Ab initio examination of ductility features of fcc metals
Texte intégral
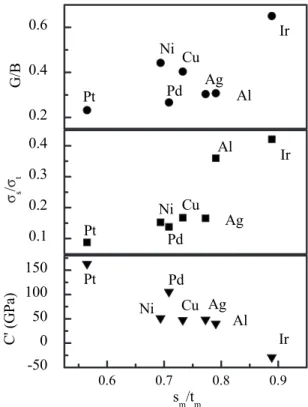
Figure
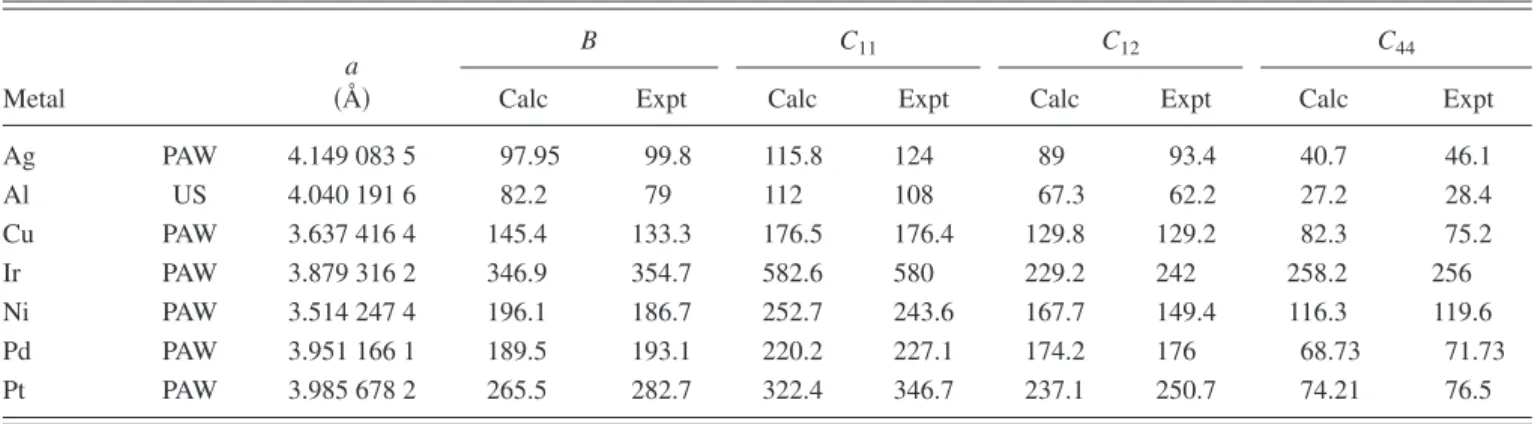
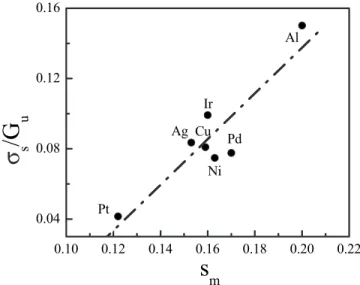
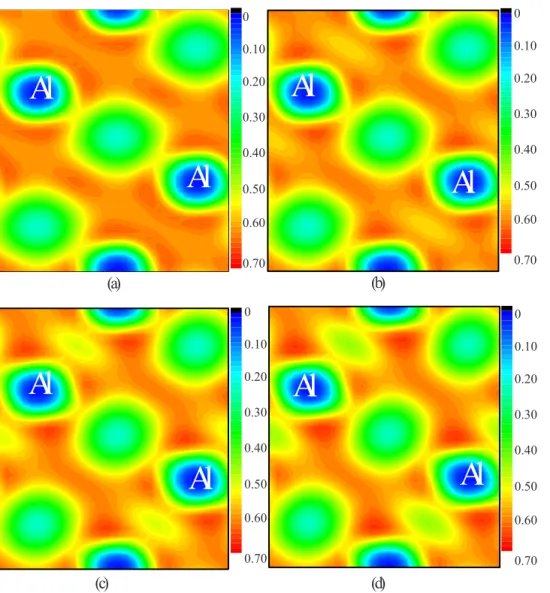
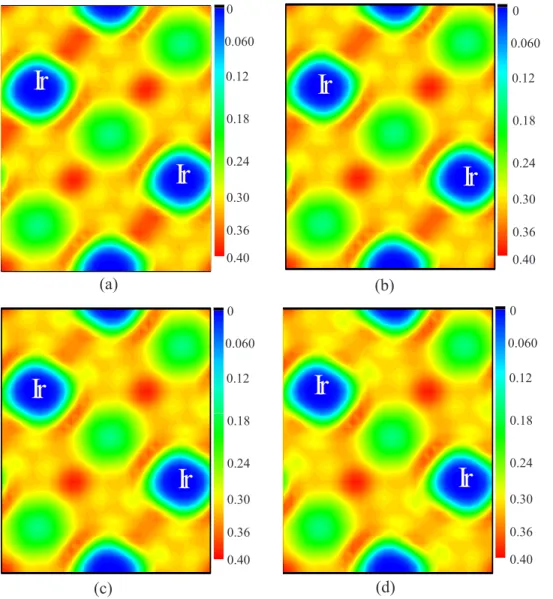


Documents relatifs
Using another value of the dielectric function as input for the model is possible but this value has to be computed for a small q 0 , meaning a fine grid to compute it,
The aim of the present study is to remedy the situation prohibiting the use of MgH 2 in devices by selective substitution of Mg with transition metals (T = Fe, Co, Ni, Pd, Pt) in a
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Our aim was to assess the prevalence and the determining factors of epileptic seizures anticipatory anxiety in patients with drug-resistant focal epilepsy, and the
Dans une maternité où l’accouchement par voie basse après césarienne est réalisé de principe pour les utérus uni-cicatriciels sauf en cas de contre-indication, on n’observe pas
Abstract.- AuFe and CuFe films, produced by quench condensation, show a round maximum of the initial susceptibility at temperatures T_ whereas annealed films and bulk alloys have
This means that the amplitudes of the wave functions should decrease at the surrounding Ni atoms to produce an electron deficit there ; the amount of the
substrate and the SFG measurements are performed in the total internal reflection configuration, drastic modifications in the C-H and N-H spectral ranges attest
