Design of an intraperitoneal drug-release device for advanced
ovarian cancer therapy
by
Laura Melanie Tanenbaum B.S. in Chemical Engineering
Rice University, 2011
Submitted to the Harvard-MIT Program in Health Sciences and Technology in partial fulfillment of the requirements for the degree of
Doctorate of Philosophy in Medical Engineering and Medical Physics at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY June 2016
C 2016 Massachusetts Institute of Technology. All rights reserved.
ACfMIVES
MASSACHUSETTS INSTITUTE OF TECHNOLOGYJUN 2
2
2016
LIBRARIES
Signature of Author: ... Certified by: ...ASi
Accepted by:...Signature redacted-
...
arvard-MI Prograpi t~ealth Sciences and Technology &# '
-3A Z / zMay 17, 2016
Signature redacted
e...
Michael J. Cima, PhDDavid H. Koch Professor of Engineering Thesis Supervisor
gr
nature
redacted
Emery N. Brown, MD, PhD (--6irector, Harvard-MIT Program in Health Sciences and Technology Professor of Computational Neuroscience and Health Sciences and Technology
'
Design of an intraperitoneal drug-release device for advanced
ovarian cancer therapy
by
Laura Melanie Tanenbaum
Submitted to the Harvard-MIT Program in Health Sciences and Technology on May 18, 2016 in partial fulfillment of the requirements for the degree of
Doctorate of Philosophy in Medical Engineering and Medical Physics
Abstract
More than 14,000 women in the United States die from ovarian cancer each year. The standard of care is tumor-debulking surgery followed by adjuvant chemotherapy. Combination intraperitoneal (IP) and intravenous (IV) chemotherapy has been shown to lengthen survival over IV therapy alone. Large-volume infusions, drug-associated toxicity, and catheter-associated complications, however, increase morbidity and limit patient adherence, often resulting in discontinuation of IP therapy. The technical skill required for catheter implantation and IP chemotherapy administration has also limited its clinical adoption.
The proposed solution is an implantable IP device capable of localized drug delivery that maintains the efficacy of the standard of care and overcomes current clinical challenges. A reservoir-based device was developed to release cisplatin at a constant rate. In vivo studies demonstrated that continuous dosing reduces tumor burden to the same extent as weekly IP injections. The implanted device induced significantly less systemic toxicity compared to IP injections, despite administration of higher cumulative doses. A subsequent in vitro study revealed that greater tumor shrinkage following continuous cisplatin exposure was achieved with smaller tumor nodules. These results support that an implanted device would be maximally effective against microscopic residual disease. In vitro results also illustrated that a human-scale device fabricated from orifice-lined silicone can be designed to release cisplatin continuously at the desired rate.
The promising preclinical results in this thesis highlight the potential for this novel IP dosing regimen to improve the treatment of late-stage ovarian cancer and set the stage for development of the proposed human device.
Thesis Supervisor: Michael J. Cima, PhD Title: David H. Koch Professor of Engineering
Acknowledgements
I would first like to thank my advisor, Michael Cima, for his support and mentorship over the last five years. Your guidance has helped me gain confidence as a scientist and an independent researcher, and I have learned an incredible amount during my time at MIT. I would also like to thank you for your invitations to Maine each summer. It was a joy to spend time with you and your family at your beautiful home away from home.
I am also extremely grateful for the mentorship of Sangeeta Bhatia and Marcela del Carmen, two inspiring women on my thesis committee. You both added unique insights to my thesis and helped it develop into this final product. I enjoyed our discussions and appreciate all
of your advice throughout my PhD, through your many roles during my time here.
My decision to pursue a PhD was heavily influenced by my incredible undergraduate research mentors. It was a joy working in the lab of Jennifer West at Rice University, and I am particularly grateful to Andy Coughlin for being an amazing role model during my two years in the lab. You continue to be a supportive mentor and friend, and I can't thank you enough for
everything.
I would like to thank the entire Cima Lab, past and present, for their guidance, friendship, and advice throughout my PhD (special shout-out to the early lunch crew: Matt, Greg, Alex, Kevin, Jay, and Rachael). I am particularly grateful to Maple Ye for all of her help during my first few years in the lab, as well as Matt Li and Jay Sy for many useful scientific discussions. The Bridge Project allowed me to be part of a great collaboration between MIT and MGH and the opportunity to work with a diverse team of physicians and scientists including Katerina Mantzavinou, Young Jeong Na, Michael Birrer, Giulia Fulci, Lorenzo Ceppi, and Sam Lauffer. I would also like to thank my two undergraduate students, Carrie DeMoulin and Stephanie Tzouanas, for their hard work, as well as Vyas Ramanan for his spheroid protocol. Thank you to Lenny Rigione and Barbara Layne for everything that you do to support the lab and make sure things run smoothly. I am also grateful for the resources at the Koch Institute and for the very knowledgeable staff. There are so many people that have helped make this thesis possible.
In addition to my amazing colleagues, I could not have made it through graduate school without my incredible friends. You have all helped me through the bad days and the long winters, and I appreciate the coffee breaks, whirlwind visits, bake-a-thons, food adventures, hugs, and laughs. Special thanks to my three loveliest ladies, Shea, MK, and Amy, for always being there (we did it, Shea!!).
I would not be the person I am today without my family. I am so lucky to have two wonderful siblings, Geoffrey and Rebecca. I'm glad that we have grown closer as we have gotten older, and I hope we can all live in the same place again one day. To my grandparents: thank you for your love and encouragement and for coming to every one of my graduations, despite the distance. And to Mom and Dad: you have encouraged me through every step of this crazy journey, and I could not have done it without you. You have always believed in me, and I'm so
thankful to have such wonderful, supportive parents. I love you all so much.
Finally, to my best friend and fiance, Abe: you have made the last 7 years the happiest of my life, and I am a better person because of you. Your love and positive encouragement have pushed me through every challenge that I have faced, and I definitely could not have made it to this point without you. You amaze me, and I am so lucky to have you by my side. I can't wait for our future adventures!
