipitation (IP), which can allow for the isolation of specific protein complexes and identification of protein-protein interactions. In IP, an antibody against the endogenous protein or a genetically fused tag can be used to purify a specific protein from cell lysate, called the "bait"'. Because gentle lysis techniques are employed during sample preparation for IP, protein complexes are not disrupted, which allows proteins that interact with the bait protein to also be isolated and identified using MS.
While IP is a powerful technique for discovering protein-protein interactions and mapping protein complexes, several studies have shown that some of the interaction partners detected using IP can result from nonspecific interactions that occur after cell lysis. For example, a study investigating the interactions of ribonucleoprotein complexes definitively showed that some of the IP-enriched interactors had resulted from rearrangement post-lysis and did not recapitulate the in
vivo state6. Upon cell lysis, biologically irrelevant interactions can form when proteins, which
could be partially unfolded by the detergents used for lysis, encounter proteins with which they would not interact under physiological conditions. This can be particularly troublesome for proteins that are intrinsically unstructured or unfolded, such as some microproteins 7. Furthermore,
dissociation of these non-specific interactions can be difficult due to the low stringency of washes that are necessary to ensure true-positive interactions are not dissociated. In addition to high rates of false-positive detections, IP has a high false-negative rate because it only allows the identification of proteins that form strong or stable interactions with the bait protein, and often misses weak or transient interactors. This false-negative rate can be reduced by employing crosslinking techniques, which stabilizes weak interactions between proteins8, but this can also further stabilize of non-specific interactions.
Enzymatic proximity labeling methods
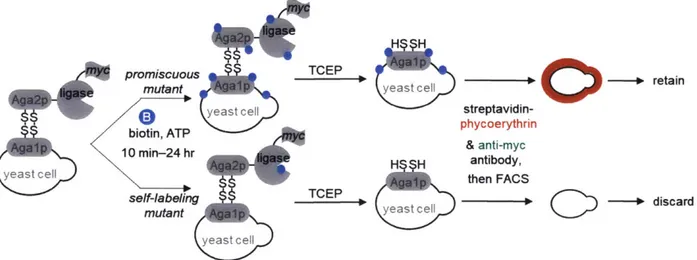
Enzyme-catalyzed proximity labeling (PL) is an alternative to immunoprecipitation and biochemical fractionation for proteomic analysis of macromolecular complexes, organelles, and protein interaction networks9. In PL, a promiscuous labeling enzyme is targeted by genetic fusion to a specific protein or subcellular compartment. Addition of a small molecule substrate, such as biotin, initiates covalent tagging of endogenous proteins within a few nanometers of the promiscuous enzyme (Figure 1-1). Subsequently, the biotinylated proteins are harvested using streptavidin-coated beads and identified by mass spectrometry (MS). Unlike biochemical fractionation and IP, PL enables the recording of proteomic information while the cell is intact,
preserving all spatial information. Due to the high-affinity interaction between biotin and streptavidin, more stringent washes can be used to dissociate non-specific binders while retaining tagged proteins, resulting in decreased false-positive rates and higher signal-to-noise ratios. Two enzymes commonly used for PL, APEX210'" and BiolD12,13, are discussed below.
distal endogenous
biotin proteins
proximal endogenous protein
Figure 1-1. Proximity-dependent biotinylation catalyzed by proximity labeling enzymes. Proximity labeling enzymes catalyze the formation of a reactive chemical species containing a chemical handle, such as biotin, which diffuses out of the active site to biotinylate proximal endogenous proteins.
APEX2 is a proximity labeling enzyme that was derived from soybean ascorbate peroxidase (APX), which acts as a cytosolic hydrogen peroxide scavenger in plants'4. The Ting lab used rational design and structure guided mutagenesis to engineer a more active and
monomeric variant of APX, called enhanced APX (APEX)1 5. Through yeast display-based
directed evolution of APEX, an additional mutation was found that further increases its activity, resulting in APEX2". In proximity labeling, APEX2 can oxidize biotin phenol to generate biotin phenoxyl radicals, which can then diffuse away from the active site and covalently react with electron rich side chains on proteins, such as tyrosine (Figure 1-2)10. Because of the radicals are very reactive and have a half-life < 1 ms, the labeling radius of APEX2 is small (< 20 nm) and only proteins proximal to APEX2 will be sufficiently tagged' 0.