TABLE OF CONTENTS
A BSTR A CT ... 2 LIST O F FIG U R ES ... 6 LIST O F TA BLES ... 8 LIST O F A BBR EV IA TIO N S ... 9 CH APTER 1: BA C K G R O UN D ... 11 1.1 CLINICAL N EED ... 11 1.2 OVARIAN CANCER... 12 1.2.1 Clinical Presentation ... 12 1.2.2 Standard of Care ... 131.2.3 Intraperitoneal Chem otherapy... 18
1.3 LOCA L D RUG D ELIVERY ... 24
1.3. 1 Rationale ... 24
1.3.2 Existing Solutions... 25
1.4 PROPOSED SOLUTION ... 27
CHAPTER 2: FABRICATION AND CHARACTERIZATION OF PRECLINICAL DEVICES.. 28
2.1 INTRODUCTION ... 28
2.2 M ATERIALS AND M ETHODS ... 28
2.2.1 Chem icals and M aterials ... 28
2.2.2 M aterial Selection... 29
2.2.3 Injection M olding... 29
2.2.4 Cisplatin Q uantification... 30
2.2.5 D evice Preparation for In Vitro and In Vivo Studies... 30
2.2.6 In Vitro Cisplatin Release ... 31
2.2.7 Animal Care, Surgery, and Euthanasia Procedures for In Vivo Experiments ... 31
2.2.8 In Vivo Cisplatin Release ... 32
2.3 RESULTS AND D ISCUSSION ... 32
2.3.1 M echanism of Release... 32
2.3.2 In Vitro Cisplatin Release from D evices... 33
2.3.3 In Vivo Cisplatin Release from D evices... 34
2.4 CONCLUSION ... 35
CHAPTER 3: COMPARISON OF CONTINUOUS AND INTERMITTENT CISPLATIN DOSING IN A PLATINUM-RESISTANT OVARIAN CANCER MOUSE MODEL... 36
3.1 INTRODUCTION ... 36
3.2 M ATERIALS AND M ETHODS ... 36
3.2.1 Chem icals and M aterials ... 36
3.2.2 Cell Line Authentication ... 37
3.2.3 Cisplatin Quantification... ... 37
3.2.4 In Vivo Pharm acokinetics ... 37
3.2.5 Efficacy Study in Orthotopic Tum or M odel... 38
3.2.6 Biolum inescence Imaging ... 38
3.2.7 Statistical Analysis ... 40
3.3 RESULTS AND D ISCUSSION ... 40
3.3.1 SK O V3 Cell Line Authentication ... 40
3.3.2 Pharm acokinetics of D evice and IP Bolus Injections... 41
3.3.3 Efficacy and Toxicity Study... 44
3.4 CONCLUSION ... 46
CHAPTER 4: DETERMINATION OF OPTIMAL CONTINUOUS CISPLATIN DOSE RANGE
IN A PLATINUM-SENSITIVE OVARIAN CANCER MOUSE MODEL... 48
4.1 INTRODUCTION ... 48
4.2 M ATERIALS AND M ETHODS ... 48
4.2.1 Chemicals and M aterials ... 48
4.2.2 Cell Line Authentication ... 49
4.2.3 In Vitro Cytotoxicity Assay ... 49
4.2.4 Animal Care, Surgery, and Euthanasia Procedures for In Vivo Experiments ... 49
4.2.5 Orifice Fabrication ... ... 50
4.2.6 Device Preparation for In Vitro and In Vivo Studies... 51
4.2.7 In Vivo Cisplatin Release ... 51
4.2.8 Efficacy Study in Orthotopic Tumor M odel ... 51
4.2.9 Statistical Analysis ... 52
4.3 RESULTS AND DISCUSSION... 52
4.3.1 In Vitro UCI10 Cytotoxicity Assay... 52
4.3.2 Matching Efficacy of IP Therapy with Multiple 1-Hole Devices... 53
4.3.3 Exceeding Efficacy of IP Therapy with Large-Orifice Devices... 62
4.4 CONCLUSION ... 77
CHAPTER 5: FACTORS AFFECTING EFFICACY OF IMPLANTED DEVICES... 79
5.1 INTRODUCTION ... 79
5.1.1 IP Drug D istribution from Implanted Devices ... 79
5.1.2 Immune Response to Continuous IP Chemotherapy... 80
5.1.3 Intratumoral Drug Penetration... 81
5.2 M ATERIALS AND M ETHODS ... 83
5.2.1 Chemicals and M aterials ... 83
5.2.2 Spheroid Culture and M aintenance ... 84
5.2.3 Spheroid Cytotoxicity Assay... 85
5.2.4 Statistical Analysis ... 86
5.3 RESULTS AND DISCUSSION... 86
5.3.1 Ovarian Cancer Spheroid Growth... 86
5.3.2 Spheroid Cytotoxicity Assay... 88
5.4 CONCLUSION ... 96
CHAPTER 6: HUM AN DEVICE DESIGN... 98
6.1 INTRODUCTION ... 98
6.2 M ATERIALS AND M ETHODS ... 99
6.2.1 Synthesis of Porous Silicone ... 99
6.2.2 Characterization of Porous Silicone... 100
6.2.3 Laser-Drilling of Orifices ... 100
6.2.4 Cisplatin Pellet Fabrication... 100
6.2.5 In Vitro Cisplatin Release ... 100
6.3 RESULTS AND DISCUSSION... 101
6.3.1 Device Functional Requirements ... 101
6.3.2 M aterial Selection... 102
6.3.3 In Vitro Cisplatin Release from Orifice-Lined Silicone Tubing ... 102
6.3.4 Proposed Device Dimensions ... 106
6.4 CONCLUSION ... 107
CHAPTER 7: CONCLUSIONS AND FUTURE DIRECTIONS ... 108
REFERENCES... 111
LIST OF FIGURES
FIGURE 1.1. M ETASTATIC OVARIAN CANCER ... 13
FIGURE 1.2. PORT-CATHETER SYSTEM USED TO DELIVER IP THERAPY ... 17
FIGURE 1.3. TWO-COMPARTMENT MODEL OF PERITONEAL DRUG TRANSPORT ... 19
FIGURE 1.4. IP THERAPY SIGNIFICANTLY INCREASES AVERAGE OVERALL SURVIVAL COMPARED TO IV T H E R A PY ... 2 2 FIGURE 2.1. DEVICE SCALE AND PREPARATION... 31
FIGURE 2.2. IN VITRO CHARACTERIZATION OF THE 180 pM ORIFICE DEVICE ... 34
FIGURE 2.3. IN VIVO CHARACTERIZATION OF THE 180 [tM ORIFICE DEVICE ... 35
FIGURE 3.1. BIOLUMINESCENCE IMAGING OF SKOV3 TUMORS IN VIVO... 39
FIGURE 3.2. COMPARISON OF 180 pM ORIFICE DEVICE AND IP BOLUS PHARMACOKINETICS... 42
FIGURE 3.3. TREATMENT EFFICACY AND TOXICITY OF A SINGLE 180 pIM ORIFICE DEVICE AND IP INJECTIONS IN AN SKOV3 OVARIAN CANCER MOUSE MODEL ... 46
FIGURE 4.1. IN VITRO CYTOTOXICITY ASSAY WITH CONTINUOUS CISPLATIN EXPOSURE ... 53
FIGURE 4.2. IN VITRO AND IN VIVO RELEASE OF CISPLATIN FROM DEVICES CONTAINING A SINGLE OR M ULTIPLE 180 pM ORIFICES... 54
FIGURE 4.3. TREATMENT EFFICACY OF 2 OR 4 MG/KG IP BOLUS INJECTIONS OR 2 OR 4 IMPLANTED 180 pM ORIFICE DEVICES IN A UCIl01 OVARIAN CANCER MOUSE MODEL... 56
FIGURE 4.4. WBC COUNTS FOLLOWING TREATMENT WITH 2 OR 4 MG/KG IP BOLUS INJECTIONS OR 2 OR 4 IMPLANTED 180 pM ORIFICE DEVICES IN A UCIl01 OVARIAN CANCER MOUSE MODEL ... 57
FIGURE 4.5. BODY WEIGHT OVER TIME FOLLOWING TREATMENT WITH 2 OR 4 MG/KG IP BOLUS INJECTIONS OR 2 OR 4 IMPLANTED 180 pM ORIFICE DEVICES IN A UCI 101 OVARIAN CANCER MOUSE M O D EL ... 5 8 FIGURE 4.6. TREATMENT EFFICACY AND TOXICITY OF 4 MG/KG IP BOLUS INJECTIONS OR 4 OR 6 IMPLANTED 180 pM ORIFICE DEVICES IN A UCIl1 OVARIAN CANCER MOUSE MODEL ... 59
FIGURE 4.7. WEIGHT LOSS FOLLOWING TREATMENT WITH 4 MG/KG WEEKLY IP BOLUS INJECTIONS OR 4 OR 6 IMPLANTED 180 IM ORIFICE DEVICES IN A UCI 10 1 OVARIAN CANCER MOUSE MODEL ... 61
FIGURE 4.8. TOTAL CISPLATIN DOSE ADMINISTERED OVER THE COURSE OF TREATMENT WITH 4 MG/KG WEEKLY IP BOLUS INJECTIONS OR 4 OR 6 IMPLANTED 180 pM ORIFICE DEVICES... 62
FIGURE 4.9. IN VITRO CISPLATIN RELEASE RATE AS A FUNCTION OF STARTING PAYLOAD IN THE 180 pM D E V IC E ... 6 4 FIGURE 4.10. IN VIVO CISPLATIN RELEASE RATE AS A FUNCTION OF STARTING PAYLOAD IN THE 180 pM D E V IC E ... 6 5 FIGURE 4.11. IN VITRO CISPLATIN RELEASE FROM DEVICES WITH VARIOUS ORIFICE DIAMETERS... 66
FIGURE 4.12. RELATIONSHIP BETWEEN ORIFICE SIZE AND CORRESPONDING IN VITRO RELEASE RATE ... 67
FIGURE 4.13. RELATIONSHIP BETWEEN MOUSE AGE AT THE START OF TREATMENT AND WEIGHT LOSS FOLLOWING WEEKLY CISPLATIN INJECTIONS ... 68
FIGURE 4.14. CISPLATIN DOSE ADMINISTERED OVER THE COURSE OF TREATMENT WITH FIXED-DOSE 3, 4, OR 5 MG/KG WEEKLY IP BOLUS INJECTIONS IN NON-TUMOR BEARING MICE ... 70
FIGURE 4.15. BODY WEIGHT OVER THE COURSE OF TREATMENT WITH FIXED-DOSE 3, 4, OR 5 MG/KG WEEKLY IP BOLUS INJECTIONS OR 2, 3, OR 4 IMPLANTED 254 JIM ORIFICE DEVICES IN NON-TUMOR B EA R IN G M IC E ... 7 1 FIGURE 4.16. WBC COUNTS FOLLOWING TREATMENT WITH FIXED-DOSE 3, 4, OR 5 MG/KG WEEKLY IP BOLUS INJECTIONS OR 2, 3, OR 4 IMPLANTED 254 [tM ORIFICE DEVICES IN NON-TUMOR BEARING M IC E ... 7 1 FIGURE 4.17. COMPARABLE TOXICITY RESULTS WITH FIXED 4 MG/KG WEEKLY IP BOLUS INJECTIONS AND 3 IMPLANTED 254 pM ORIFICE DEVICES IN NON-TUMOR BEARING MICE... 72
FIGURE 4.18. CISPLATIN RELEASE RATES ADMINISTERED WITH FIXED-DOSE 3, 4, OR 5 MG/KG WEEKLY IP BOLUS INJECTIONS OR 2, 3, OR 4 IMPLANTED 254 pM ORIFICE DEVICES IN NON-TUMOR BEARING M IC E ... 7 3 FIGURE 4.19. TOXICITY AS A FUNCTION OF RELEASE RATE FOR NON-TUMOR BEARING MICE IMPLANTED
W ITH 2, 3, OR 4 254 IM ORIFICE DEVICES... 74
FIGURE 4.20. CISPLATIN MASS ADMINISTERED PER DEVICE WITH MULTIPLE IMPLANTED 254 pM-ORIFICE DEVICES IN MICE WITH AND WITHOUT TUMORS ... 76
FIGURE 4.21. EXPERIMENTAL PREDICTION OF MAXIMUM-TOLERATED DOSE (MTD)... 77
FIGURE 5.1. EXPERIMENTAL SETUP FOR SPHEROID CYTOTOXICITY ASSAY... 85
FIGURE 5.2. SPHEROIDS CULTURED IN AGGREWELL PLATES FOR 24 HOURS ... 87
FIGURE 5.3. RELATIONSHIP BETWEEN SPHEROID DIAMETER AND CELL NUMBER ... 88
FIGURE 5.4. REPRESENTATIVE IMAGES OF UNTREATED SPHEROIDS AND THOSE TREATED WITH AUC-MATCHED IP AND DEVICE DOSES OF CISPLATIN ... 90
FIGURE 5.5. AVERAGE SPHEROID DIAMETER OF UNTREATED SPHEROIDS AND THOSE TREATED WITH AUC-MATCHED IP AND DEVICE DOSES OF CISPLATIN ... 91
FIGURE 5.6. REPRESENTATIVE IMAGES OF UNTREATED SPHEROIDS AND THOSE TREATED WITH IP AND HIGH-A U C DEVICE DOSES OF CISPLATIN ... 93
FIGURE 5.7. AVERAGE SPHEROID DIAMETER OF UNTREATED SPHEROIDS AND THOSE TREATED WITH IP AND HIGH-AUC DEVICE DOSES OF CISPLATIN... 94
FIG URE 6.1. POROU S SILICONE ... 99
FIGURE 6.2. SUGAR LEACHING FROM PDM S... 102
FIGURE 6.3. EFFECT OF LASER-DRILLED ORIFICE DISTANCE ON IN VITRO CISPLATIN RELEASE FROM SILIC O N E TU B IN G ... 104
FIGURE 6.4. RELATIONSHIP BETWEEN NUMBER OF LASER-DRILLED ORIFICES ON IN VITRO CISPLATIN RELEA SE FROM SILICONE TUBING... 105
LIST OF TABLES
TABLE 3.1. EVALUATION OF VARIOUS GENES FOR KNOWN MUTATIONS IN SKOV3 CELL LINE. ... 41 TABLE 3.2. AUC AND CMAX COMPARISON FOR 180 pM ORIFICE DEVICE AND IP BOLUS INJECTIONS ... 43 TABLE 4.1. QUANTIFICATION OF ERROR ASSOCIATED WITH HPLC DETECTION OF CISPLATIN MASS ... 63 TABLE 4.2. PREDICTED AND MEASURED INCREASE IN IN VITRO CISPLATIN RELEASE RATE AS A FUNCTION
O F O R IFIC E A R EA ... 66 TABLE 5.1. AVERAGE SPHEROID DIAMETER (D) FOR VARIOUS OVARIAN CANCER CELL LINES... 88 TABLE 5.2. AVERAGE SPHEROID DIAMETER (D) FOR UNTREATED SPHEROIDS AND THOSE TREATED WITH
AUC-MATCHED IP AND DEVICE DOSES OF CISPLATIN AFTER 72 HOURS ... 91 TABLE 5.3. AVERAGE SPHEROID DIAMETER (D) FOR UNTREATED SPHEROIDS AND THOSE TREATED WITH
IP AND HIGH-AUC DEVICE DOSES OF CISPLATIN AFTER 72 HOURS ... 94
TABLE 6.1. COMPARISON OF PREDICTED AND MEASURED CISPLATIN RELEASE RATES FROM SILICONE TUBING. FICK'S FIRST LAW WAS USED TO CALCULATE THE PREDICTED VALUES ... 106
TABLE 6.2. REQUIRED DIMENSIONS, ORIFICE NUMBER, AND PAYLOAD OF A HUMAN-SCALE DEVICE... 107
AUC BLI BSA CBC CCLE Cmax DDTC FBS GOG HPLC ICP-MS IP IV LCP MCL MCS MED MTD OS PBS PDMS Pen-strep-glut PFS PLLA WBC
LIST OF ABBREVIATIONS
Area under the concentration-time curve Bioluminescence intensity
Body surface area Complete blood count
Cancer Cell Line Encyclopedia Peak cisplatin concentration
Sodium diethyldithiocarbamate trihydrate Fetal bovine serum
Gynecologic Oncology Group
High-performance liquid chromatography Inductively-coupled plasma mass spectrometry Intraperitoneal
Intravenous
Liquid crystal polymer Multicellular layer Multicellular spheroid Minimum effective dose Maximum tolerated dose Overall survival Phosphate-buffered saline Polydimethylsiloxane Penicillin-streptomycin-glutamine Progression-free survival Poly-L-lactic acid White blood cell
THIS PAGE INTENTIONALLY LEFT BLANK
Chapter 1: Background
1.1 Clinical Need
Approximately 22,000 women in the United States are diagnosed with ovarian cancer each year, with annual death rates of over 14,000 (3). The majority of ovarian cancer patients (approximately 61%) have metastatic disease at presentation (3). The current standard of care is tumor-debulking surgery to remove all tumors larger than 1 cm in diameter, followed by intravenous (IV) or intraperitoneal (IP) chemotherapy with a platinum-based agent (4-6). Combination IP and intravenous IV chemotherapy has been shown to improve progression-free survival (PFS) and overall survival (OS) over conventional IV therapy (7). Chemotherapy-induced toxicity and complications of the indwelling catheter used to deliver IP therapy, however, often result in patient discontinuation of treatment and have limited physician adoption of this technique (7-9). An implantable device that administers lower drug doses over a longer period of time will maximize the benefits of IP therapy while lowering its associated morbidity, improving patient quality of life and significantly impacting the clinical management of ovarian cancer.