One advantage of APEX2 is its speed: with a kcat = 299 s-1, APEX2 can quickly generate sufficient quantities of biotin phenoxyl radicals to tag proximal proteins in 1 min or less. This feature of APEX2 enables dynamic analysis of protein interaction networks, such as the mapping of GPCR signaling pathways in real time 16,". APEX2 is also versatile: the biotin phenoxyl radicals not only react with proteins, but also nucleic acids, enabling the spatial distribution of endogenous RNAs throughout the cell to be mapped18 (unpublished work from the Ting lab).
The versatility of APEX2 also lies in its promiscuous substrate specificity, which allows for applications outside of proteomics. For example, APEX2 can oxidize the fluorogenic dye Amplex red in the presence of hydrogen peroxide, allowing it to be used as a genetically-encoded hydrogen peroxide sensor with a colorimetric readout19. APEX2 can also catalyze the H202
-dependent polymerization of 3,3'-diaminobenzidine (DAB) into a localized precipitate that gives contrast in electron microscopy (EM) after treatment with OsO415, allowing it to be used as a genetically-encoded reporter for EM.
APEX and APEX2 have been used in cell culture to map various subcellular structures, such as mitochondrial sub-compartments10,2022, the ER membrane2 2, and the neuronal synaptic cleft2 3, as well as protein interaction networks1'6"7. While APEX has been implemented
proximity labeling pose challenges for its use in vivo. APEX labeling requires the use of hydrogen peroxide, which is toxic to cells and difficult to deliver into live organisms without causing severe
0 0 HN NH biotin phenol HN NH H H H H H H N N 0 s HO HO OH HN ) NH
APEX peroxidase 6 0proximal
endogenous protein
Figure 1-2. Proximity-dependent biotinylation catalyzed by APEX peroxidases. Peroxidases catalyze the formation of biotin phenoxyl radicals, which diffuses out of the active site to biotinylate proximal endogenous proteins on electron-rich residues such as tyrosine.
tissue damage. Furthermore, biotin phenol has limited cell permeability and requires long incubation times to diffuse through tissues. These limitations have resulted in APEX peroxidase being used in only three in vivo studies, and in each case, genetic modification to compromise cuticle integrity242 5 or manual dissection of tissue had to be performed26 to deliver APEX2
chemical substrates to the relevant cells.
BiolD is a second proximity labeling enzyme, derived from Escherichia coli biotin ligase (BirA). Like other biotin ligases, E. coli BirA covalently attaches the cofactor biotin to the active site of enzymes whose chemistry is biotin-dependent, such as carboxylases involved in fatty acid metabolism27. To carry out biotinylation of biotin-dependent enzmyes, wild-type BirA adenylates biotin to create a reactive intermediate, biotin adenosine monophosphate (biotin-5'-AMP), which is held tightly in its active site until it contacts a target protein. Upon contact, BirA transfers the biotin to a specific lysine side-chain on the target protein. In the entire E. coli proteome, BirA recognizes and biotinylates only a single lysine residue - lysine 122 of the biotin carboxyl carrier protein subunit of acetyl-coenzyme A (CoA) carboxylase2 8,2 9
In addition to catalyzing the biotinylation of acetyl-CoA carboxylase, E. coli BirA is also a biotin sensor and can repress transcription of the biotin biosynthetic operon30. When all biotin-dependent enzymes are loaded with biotin, BirA will retain the biotin-5'-AMP intermediate in its active site, which causes the ligases to homodimerize31. The homodimer can then bind the promoter of the biotin biosynthetic operon through their N-terminal domains, which are DNA-binding domains3 2, and repress transcription of biotin biosynthetic machinery. This bifunctionality
of E. coli BirA allows the cell to negatively regulate the energetically costly biosynthesis of biotin when it is present in excess. Several mutations in the dimer interface have been found to repress dimerization and DNA binding, such as the deletion of the alanine at position 146 (A146A)31 which is included in all biotin ligase mutants used in this study.