* Portions of this chapter were reproduced from:
1. Cima MJ, Lee H, Daniel K, Tanenbaum LM, Mantzavinou A, Spencer KC, Ong
Q,
Sy JC, Santini J, Jr., Schoellhammer CM, Blankschtein D, Langer RS. Single compartment drug delivery. Journal of controlled release : official journal of the Controlled Release Society. 2014;190:157-71. doi: 10.1016/j.jconrel.2014.04.049. PubMed PMID: 24798478; PMCID: PMC4179298, 2. Ye H, Tanenbaum LM, Na YJ, Mantzavinou A, Fulci G, Del Carmen MG, Birrer MJ, Cima MJ. Sustained, low-dose intraperitoneal cisplatin improves treatment outcome in ovarian cancer mouse modelsibid.2015. doi: 10.1016/j.jconrel.2015.11.001. PubMed PMID: 26548976.1.2 Ovarian Cancer
1.2.1 Clinical Presentation
Epithelial ovarian cancer is the most prevalent form of ovarian cancer, occurring in approximately 80% of cases (10). This subset of the disease is characterized by tumors that arise from the epithelial cells covering the ovary. The other 20% of ovarian cancers arise in the fallopian tube epithelium or the peritoneal mesothelium (10). The majority of patients with epithelial ovarian cancer present with non-specific symptoms that make diagnosis difficult (11). Metastasis to the peritoneal cavity is most common, and, in contrast to most other cancers, metastatic spread via lymphatics is rare. Ovarian cancer cells spread instead by direct contact with adjacent organs or by detachment from the primary tumor and regional seeding via the peritoneal fluid (10) (Figure 1.1). Patients rarely present with symptoms prior to metastasis, and no effective screening tests exist for ovarian cancer. Serum antigen CA-125 is elevated in the majority of ovarian cancer patients, although it is not specific to the disease (11). Transvaginal ultrasound indicates the presence of ovarian masses, but a high false positive rate leads to unnecessary surgeries and associated complications in many cases (12). Once the cancer progresses, symptoms may include fullness, abdominal pain, indigestion, and bloating from the development of ascites, or the buildup of fluid in the peritoneal cavity (11, 13). These symptoms are also non-specific to ovarian cancer and further contribute to the likelihood of late diagnosis. Diagnosis at a late stage (III or IV) reduces patient five-year survival from 90% for patients diagnosed at an early stage (I or II) to 10-30% (14).
Lungs Diaphragm Liver serosa 7/0 Bowel serosa P Pelvic J peritoneum! P PP ~~yy Fallopian Ovary tube Uterus Broad tomach Omentum araaortic mph nodes elvic mph nodes ligament
Figure 1.1. Metastatic ovarian cancer. Schematic illustrating the pattern of ovarian cancer tumor spread
to adjacent tissues throughout the abdomen, as opposed to systemic metastasis via lymphatics. Tumors are represented as yellow masses. Reproduced with permission from (1), Copyright Elsevier.
1.2.2 Standard of Care
1.2.2.1 Surgical Tumor Debulking
The presence of ovarian cancer tumors throughout the abdominal cavity at the time of
diagnosis has made tumor-debulking surgery the clinical standard of treatment prior to adjuvant
chemotherapy. The technique was adopted in the 1970s after Dr. C. Thomas Griffiths published
seminal work indicating its efficacy (15). Subsequent studies performed shortly thereafter
confirmed the improved survival benefits. Ovarian cancer still remains the only malignancy where surgery has proved beneficial and is performed in the majority of cases (13). The surgery
13
itself consists of removing the bulk of the tumor cells such that no masses greater than 1 cm in size are left behind (13). The hope is that by removing large aggregations of cells, subsequent chemotherapy can more effectively target the remaining cancer cells. One or both ovaries, the uterus, cervix, and/or fallopian tubes may also be removed, depending on the severity of the individual case.
The ability of surgical debulking to lengthen patient survival following treatment depends on a number of other factors. Optimal resection is defined by no residual tumors < 1 cm in diameter following surgery and is technically difficult to achieve. Only 20-40% of ovarian cancer patients have access to surgeons skilled in surgical cytoreduction, such as gynecologic oncologists, who can improve rates of optimal resection in patient cohorts from less than 25% to
75% (16). Access to a skilled surgeon was shown to increase average OS from 23 to 36.8 months
(16). There is still some debate over the extent of surgical debulking required for good prognosis, despite the Gynecologic Oncology Group (GOG) recommendation for optimal debulking to < 1 cm. Data published in 2006 demonstrated significant differences in OS between patients with microscopic residual disease, gross disease < 1 cm in diameter, and gross disease > 1 cm in diameter after debulking surgery (17). Another study published in 2008, however, found no difference in PFS or OS between patients with 0.1-1 or 1.1-5 cm residual disease (18). Despite disagreement among the exact diameters of tumors required for "optimal" debulking, it has been shown that achieving microscopic residual disease results in the best outcomes for patients in the long-term, particularly for patients receiving IP chemotherapy (17-22). The extent of a patient's tumor prior to debulking surgery is also an important factor of patient survival after treatment (19, 23). Horowitz, et al. assigned a disease score to patients before surgery using the following criteria: low for patients with pelvic and retroperitoneal spread, moderate for patients with
additional disease spread to the abdomen but sparing the upper abdomen, and high for patients with upper abdominal disease affecting the diaphragm, spleen, liver, or pancreas (19). Patients with high starting disease scores and microscopic residual disease after tumor debulking had significantly lower survival rates than patients with low to moderate disease scores and microscopic residual disease (19). Neoadjuvant chemotherapy for the patient population with a high disease score was, therefore, suggested as a strategy to reduce tumor burden before debulking surgery.
1.2.2.2 Adjuvant Chemotherapy
Platinum-based therapies have been used over the past 30 years for the treatment of ovarian cancer and, in combination with other chemotherapies, continue to provide the most effective clinical results. Cisplatin was found to arrest cell division in bacteria in 1965 and was first used in a clinical trial to treat ovarian cancer in the late 1970s (8, 24). The drug works by creating intrastrand DNA adducts, or covalent bonds, between adjacent guanines or guanine and adenine (25). This linkage activates a DNA repair mechanism, but because the cell cannot repair this crosslink, apoptosis ensues. Carboplatin, another platinum-based therapy, was introduced in the 1990s. The drug is a cisplatin analog that achieves lower overall toxicity and comparable survival rates to cisplatin, but patients often suffer from increased myelosuppression (8). Both platinum agents are used clinically, although cisplatin is more common as a first-line therapy. Cisplatin is administered in combination with other drugs because chemotherapies often work synergistically when coupled (26). Cyclophosphamide was approved for use with cisplatin in the late 1980s, but in the 1990s, clinical studies demonstrated the superior efficacy of paclitaxel and it became the new clinical standard (8). Paclitaxel binds preferentially to microtubules, promoting microtubule assembly and stability. This binding inhibits microtubule shortening,
which prevents the formation of the mitotic spindle and slows the cell cycle, ultimately causing cell death (27).
Cisplatin and paclitaxel are the only two chemotherapy agents currently approved for IP infusion. Cisplatin was, therefore, chosen for the proof-of-concept experiments performed in this thesis to ease the clinical translation of a novel IP dosing regimen. Prior to IP therapy, a port is implanted in a subcutaneous pocket on the chest wall and a catheter is tunneled through the abdominal wall, inserted into the peritoneal cavity, and directed towards the pelvis (28). The port-catheter system allows for intermittent infusions of drug directly into the peritoneal cavity. A number of port-catheter systems have been FDA approved for use in IP chemotherapy, all of which contain a titanium port and a silicone or polyurethane catheter (Figure 1.2A) (29). Catheters containing a cuff are sutured to the abdominal fascia to prevent migration, but literature suggests that these catheters lead to higher infection rates (28, 29). During the infusion procedure, the drug is dissolved in 2 liters of saline solution and infused through the catheter (Figure 1.2B) (8). Large infusion volumes are employed to distend the abdomen and attempt sufficient distribution of agents. Patients are also instructed to turn from side to side every 15 minutes over a 1-2 hour period to further distribute drugs throughout the abdominal cavity (8). The infusion of such a high volume of fluid can cause severe abdominal discomfort, and increased pressure inside the abdomen can result in respiratory distress (8). Additionally, patients face the possibility of severe nausea, diarrhea, cramps and gastric symptoms during treatment due to the increased local concentration of drug (8).
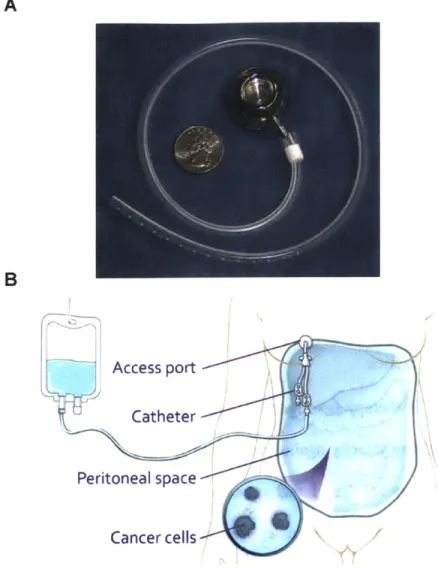
A B Access port Catheter Peritoneal space Cancer cells
Figure 1.2. Port-catheter system used to deliver IP therapy. (A) Example of an FDA-approved
port-catheter system for IP therapy. The device shown is from Bard Access Systems and contains a fenestrated silicone-based, cuffless catheter and titanium access port. Reproduced with permission from (29), Copyright John Wiley and Sons. (B) Schematic illustrating the infusion of chemotherapy directly into the peritoneal cavity during treatment. Reproduced with permission from (30), Copyright Memorial Sloan Kettering Cancer Center.
1.2.2.3 Platinum Resistance
Three-quarters of women treated for ovarian cancer relapse within the first two years of therapy from platinum sensitivity (31). Platinum sensitivity is defined in patients that relapse more than 6 months after treatment with a platinum-based agent, whereas platinum resistance is defined as occurring within the first 6 months (32). Platinum sensitivity poses a significant clinical problem, as the majority of patients who initially respond to surgery and chemotherapy
ultimately relapse (31). Proposed mechanisms of acquired resistance include changes in the mechanism of cellular uptake of cisplatin into the cell, the development of a more robust drug detox system, changes in the DNA repair system that make cells more robust, and changes in the apoptotic pathway (25, 33). These adaptations allow some tumor cells to survive and proliferate in the presence of cisplatin, which leads to recurrence after the treatment cycle ends. The clinical importance of platinum resistance has made the standard in ovarian cancer research to evaluate new therapies against both platinum sensitive and platinum resistant cell lines (34-36).
1.2.3 Intraperitoneal Chemotherapy
1.2.3.1 Pharmacokinetics
The goal of IP therapy is to maintain high concentrations of drug in the vicinity of the tumor while minimizing toxicity against non-tumor tissues, which are typically affected during conventional IV therapy (7). Drugs that are cleared more slowly from the peritoneal cavity, or have high cavity-to-plasma area under the concentration-time curve (AUC) ratios, are advantageous for use in IP chemotherapy due to increased direct tumor exposure (37). IP therapy with cisplatin achieves peritoneal concentrations 10-20 times greater than those in the plasma, whereas IP infusion of paclitaxel achieves concentrations 1000 times greater (37). The peritoneum is very permeable to the majority of drugs with molecular weight < 20 kD (38).
These drugs are then transported across capillary membranes into the systemic circulation, where they enter tumor tissue via vasculature, enter non-tumor tissues and potentially cause side effects, or are cleared from the body. Alternate routes of peritoneal clearance include the vascular and lymphatic systems. Large molecular weight and highly lipophilic compounds are traditionally drained via lymph nodes (38, 39).
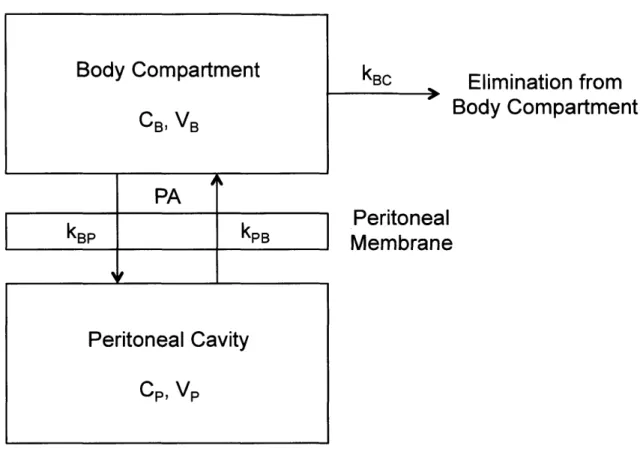
PA
kp
kPPeritoneal Cavity
CP, VP kBCElimination from
Body Compartment
Peritoneal
Membrane
Figure 1.3. Two-compartment model of peritoneal drug transport. Drug is transported from the
peritoneal cavity into the systemic circulation (body compartment) through the peritoneal membrane. The drug clearance rate (PA) controls the rate of this transfer. Concentrations and volumes of the body compartment and peritoneal cavity are represented with the subscripts B and P, respectively. Adapted from (40).
A two-compartment model is traditionally employed to describe peritoneal transport
(Figure 1.3) (40, 41). The peritoneal cavity is treated as one compartment, and the blood (body
compartment) is treated as a second. Drug clearance from the peritoneal cavity depends on both
the concentrations of drug in the blood and peritoneal fluid, as well as the clearance rate through
the peritoneal membrane (40). The clearance term is expressed as a permeability-area product
[cm3/min] and is drug-specific (40). This model can be described with the following differential
equation for the rate of mass transfer from the peritoneal cavity:
d(VPC)= PA(CP - C B) (Eqn 1)
dt
19
Body Compartment
where Vp is the volume of the peritoneal cavity, Cp is the concentration of drug in the peritoneal fluid, PA is the permeability-area product or drug clearance rate, and CB is the concentration of drug in the blood (40). Dedrick et al. solved this equation at steady-state, yielding the following:
C (x) = CB + P B D (Eqn 2)
where x is the distance from the fluid in the peritoneal cavity, Ce is the concentration of drug in the capillary bed (which approaches the concentration of drug in the blood, CB, as x approaches infinity), k is the rate of drug removal from the tissue by diffusion into capillaries (assumed to be constant), and D is the diffusivity of drug in the peritoneal tissue (40). The concentration of drug in the tissue space adjacent to the peritoneal cavity decreases exponentially as a function of the distance from the peritoneal fluid.