Cronan et al. reported that the high sequence-specificity of BirA biotinylation could be abolished by a single point mutation at arginine 118 to glycine12, which was incorporated into BirA by Roux et al. to develop BioID13. The RI 18G mutation weakens the affinity of the enzyme for
the reactive biotin-5'-AMP species, allowing it to diffuse out and react with nearby nucleophiles, such as deprotonated lysine side-chains (Figure 1-3). The rate of biotin-5'-AMP release from BiolD increases approximately 440-fold from that of wild-type33,34. However, the rate of biotin-5'-AMP formation by BiolD also decreases approximately 5-fold relative to wild-type33'34,
suggesting that the RI 18G mutation also hinders the ability to generate the reactive species. The half-life of adenylates such as biotin-5'-AMP have been measured to be on the order of several minutes in vitro3, giving it a potential reactive radius that is on the order of millimeters. Although the reactive biotin-5'-AMP intermediate is likely hydrolyzed slowly in solution, the high concentrations of nucleophilic protein side-chains and small molecules inside cells are likely to shorten its lifetime. Roux et al. experimentally approximated the reactive radius of BioID-generated biotin-5'-AMP in living cells to be roughly 10 nm36.
ATP 0 0 0 N\ 0 P P.P, N=\ O 0 1 LO OH biotin H2N0 0 0 0 NttN HO OH H HN-H NH 0 s HN HN H 01 HH N N 0 1-0 A BiolD H2N H 0 S H NyN HO proximal endogenous protein
Figure 1-3. Proximity-dependent biotinylation catalyzed by promiscuous biotin ligases. Ligases catalyze the formation of biotin-5'-AMP anhydride, which diffuses out of the active site to biotinylate proximal endogenous proteins on nucleophilic residues such as lysine.
In contrast to APEX peroxidases, BiolD labeling is simple and non-toxic: only biotin needs to be supplemented to initiate labeling. This feature has resulted in >100 applications of BiolD since its introduction 5 years ago, in cultured mammalian cells'3,36,37, plant protoplasts38,
parasites39-47, slime mold4849, mouse50, and yeastl. Bio ID has been used, for example, to map the
protein composition of the centrosome-cilium interface37 and the inhibitory post-synaptic region50,
each with nanometer spatial specificity. Because biotin is non-toxic and easily deliverable, BiolD is more amenable for proximity labeling in vivo. Despite this, however, there have been only two
in vivo demonstrations to date5052. This is likely related to BioID's low catalytic activity, which makes BiolD difficult or impossible to apply in some contexts -such as in worms, flies, or the ER lumen. Furthermore, its slow kinetics necessitates labeling with biotin for 18-24 hours (and sometimes much longer 0) to produce sufficient biotinylated material for proteomic analysis. This precludes the use of BiolD for studying dynamic processes that occur on the timescale of minutes or even a few hours.
Recently, two new promiscuous biotin ligase variants, BioID25 3 and BASU54, have been reported. BioID2 was derived from Aquifex aeolicus biotin ligase and made promiscuous by introducing the point mutation R36G, which is analogous to the RI 18G mutation in BiolD. The A.
smaller than E. coli BirA53. Because BioID2 is a smaller tag than BiolD, it could result in less
perturbation of protein folding or trafficking when fused to a bait protein. Additionally, because A. aeolicus is a thermophilic bacterium, BioID2 can be used at temperatures higher than 37'C, which may be useful for some applications.
BioID2 has a higher affinity for biotin than BiolD, and can carry out promiscuous biotinylation with lower concentrations of biotin 3. While this may be advantageous when performing proximity labeling in organisms with poor biotin uptake mechanisms, it can be very disadvantageous in contexts such as cell culture where free biotin concentrations in the media and biotin uptake are high enough to induce promiscuous biotinylation by BioID2 intracellularly. The use of endogenous biotin present in media by BioID2 means that it can carry out biotinylation of endogenous proteins the entire time it is being expressed, which removes any temporal control of labeling by the user (through the addition of exogenous biotin to initiate labeling). This lack of temporal control prevents the use of BioID2 to probe dynamic proteomes, limiting its application in certain contexts. Users can regain temporal control of the labeling window by depleting biotin from the media, however this starves cells of biotin and can lead to poor cell health and proteome perturbation. Furthermore, even when user-control of the labeling window is regained, BioID2
still requires long labeling times of over 16 hr53'55-57, therefore the scope of dynamic processes
that can be probed using BioID2 is still narrow.