The two-compartment model also allows calculation of the relative masses of drug in the peritoneal fluid and blood as a function of time following the administration of an IP injection. Assuming the rate of drug transfer from the peritoneal fluid to the blood and vice versa are represented with the rate constants kPB and kBp, respectively, and that the rate of clearance of
drug from the blood is represented with the rate constant kBC, the following differential equations can be derived (Figure 1.3):
dC B) = -(k BC +k BP)CB(t) +k PBCP(t) (Eqn 3) dt
dt) -kPBCP(t)+kBPCB(t) (Eqn 4)
dt
One can then solve for CB(t) and Cp(t) using the following initial conditions: CB(O) = 0 and Cp(O)
= M, where M is the mass of drug in the administered IP injection.
1.2.3.2 Clinical Advantage
The therapeutic advantage of IP therapy in ovarian cancer has been well established. Multiple randomized phase III clinical trials comparing IV and IP therapies have demonstrated a survival advantage for women receiving IP therapy (7, 42, 43). The risk of relapse also decreases by over 20% with the use of combination IP and IV chemotherapy, as compared to IV chemotherapy alone (44).
The first phase III clinical trial documenting the advantage of IP therapy (GOG 104) was published in 1996 (42). The delivery of IV cyclophosphamide with IV or IP cisplatin yielded a 20% increase in median OS in the IP arm (P = 0.02). Paclitaxel proved more effective than cyclophosphamide in combination with cisplatin, however, so an additional phase III trial (GOG 114) was completed in 2001 (8, 43). IP cisplatin again increased progression-free survival (PFS) and OS compared to the IV treatment arm. Toxicity was higher in the IP group, but the study was biased due to high-dose chemical tumor debulking with carboplatin in the IP arm alone.
The final, and most important, landmark clinical trial (GOG 172) was published in 2006 (7). Patients were first surgically debulked to leave no residual tumor mass greater than 1 cm. Patients were then administered either (i) IV paclitaxel on day 1 (135 mg/m2 body surface area
(BSA), 24 hr infusion) and IV cisplatin (75 mg/m2 BSA) on day 2, or (ii) IV paclitaxel (135
mg/m2 BSA, 24 hr infusion) on day 1, IP cisplatin (100 mg/m2 BSA) on day 2, and IP paclitaxel
on day 8 (60 mg/M2 BSA). Treatments were given every 3 weeks for 6 cycles. Median PFS
increased from 18.3 months in the IV treatment group to 23.8 months in the IP group (P = 0.05),
and OS increased from 49.7 to 65.6 months (P = 0.03) (Figure 1.4). Patients receiving IP therapy experienced an increased incidence of side effects that included: fatigue; pain; and hematologic (leukopenia, thrombocytopenia), gastrointestinal, metabolic, and neurologic toxicities.
1.0-
0.9-0.8-
Intraperitoneal therapy
U) 0.7- 0.6-C 0.5-0 vi0.4-0.
Intravenous therapy
0. e0.3-
0.2-0.1-
P=0.03
0.00
6
12
18
24
30
36
42
48
54
60
Months of Study
Figure 1.4. IP therapy significantly increases average overall survival compared to IV therapy.
Overall survival was shown to increase by 32% from 49.7 months to 65.6 months. Reproduced with permission from (7), Copyright Massachusetts Medical Society.
A recently published retrospective analysis of GOG studies 114 and 172 evaluated
long-term patient survival following treatment (45). The study concluded that IP therapy is associated
with a 23% decreased risk of death compared to IV therapy. A survival improvement was seen
following IP therapy in patients with both gross residual (< 1 cm) and no visible disease
following surgical debulking, despite a 1.89-fold increased risk of death for patients with gross
residual disease. A positive correlation between OS and the number of IP cycles administered
was also established. The study confirms the long-term benefits of IP therapy and encourages its
use in the clinic, even for patients with gross residual disease or those who cannot complete the
full prescribed 6 cycles of IP therapy.
The promising results of the aforementioned clinical trials have also been validated in
22 - ---- 190mr-_
-clinical practice. Wright et al. evaluated the treatment of optimally debulked patients in six National Cancer Institute Network institutions with either combination IP/IV therapy or IV therapy between 2003 and 2012 (46). The study confirmed that IP/IV therapy significantly improves OS compared to IV therapy alone, despite variations in the IP regimens administered across institutions.
A recently completed clinical trial, GOG 252, concluded that IP chemotherapy does not increase PFS compared to IV therapy (47). The treatment arms were as follows: 1) Dose-dense IV paclitaxel (80 mg/m2 BSA on days 1, 8, and 15), IV carboplatin (AUC 6 on day 1), and IV
bevacizumab (15 mg/kg on day 1, starting on cycle 2); 2) Dose-dense IV paclitaxel (80 mg/m2
BSA on days 1, 8, and 15), IP carboplatin (AUC 6 on day 1), and IV bevacizumab (15 mg/kg on day 1, starting on cycle 2); or 3) IV paclitaxel (135 mg/m2 BSA on day 1), IP cisplatin (75
mg/M2 BSA on day 2), and IV bevacizumab (15 mg/kg on day 1, starting on cycle 2), every 3
weeks for 6 cycles. Patients also received an additional 16 cycles of maintenance bevacizuinab. A number of dose modifications, including reduction of the IP cisplatin dose from 100 to 75 mg/M2 BSA, were, however, made in this trial and likely contributed to the limited increase in
PFS. The 1560 participants in the study were also recruited from 498 institutions, likely adding significant variability in the degree of surgeon expertise and implemented techniques, both of which could affect IP PFS outcomes. OS has also not yet been established across the applied treatment arms. The highest incidence of dose-related toxicity was also observed in the IP cisplatin arm, motivating the need to reduce side effects while still delivering sufficient drug to achieve tumor kill.
1.3 Local Drug Delivery
1.3.1 Rationale
The aforementioned clinical results demonstrate a strong advantage of IP therapy. Very few physicians, however, administer IP therapy in the clinic (48). Despite the significant survival benefit offered by the treatment regimen in the GOG 172 trial, only 42% of patients were able to complete all intended cycles of the IP therapy (7). The primary reasons for early termination were catheter-related complications including infections, blockages, and leaks, and drug-related toxicities (7, 49). Lack of adoption of IP therapy can be attributed to a number of reasons: it increases morbidity, has higher associated costs, and requires more provider time and technical skill (48). Recently published results of a retrospective study that evaluated the use of IP chemotherapy in six National Comprehensive Cancer Network institutions found that adoption plateaued after reaching 50% in 2008, and that its use has varied widely across institutions from 4% to 67% (9). The significant survival benefits of IP therapy should, however, outweigh the greater toxicity and lower patient quality of life (50). Outside large cancer centers and hospitals, access to care also varies widely. A recent report by the National Academies of Sciences, Engineering, and Medicine found that less than half of women with ovarian cancer receive treatment according to national standard-of-care guidelines (51). A device that can be implanted via a standard surgical procedure has the potential to significantly improve ovarian cancer patient
access to IP therapy.