BASU is a promiscuous biotin ligase derived from the Bacillus subtilis biotin ligase54. To develop BASU, the N-terminal domain was removed, and 4 mutations were introduced: R124G, which is analogous to the E. coli BirA RI 18G mutation to impart promiscuity, and 3 additional mutations in the C-terminal domain that are analogous to residues in other biotin ligases and are thought to improve ATP binding 4.
While the creators of BASU claim that it has >1000-fold faster kinetics than BioLD and requires a 1-minute labeling window, there is only a single side-by-side comparison of BASU and BiolD published54. For this comparison, exogenous biotin was added to cells expressing BASU
and BiolD for one hour, then the whole cell lysates were blotted with streptavidin to visualize the extent of biotinylation on endogenous proteins. This comparison showed that BASU gives approximately 7-fold more higher streptavidin signal than BiolD after one hour of labeling54;
however, the comparison show only this single time point, and did not show how BASU labeling compared to the labeling yield from BiolD after the standard labeling window of 18 hr (which indicates the approximate yield of biotinylated material required for proteomic analysis). Furthermore, this comparison was performed in biotin-starved cells, which could have affected cell health and ligase expression, stability, or activity.
BASU was also used in the context of RaPID, which is a method that allows for the identification of RNA-binding proteins54. In RaPID, BASU is fused to the protein ?N, which can
bind to the RNA stem loop BoxB. This RaPID-BASU construct is co-transfected with an RNA component that contains the BoxB stem loop flanking an RNA motif of interest. XN binding to BoxB brings BASU into proximity with the RNA motif of interest, allowing biotinylation of RNA-binding proteins that interact with the RNA motif of interest. The creators of BASU claim that it has 1-minute labeling window and >1000-fold faster kinetics because they were able to detect biotinylation of a single known RNA-binding protein after pulsing with biotin for 1 minute. However, biotinylation of a single protein does not reflect the ligases capability of generating enough biotinylated material for proteomics during the I -minute labeling window. In fact, in a proteomics experiment using BASU-RaPID labeling for 30 minutes, only two proteins were enriched54, suggesting the biotinylation yield is very low after 30 minutes and is insufficient for
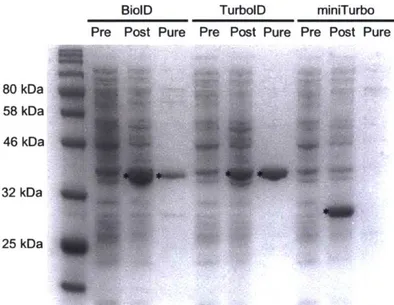
proteomic analysis. Additional comparisons or proteomic studies using BASU have yet to be published. Further characterization shown here indicates that the activities of BiolD, BioID2, and BASU are all comparable (Chapter 2).
Conclusion
Knowing the localization, trafficking, and interactions of a protein are key to understanding its function and its role in a biological process. MS-based proteomic analysis allows high throughput identification of proteins from complex samples such as cell lysates or tissue homogenate, however traditional techniques to retain spatial information have limitations. For example, biochemical fractionation allows organelles to be isolated for MS analysis of subcellular proteomes, but purifications are often incomplete from loss of material and contain contaminants. IP is a widely used technique that allows the identification of protein-protein interactions and cataloging of protein complexes, but transient and weak interactions are often missed, and the nature of the protocol can result in high rates of false-positives.
Enzyme-catalyzed proximity labeling has emerged as an alternative to these traditional techniques, allowing proteomic inventorying of subcellular compartments as well as identification of protein-protein interactions. The enzymes employed in this technique catalyze the covalent tagging of endogenous proteins with a chemical handle, such as biotin, within tens of nanometers of the enzyme. Because the enzymes are genetically encoded, they can be targeted to the compartment of interest or fused to the protein of interest, and the tagging of proximal endogenous proteins can be catalyzed by the enzyme while the cell is live. Because the cell is intact when labeling takes place, physiologically relevant spatial information is preserved. Furthermore, because tagging of endogenous proteins is based on proximity and not affinity, weak and transient protein-protein interactions are captured.
Existing proximity labeling methods, such as APEX2 and BioLD, have resulted in over a hundred proteomic applications over the past several years; yet despite their wide adoption, both methods have shown to have limitations. APEX2 catalyzes labeling of endogenous proteins on the order of minutes, allowing dynamic proteomes to be probed with high temporal resolution; however, the substrates required are toxic and have poor cell permeability, limiting its application
in vivo. BiolD has simple and non-toxic labeling conditions, however its low catalytic activity
limits its application in several biological contexts and prevents its use in analyzing many dynamic proteomes.
A new proximity labeling method that combines the catalytic efficiency of APEX2 and the non-toxic labeling conditions of BiolD would open much more biological space to exploration, particularly in vivo. In this thesis, we discuss the development of such a method, which we have named TurbolD, and its characterization in several biological contexts, and its applications for proteomics. Chapter 2 will describe how rational design and yeast display-based directed evolution was used to engineer E. coli biotin ligase (BirA) to generate two promiscuous variant of
the ligase, TurbolD and miniTurbo, which both carry out proximity labeling much more efficiently than any existing biotin ligase-based proximity labeling method. Chapter 3 will describe the characterization of TurbolD and miniTurbo in vitro and in mammalian cells, yeast, bacteria,
Drosophila melanogaster, and C. elegans. Chapter 4 will describe the application of TurboLD and
miniTurbo for proteomic mapping in living cells and will show that proteome quality and labeling radius remain the same as BiolD despite having >100-fold shorter labeling times.
The appendix will discuss additional projects. Part I describes how APEX2 could be used to map mitochondrial cristae junctions and will show EM imaging of mitochondria with APEX2 targeted to cristae junctions. Part II describes the development of a platform to control genomic editing with light.
References
1. Braakman, I. & Hebert, D. N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. (2013). doi:10.1101/cshperspect.a013201
2. Alberts, B. et al. Molecular Biology of the Cell. 4th Edition, New York (2002).
doi:10.1091/mbc.E14-10-1437
3. Aebersold, R. & Mann, M. Mass spectrometry-based proteomics. Nature (2003).
doi: 10.103 8/nature015 11
4. Lee, Y. H., Tan, H. T. & Chung, M. C. M. Subcellular fractionation methods and strategies for proteomics. Proteomics (2010). doi: 10.1002/pmic.201000289
5. Ten Have, S., Boulon, S., Ahmad, Y. & Lamond, A. I. Mass spectrometry-based immuno-precipitation proteomics -The user's guide. Proteomics (2011). doi:10.1002/pmic.201000548 6. Mili, S. & Steitz, J. A. Evidence for reassociation of RNA-binding proteins after cell lysis:
Implications for the interpretation of immunoprecipitation analyses. RNA (2004).
doi:10.1261/ma.7151404
7. Chu,
Q.
et al. Identification of Microprotein-Protein Interactions via APEX Tagging. Biochemistry (2017). doi:10.1021/acs.biochem.7b002658. Rappsilber, J. The beginning of a beautiful friendship: Cross-linking/mass spectrometry and
modelling of proteins and multi-protein complexes. J. Struct. Biol. (2011).
doi:10.1016/j.jsb.2010.10.014
9. Kim, D. I. & Roux, K. J. Filling the Void: Proximity-Based Labeling of Proteins in Living Cells. Trends in Cell Biology 26, 804-817 (2016).
10. Rhee, H.-W. et al. Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging. Science (80-. ). 339, 1328-1331 (2013).
11. Lam, S. S. et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51-54 (2014).
12. Choi-Rhee, E., Schulman, H. & Cronan, J. E. Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci. 13, 3043-50 (2004).
13. Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801-810 (2012).
14. Raven, E. L. Understanding functional diversity and substrate specificity in haem peroxidases:
What can we learn from ascorbate peroxidase? Natural Product Reports (2003).
doi: 10.1039/b210426c
15. Martell, J. D. et al. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 30, 1143-1148 (2012).
16. Paek, J. et al. Multidimensional Tracking of GPCR Signaling via Peroxidase-Catalyzed Proximity Labeling. Cell 169, 338-349.el 1 (2017).
17. Lobingier, B. T. et al. An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 169, 350-360.e12 (2017).
18. Kaewsapsak, P., Shechner, D. M., Mallard, W., Rinn, J. L. & Ting, A. Y. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. Elife 6, (2017).
19. Dwyer, D. J. et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. (2014). doi:10.1073/pnas.1401876111
20. Hung, V. et al. Proteomic Mapping of the Human Mitochondrial Intermembrane Space in Live Cells via Ratiometric APEX Tagging. Mol. Cell 55, 332-341 (2014).