Metronomic chemotherapy, or "the chronic administration of chemotherapeutic agents at relatively low, minimally toxic doses, and with no prolonged drug-free breaks," is an emerging new strategy in the treatment of cancer (52, 53). Maximum tolerated doses (MTDs) presently administered cause significant drug-induced toxicity in patients. Rest periods between therapy
cycles are required to allow patients to recover, during which clones of resistant tumor cells have time to grow (52). Administration of more frequent, better-tolerated chemotherapy doses could therefore maintain the same level of efficacy while minimizing the regrowth periods. Multiple studies advocate more frequent, low-dose IP bolus injections to reduce toxicity and increase antitumor efficacy by maintaining a high AUC, and various preclinical and clinical efforts have demonstrated the potential for success of metronomic chemotherapy (52-55). The Japanese GOG, in particular, showed in a phase III ovarian cancer clinical trial that dose-dense paclitaxel (weekly IV administration instead of once every three weeks) in combination with carboplatin increased PFS and OS compared to the current standard of care (56). A higher incidence of anemia was observed in the dose-dense arm, but no dose optimization had yet been performed. Long-term follow-up confirmed the survival benefits of dose-dense therapy, which increased OS from 62.2 to 100.5 months (57). Previous attempts at paclitaxel dose escalation, on the other hand, resulted in no survival benefit (56). MITO-7 was a similar phase III clinical trial comparing weekly to tri-weekly IV paclitaxel but with approximately equivalent total paclitaxel doses administered in the two treatment arms. This study highlighted that ovarian cancer patients receiving the weekly dose had comparable PFS with reduced toxicity and improved quality of life compared to the patients receiving tri-weekly treatment (58). Prolonged exposure to chemotherapy could, therefore, improve the efficacy of IP bolus therapy while reducing the incidence of tumor recurrence and drug-induced side effects.
1.3.2 Existing Solutions
Improved efficacy of continuous IP dosing has been demonstrated previously in ovarian cancer models, but published work faces significant limitations in (i) the clinical relevance of the evaluated treatment periods and (ii) the translatability of the proposed delivery systems to
humans. Work by Piquette-Miller and Allen has focused on eliminating drug-free periods between IP infusions of chemotherapy for ovarian cancer, and they have published results to support superior antitumor response of continuous delivery compared to intermittent dosing
(59-63). Commercially available osmotic pumps and custom-made injectable gels were used as vehicles to deliver drug locally to the peritoneal cavity. Such vehicles are ill-suited for clinical translation because of the impractical volumes required to contain the therapeutic payload when scaled for human use. Injected gels additionally achieve poorly controlled release rates as the polymer degrades. De Souza et al. demonstrated improved therapeutic efficacy of continuous IP docetaxel delivery in vivo in murine ovarian cancer models. The drug was contained in an injectable biodegradable chitosan egg-phosphatidylcholine (PoLigei) matrix developed by the authors, which was injected in the peritoneal cavity (59). The matrix had a final drug to material ratio of approximately 1:8 w/w. This kind of drug-polymer composite is not reasonable for human dosing because of the large volume assumed by the polymeric matrix. De Souza et al. injected a 30 gL depot in each mouse to deliver docetaxel over a 3-week period, which would scale to a 100 mL depot in a 70 kg human. This volume is an underestimate because the standard clinical chemotherapy regimen is 18 weeks (6 infusions separated by 3-week recovery periods). The viscosity of the docetaxel-PoLigei was also shown to increase with drug payload, so it is unlikely that such a large depot could feasibly be injected into the IP space (64). Previous formulations of the PoLigei in the form of a surgically implantable film, as in the work of Vassileva et al., face similar issues of scalability (63). A lOxlO mm2 film was used to deliver 4.5
mg of paclitaxel in each mouse. The film would have to be more than 3x3 m2 to deliver the MTD
of 280 mg/kg/wk in humans over a 3-week period. Zhidkov et al. demonstrated the efficacy of continuous carboplatin delivery in vivo in murine ovarian cancer models, as compared to
intermittent bolus injections. Carboplatin has not been approved for IP chemotherapy and has exhibited significantly less tumor penetration than cisplatin in vivo (65, 66). Zhidkov et al. also limited the duration of treatment to 2 weeks, allowing the administration of only 2 IP bolus injections (61). The size of the commercially available osmotic pump used by Zhidkov et al. may also cause significant morbidity to the animals if the treatment period is extended. Any implant-generated fibrous capsule can take at least 4 weeks to develop (67). This may affect the in vivo release of drug from the pump, which will likely be compromised if treatment is administered for a longer period. Zhidkov et al., finally, limited their studies to a platinum-resistant model.
1.4 Proposed Solution
The proposed solution is an implantable IP device capable of localized drug delivery that maintains the efficacy of the standard of care, lowers the associated morbidity, and increases clinical adoption of IP therapy. These goals can be achieved through localized drug delivery via an implantable device that eliminates the current port-catheter system, lowers systemic drug concentrations, eliminates the current large infusion volume required for IP therapy, and requires a simple surgical procedure for device deployment and retrieval. Implementation of such a device, however, first requires evidence that the high intermittent drug concentrations of the current IP dosing regimen are not essential to achieve successful treatment outcomes. This thesis work demonstrates that (i) continuous dosing is as effective and less toxic than the current standard of care, (ii) low doses of cisplatin more effectively treat small tumor nodules, and (iii) a tunable, human-scale device of feasible dimensions can be designed to deliver the desired dose as predicted by our preclinical in vivo studies.
Chapter 2: Fabrication and characterization of preclinical
devices*
2.1 Introduction
The goal of controlled release systems is to deliver therapeutic agents at a desired rate for an extended period of time. The release rate is chosen to maintain a sustained in vivo concentration of drug between the minimum effective dose (MED) and MTD. A controlled release system is able to maintain concentrations inside this therapeutic range with a single administration (68). Examples of controlled release systems with ovarian cancer applications are discussed in section 1.3.2, although controlled release systems have been used extensively to treat many physiologic targets (1, 69, 70). Reservoir-based systems are desirable because they allow prolonged containment of therapeutic agents in vivo while maximizing the therapeutic payload per volume. Loading the reservoir with drug in powder form, as opposed to drug in solution, further minimizes the required size of the drug reservoir and prolongs the stability of drugs that degrade quickly in solution. Controlled release from reservoir-based systems has been previously characterized in our lab with demonstrated efficacy in vivo (71-74). A reservoir-based system is shown here to release cisplatin continuously with a zero-order release profile, as predicted by Fick's Law.
2.2 Materials and Methods
2.2.1 Chemicals and Materials
Phosphate-buffered saline (PBS), sodium diethyldithiocarbamate trihydrate (DDTC), nickel (II) chloride, sodium hydroxide, high-performance liquid chromatography (HPLC)-grade
* Portions of this chapter were reproduced from 2. ibid.
water, HPLC-grade methanol, and cisplatin powder were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2.2 Material Selection
Reservoir-based devices were used for both in vitro and in vivo testing, requiring the use of biocompatible materials. It was also important to select materials that would not degrade over the evaluated treatment period, in order to achieve continuous drug release profiles. Injection molding was used to maintain reproducibility with a high degree of precision, so the selected materials needed to be able to undergo this fabrication process. Devices were therefore fabricated from poly-L-lactic acid (PLLA) (SurModics Inc., Eden Prairie, MN, USA) and Vectra MT1300 liquid crystal polymer (LCP) (microPEP, East Providence, RI, USA) because they fulfilled these requirements. PLLA is known to be biocompatible, and although it typically degrades over time, it was shown that this material did not degrade over the 6-week implantation period evaluated in these studies (75, 76). The medical-grade LCP used for the devices has been shown to be biocompatible via USP Class VI biocompatibility testing. This material is non-degradable and was, therefore, suitable for our in vivo dosing studies. These materials were used interchangeably, because it was shown that drug release from the devices does not depend on the material used (data not shown).
2.2.3 Injection Molding
The devices used in this study were fabricated by microPEP and consist of an injection-molded reservoir and cap. The devices are injection-injection-molded into a cylindrical reservoir of approximately 3 mm in diameter, 3.5 mm in height, and 0.3 mm in thickness. The cap is a thin
disk, 3 mm in diameter and 400 pm in thickness, with a 180 ptm orifice in the center (Figure 2.1 A).