21. Han, S. et al. Proximity Biotinylation as a Method for Mapping Proteins Associated with mtDNA in Living Cells. Cell Chem. Biol. 24, 404-414 (2017).
22. Hung, V. et al. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6, (2017).
23. Ting, A. Y. et al. Proteomic Analysis of Unbounded Cellular Compartments: Synaptic Clefts. Cell (2016). doi:10.1016/j.cell.2016.07.041
24. Reinke, A. W., Balla, K. M., Bennett, E. J. & Troemel, E. R. Identification of microsporidia host-exposed proteins reveals a repertoire of rapidly evolving proteins. Nat. Commun. 8, 14023 (2017). 25. Reinke, A. W., Mak, R., Troemel, E. R. & Bennett, E. J. In vivo mapping of tissue- and
subcellular-specific proteomes in Caenorhabditis elegans. Sci. Adv. 3, e1602426 (2017).
26. Chen, C.-L. et al. Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc. Natl. Acad. Sci. U. S. A. 112, 1-6 (2015).
27. Tong, L. Structure and function of biotin-dependent carboxylases. Cellular and Molecular Life Sciences (2013). doi: 10.1007/sOO 18-012-1096-0
28. Chapman-Smith, A. & Cronan, J. E. The enzymatic biotinylation of proteins: A post-translational
modification of exceptional specificity. Trends in Biochemical Sciences (1999).
doi:10.1016/SO968-0004(99)01438-3
29. Cronan, J. E. & Waldrop, G. L. Multi-subunit acetyl-CoA carboxylases. Progress in Lipid Research (2002). doi:10.1016/SO163-7827(02)00007-3
30. Streaker, E. D., Gupta, A. & Beckett, D. The biotin repressor: Thermodynamic coupling of corepressor binding, protein assembly, and sequence-specific DNA binding. Biochemistry (2002). doi: 10.102 1/bi0203839
31. Wood, Z. A., Weaver, L. H., Brown, P. H., Beckett, D. & Matthews, B. W. Co-repressor induced order and biotin repressor dimerization: A case for divergent followed by convergent evolution. J. Mol. Biol. 357, 509-523 (2006).
32. Buoncristiani, M. R., Howard, P. K. & Otsuka, A. J. DNA-binding and enzymatic domains of the bifunctional biotin operon repressor (BirA) of Escherichia. Gene (1986).
doi:10.1016/0378-1119(86)90189-7
33. Xu, Y. & Beckett, D. Kinetics of Biotinyl-5'-adenylate Synthesis Catalyzed by the Escherichia coli Repressor of Biotin Biosynthesis and the Stability of the Enzyme-Product Complex. Biochemistry 33, 7354-7360 (1994).
34. Kwon, K. & Beckett, D. Function of a conserved sequence motif in biotin holoenzyme synthetases. Protein Sci. 9, 1530-1539 (2000).
35. BERG, P. Studies on the enzymatic utilization of amino acyladenylates; the formation of adenosine triphosphate. J. Biol. Chem. (1958).
36. Kim, D. I. et al. Probing nuclear pore complex architecture with proximity-dependent
biotinylation. Proc. Natl. Acad. Sci. 111, E2453-E2461 (2014).
37. Gupta, G. D. et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 163, 1483-1499 (2015).
38. Lin, Q. et al. Screening of Proximal and Interacting Proteins in Rice Protoplasts by Proximity-Dependent Biotinylation. Front. Plant Sci. 8, (2017).
39. Morriswood, B. et al. Novel bilobe components in Trypanosoma brucei identified using proximity-dependent biotinylation. Eukaryot. Cell 12, 356-367 (2013).
40. Chen, A. L. et al. Novel insights into the composition and function of the Toxoplasma IMC sutures. Cell. Microbiol. 19, (2017).
41. Nadipuram, S. M. et al. In Vivo biotinylation of the toxoplasma parasitophorous vacuole reveals novel dense granule proteins important for parasite growth and pathogenesis. MBio 7, (2016). 42. Chen, A. L. et al. Novel components of the toxoplasma inner membrane complex revealed by
BioID. MBio 6, (2015).
43. Long, S. et al. Calmodulin-like proteins localized to the conoid regulate motility and cell invasion by Toxoplasma gondii. PLoS Pathog. 13, (2017).
44. Zhou, Q., Hu, H. & Li, Z. An EF-hand-containing protein in Trypanosoma brucei regulates cytokinesis initiation by maintaining the stability of the cytokinesis initiation factor CIF 1. J. Biol. Chem. 291, 14395-14409 (2016).
45. Dang, H. Q. et al. Proximity interactions among basal body components in trypanosoma brucei identify novel regulators of basal body biogenesis and inheritance. MBio 8, (2017).
46. Kehrer, J., Frischknecht, F. & Mair, G. R. Proteomic Analysis of the Plasmodium berghei Gametocyte Egressome and Vesicular bioID of Osmiophilic Body Proteins Identifies Merozoite TRAP-like Protein (MTRAP) as an Essential Factor for Parasite Transmission. Mol. Cell. Proteomics 15, 2852-2862 (2016).
47. Gaji, R. Y. et al. Phosphorylation of a Myosin Motor by TgCDPK3 Facilitates Rapid Initiation of Motility during Toxoplasma gondii egress. PLoS Pathog. 11, (2015).
48. Batsios, P., Ren, X., Baumann, 0., Larochelle, D. & Graf, R. Srcl is a Protein of the Inner Nuclear Membrane Interacting with the Dictyostelium Lamin NE81. Cells 5, 13 (2016).
49. Meyer, I. et al. CP39, CP75 and CP91 are major structural components of the Dictyostelium centrosome's core structure. Eur. J. Cell Biol. 96, 119-130 (2017).
50. Uezu, A. et al. Identification of an elaborate complex mediating postsynaptic inhibition. Science (80-. ). 353, 1123-1129 (2016).
51. Opitz, N. et al. Capturing the Asclp/ R eceptor for A ctivated C K inase 1 (RACKI) Microenvironment at the Head Region of the 40S Ribosome with Quantitative BioID in Yeast. Mol. Cell. Proteomics 16, 2199-2218 (2017).
52. Dingar, D. et al. BiolD identifies novel c-MYC interacting partners in cultured cells and xenograft tumors. J. Proteomics 118, 95-111 (2015).
53. Kim, D. I. et al. An improved smaller biotin ligase for BiolD proximity labeling. Mol. Biol. Cell 27, 1188-1196 (2016).
54. Ramanathan, M. et al. RNA-protein interaction detection in living cells. Nat. Methods (2018). doi: 10.103 8/nmeth.4601
55. Birendra, K. C. et al. VRK2A is an A-type lamin-dependent nuclear envelope kinase that phosphorylates BAF. Mol. Biol. Cell mbc.E17-03-0138 (2017). doi:10.1091/mbc.E17-03-0138 56. Redwine, W. B. et al. The human cytoplasmic dynein interactome reveals novel activators of
motility. Elife 6, (2017).
57. Jung, E. M. et al. Aridlb haploinsufficiency disrupts cortical interneuron development and mouse behavior. Nat. Neurosci. 20, 1694-1707 (2017).
Chapter 2: Directed evolution of TurbolD and miniTurbo
The text and figures in this chapter were reproduced and adapted from Branon, T. et al. Efficient proximity labeling in living cells and organisms with TurboID, Nature Biotechnology (2018).
Introduction
Enzyme-catalyzed proximity labeling has emerged as a powerful technique to map subcellular proteomes and identify protein-protein interactions in living cells. Existing methods, such as APEX2"2 and BioID3'4, have limitations that preclude their use in many biological
contexts, particularly in vivo, such as substrate toxicity and slow labeling kinetics. Therefore, we set out to develop a new method for proximity labeling that is more amenable for in vivo applications, with non-toxic labeling conditions, easily deliverable substrates, and high catalytic efficiency. In this chapter, we will describe how we developed such a method using rational design and directed evolution.
Because of the non-toxic and simple labeling conditions, we chose to develop our new method based on a biotin ligase. The challenge we faced is, how we can overcome the slow kinetics of these enzymes and engineer a more catalytically efficient enzyme? There are generally two approaches for protein engineering: rational design and directed evolution. These strategies and their limitations are discussed below.