2.2.4 Cisplatin Quantification
The HPLC method was modified from the method published by Augey et al. (77). The samples were diluted in PBS to a final volume of 450 ptL before adding 50 ptL of nickel chloride (0.1 mg/mL in PBS) and 100 iL of DDTC (0.1 g/mL in 0.1 M sodium hydroxide) solutions. The samples were then incubated at 37'C for 30 minutes before passing them through the HPLC column. An Agilent 1200 LC was used for cisplatin quantification. An ODS Hypersil 250 x 4.6 mm column (Thermo Scientific, Waltham, MA, USA) was heated to 30'C and the sample holder was cooled to 4'C prior to the run. A mobile phase of 75% methanol in water (HPLC-grade) was used at a flow rate of 1.4 mL/min. 100 ptL of cisplatin-containing sample were injected into the column. The AUC of the cisplatin peak that appeared at 5.1 min on the 254 nm spectrum was normalized to the AUC of the internal standard peak at 6.0 min on the 250 nm spectrum. A linear calibration curve was obtained by plotting the ratio of the cisplatin AUC to the internal standard AUC against a concentration range of 0.1 to 20 tg/mL (r2 =0.9996).
2.2.5 Device Preparation for In Vitro and In Vivo Studies
Each device was loaded with 5 mg of cisplatin powder and the cap-reservoir interface was sealed using Loctite Hysol M-2 1 HP Medical Device Epoxy Adhesive (McMaster-Carr, Elmhurst, IL, USA) (Figure 2. 1B). Cisplatin release was activated by placing the devices in PBS solution under vacuum to replace the air in the device with PBS and form a supersaturated solution within the reservoir.
A
Figure 2.1. Device scale and preparation. (A) The devices consist of a cylindrical reservoir of
approximately 3 mm in diameter, 3.5 mm in height, and 0.3 mm in thickness, and a cap of 3 mm in diameter and 400 tm in thickness. The cap contains a 180 ptm orifice in the center to control the rate of drug release from the device. A penny is included for scale. (B) The devices are filled with cisplatin powder and medical device epoxy is used to seal the cap-reservoir interface before implantation.
Reproduced with permission from (2), Copyright Elsevier.
2.2.6 In Vitro Cisplatin Release
The devices released drug into a PBS solution at 37'C, which was replaced by fresh PBS
at various time points to maintain a constant sink condition around the device. These PBS
solutions were then assayed with HPLC to measure the mass of drug that had been released up until each time point. The cumulative mass of drug released was plotted versus time to obtain the
in vitro release profile for the device. In vitro release rate was estimated with a linear best-fit
through the origin.
2.2.7 Animal Care, Surgery, and Euthanasia Procedures for In Vivo Experiments
Animals were housed in a specific pathogen free facility. Veterinary staff monitored
animals frequently for changes in health status throughout the duration of the study. Mice were
weighed daily to monitor overall health and euthanized upon weight loss greater than 20% or
poor body condition. All listed procedures were in accordance with the MIT Committee on
Animal Care (CAC), as well as the NIH Guide for the Care and Use of Laboratory Animals.
31
Animal surgeries to implant cisplatin-loaded devices were performed within a sterile field and all surgical equipment was autoclaved. Inhalation anesthesia was maintained with isoflurane. Pre- and post-surgical analgesics were used as per recommendation by the animal care committees. The incision site was sterilized with iodine and isopropyl alcohol. A small abdominal incision of about 5 mm was made through the skin and peritoneum to implant the devices. Devices were not fixed to the abdominal wall and were allowed to freely move within the peritoneal cavity, to minimize trauma to the surrounding tissues. The devices migrated towards the pelvic region during the course of treatment. Mice were weighed daily to monitor overall health and euthanized upon weight loss greater than 20% or poor body condition. All animals were euthanized via carbon dioxide inhalation at a steadily increasing flow rate to minimize distress.
2.2.8 In Vivo Cisplatin Release
Healthy, tumor-free female nu/nu nude mice of 25-30 g from Charles River Laboratories (Wilmington, MA, USA) were used. Devices were sterilized with ethylene oxide prior to implantation. Animals were euthanized at various time points (2, 4, 7, 10, 14, 21, 35 and 42 days) and devices explanted after euthanasia. The amount of drug remaining in each explanted device at each time point was measured using HPLC. The mass of drug released by each device was calculated by subtracting the mass of drug remaining from the mass originally loaded.
2.3 Results and Discussion
2.3.1 Mechanism of Release
An implantable device was conceived to replace the current technology for IP chemotherapy delivery in ovarian cancer. A device for mice was designed to continuously
deliver drug over a prolonged period through a circular orifice on its cap (Figure 2. 1A and B). The device is diffusion-controlled, and the area of the orifice is directly proportional to the rate of release of agents as predicted by Fick's first law:
ADC
M= - Ax s (Eqn 5)
Ax
where rh is the mass diffusion rate (mass per unit time), A is the area of the orifice, D is the diffusion coefficient of the drug in the diffusion medium, Cs is the solubility of the drug, and Ax is the diffusion length, assumed to be the depth of the orifice. This equation assumes that the solution surrounding the device maintains sink conditions, with a drug concentration of approximately 0. The device is designed to contain an excess of drug. This maintains a saturated drug solution within the device reservoir, achieving a constant diffusion gradient and continuous, zero-order release throughout the treatment period.
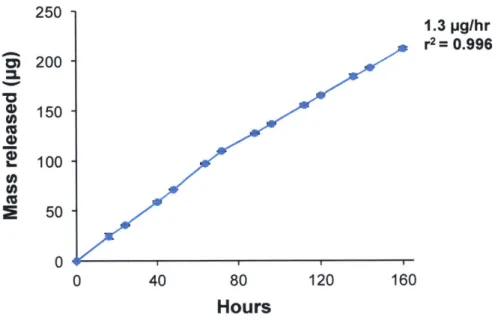
2.3.2 In Vitro Cisplatin Release from Devices
Figure 2.2 shows that the device is able to release cisplatin at 370C in vitro in a linear and reproducible fashion, without an initial burst (r2= 0.996, n = 3). The average release rate as determined by a linear fit through the origin is 1.3 p.g/hr. This value is very close to the rate of 1.4 pg/hr predicted by Fick's first law (Eqn 5), which was calculated using the following values: A = 0.00025 cm2, D = 2.1
x 10- cm2/s (diffusivity of cisplatin in water at 370C (78)), Cs = 2.87 mg/mL (experimental value of cisplatin solubility in PBS, determined via HPLC), and Ax = 0.4 mm).
250 1.3 pg/hr r2= 0.996 Si 200 150 100 c, E 50 0 40 80 120 160 Hours
Figure 2.2. In vitro characterization of the 180 lam orifice device. The in vitro release profile of the
device at 37'C in PBS is linear with a release rate of 1.3 ptg/hr (n = 3, r2= 0.996). Reproduced with
permission from (2), Copyright Elsevier.
2.3.3 In Vivo Cisplatin Release from Devices
The IP device was implanted surgically in the peritoneal cavity of mice to determine the
in vivo release profile of cisplatin. The in vivo release rate was determined by quantifying the mass of drug remaining in the device at various time points. The device is able to release
cisplatin continuously throughout the period of 42 days in vivo with a linear profile (Figure 2.3,
2
r2= 0.965, n = 3). The in vivo release rate of 1.0 ptg/hr, or 172.2 pig/wk, is very similar to the in
vitro release rate of 1.3 pig/hr (Figure 2.2). Approximately 20% of the initial 5 mg drug payload
was released from each device in vivo. Validation of the sustained-release behavior of the device
in vivo over six weeks allowed us to use this device as a tool to evaluate the efficacy and toxicity
of continuous dosing.
34