In rational design, researchers utilize biochemical data, protein structure, and sometimes molecular modeling data to propose mutations that can be introduced into the protein using site-directed mutagenesis 5. Because these mutations are focused in regions of the protein that are related to the activity researchers seek to modify, there is higher probability of discovering beneficial mutations as well as smaller library sizes. This potentially requires much less time and effort to screen mutations, which can be particularly useful if there is no high throughput assay in place to screen for beneficial mutations. Rational design, however, is severely limited by our incomplete understanding of the protein functions and folding. We have yet to understand the properties that underly protein dynamics, such as protein flexibility and conformational changes6'; and it is still very difficult to predict the effects of mutations on protein expression and stability7'8, the effects of distant mutations that appear unrelated9, or how mutations can interact with one
another'-1
Directed evolution is a protein engineering method that mimics the process of natural selection to evolve a protein toward a user-defined goal. In directed evolution, a library of genetic mutants is generated that give rise to a population of proteins with phenotypic variation. This population can be subjected to selective pressure for a particular phenotype, which allows for amplification of the respective genotype within the population13. In contrast to rational design,
preexisting understanding of the protein's structure or biochemical mechanisms are not required for directed evolution, thus allowing researchers to overcome the great knowledge gap we have in protein folding and function. Because of the black-box nature of the selections employed in directed evolution, researchers can discover of beneficial mutations that are distant from the perceived region of interest and that are seemingly unrelated to the properties researchers seek to modify, or combinations of mutations that affect multiple protein properties such as activity and stability. However, because the selections are experimentally performed, directed evolution is limited by the quality and size of the libraries generated, the throughput of the selection and screening processes, and the ability to couple the desired protein modification with easily detectable signal during the selections'4.
In this chapter, we will discuss how both rational design and directed evolution were implemented to engineer Escherichia coli biotin ligase (BirA) to generate two promiscuous, highly catalytically efficient biotin ligase variants, which we have named TurbolD and miniTurbo.
Rational mutagenesis of E. coli biotin ligase (BirA)
As discussed in chapter 1, proximity labeling of endogenous proteins catalyzed by promiscuous biotin ligases occurs in two steps3: in the first step, the enzyme catalyzes the adenylation of biotin using ATP to generate the biotin-5'-AMP reactive intermediate; and in the second step, the reactive biotin-5'-AMP species is released from the enzyme's active site, and can then go on to covalently react with nucleophilic side chains on proteins. To engineer a more catalytically active promiscuous variant of E. coli biotin ligase (BirA), we can attempt to either increase the rate of biotin-5'-AMP synthesis, which we know is compromised by the RI 18G mutation that imparts promiscuity 5'"6, or we can attempt to further increase the rate of biotin-5'-AMP release.
We first examined the crystal structure of wild-type BirA'7-19 to rationally select residues to mutagenize. Two disordered loops near the BirA active site become ordered upon biotin binding (residues 115-125) and ATP binding (residues 212-221)18,20. Upon ordering, these loops surround biotin and ATP, partially enclosing the active site and forming several stabilizing interactions that facilitate the synthesis of biotin-5'-AMP. The RI 18G mutation that imparts promiscuity in BiolD resides in the biotin binding loop3'4. Removal of this arginine weakens the enzyme's affinity for biotin and biotin-5'-AMP 5, likely opening the active site and allowing biotin-5'-AMP to diffuse away from the enzyme. While this property allows promiscuous labeling of surrounding proteins, the extreme arginine to glycine mutation significantly compromises the efficiency of biotin-5'-AMP synthesis 5"6.
a
b
untransfected N NNNR118: G S ' T C N V P K 80-860UE 80 "0- n 503 40-3P
w4BirA 4. m o .4 BirA -40 4 CFP-AP 30 M wo ow q q m .4 CFP-AP 30 streptavidin-HRP streptavidin-HRP kD anti-myc kD anti-mycFigure 2-1. Testing mutations in the active site E. coli biotin ligase (BirA). The indicated mutants were transiently expressed as NES (nuclear export signal) fusions in the HEK cytosol. All samples were co-transfected with AP-CFP (biotin ligase acceptor peptide (AP) fused to cyan fluorescent protein), a known peptide substrate for wild-type BirA21. 50 pIM biotin was added to
the cells for 18 hr, then whole cell lysates were analyzed by streptavidin-HRP blotting. Ligase expression was detected by anti-myc blotting. wt, wild-type BirA. U, untransfected. Based on these results, we selected BirA-R 118S as our template for directed evolution of TurbolD.