HAL Id: tel-02078770
https://tel.archives-ouvertes.fr/tel-02078770
Submitted on 25 Mar 2019HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Étude de la spécifité des lgA intestinales humaines
Delphine Sterlin
To cite this version:
Delphine Sterlin. Étude de la spécifité des lgA intestinales humaines. Immunologie. Sorbonne Uni-versité, 2018. Français. �NNT : 2018SORUS006�. �tel-02078770�
Université Pierre et Marie Curie
Ecole doctorale Physiologie et Physiopathologie (ED394)
Centre d’Immunologie et des Maladies Infectieuses – UMR INSERM/UPMC 1135 – Equipe 7
Etude de la spécificité des IgA intestinales humaines
Par Delphine Sterlin
Thèse de doctorat d’Immunologie
Dirigée par le Pr Guy Gorochov et le Dr Hans Yssel
Présentée et soutenue publiquement le 30 janvier 2018Devant un jury composé de :
SOKOL Harry, Professeur des Universités, Paris 6, président du jury MONTEIRO Renato, Professeur des Universités, Paris 7, rapporteur VAN DE GUCHTE Maarten, Directeur de recherche, INRA, rapporteur ANDRE Sébastien, Maître de conférences, Paris 6, examinateur
BLOTTIERE Hervé, Directeur de recherche, INRA, examinateur YSSEL Hans, Directeur de recherche, CIMI, directeur de thèse
Remerciements
« Il n’est qu’un luxe véritable et c’est celui des relations humaines ».
Antoine de Saint-Exupéry, Terre des hommes, 1939
Les lignes qui suivent sont sûrement les plus à même de traduire à quel point cette thèse n’a pas seulement été une aventure scientifique mais aussi une expérience humaine enrichissante. Trois ans pour apprendre un peu de patience et de persévérance, à la faveur de la rigueur et du sens critique scientifique, des échecs et des aléas inhérents aux mises au point techniques. Trois ans pour goûter la capacité créatrice et motrice d’une équipe, sans laquelle je ne serais probablement pas allée jusqu’au bout et à laquelle je tiens à exprimer toute ma gratitude.
Je remercie chacun des membres du jury de me faire l’honneur d’évaluer ce travail :
- Mr le Professeur Harry Sokol, merci de présider ce jury et de partager votre expertise. - Mr le Professeur Renato Monteiro et Mr Maarten Van de Guchte, merci de l’attention
et de l’intérêt avec lesquels vous avec lu et corrigé ce travail afin de l’améliorer. - Mrs Hervé Blottière et Sébastien André, merci d’avoir accepté d’être examinateurs de
ce travail, d’apporter vos regards de microbiologiste et immunologiste.
- Mr le Professeur Guy Gorochov et Mr Hans Yssel, mes directeurs de thèse. De la culture cellulaire à la culture générale, tu n’as pas été avare de temps ni de conseils pendant cette thèse, Hans, je t’en remercie vivement. Merci Guy de m’avoir proposé ce travail plutôt B que T. Je te remercie de m’avoir dirigée tout en me laissant autonome, d’avoir guidé ce travail tout en acceptant les refus que j’ai parfois opposés à certaines de tes idées. Merci de m’avoir fait confiance et de me faire confiance pour la suite.
Je remercie chaleureusement tous les membres du laboratoire, qui m’ont accueillie dans l’équipe et m’ont permis de m’y sentir comme un poisson dans l’eau. Un merci tout particulier aux ingénieurs Karim et Christophe, qui ont su me partager patiemment tout leur savoir-faire : sans vous, je ne saurai ni faire fonctionner une pompe, ni faire un panel de cytométrie ou un western-blot. Toujours à l’écoute, avec vous, tout problème trouve sa
à toutes celles qui ont travaillé dans ce fameux « bureau des filles » : Jehane, tes playlists et tes idées « c’est bon pour le moral » ; Laetitia, ta bonne humeur et ton amitié ont dépassé tous nos aléas «cytof», bravo et merci ; Yurdagul & Alicia, vous avez trouvé l'équilibre entre vie perso et vie pro HU, merci de me montrer une voie ; Audrey, merci d'être une preuve bien vivante que la recherche à temps plein, c'est possible et ça rend heureux (!) ; Gaëlle, merci pour ton aide précieuse en biologie moléculaire ; Delphine, c’est un plaisir de travailler avec toi et de savoir que les PCA sont entre de bonnes mains ; Hela pour tes talents de bio-informaticienne et Martin pour ta capacité à toujours générer de nouvelles idées.
Je remercie les stagiaires de M1 ou d’été, Khadija, Céline, Pauline, Rosalie, Asok et Marina, qui ont essuyé les plâtres de mon encadrement, attisé ma curiosité, avancé sur un bout de ce projet. Merci de m’avoir fait découvrir vos talents culinaires ou des bonnes adresses de chocolatiers !
Un pied dans la thèse, un pied dans la réalité hospitalière du département d’Immunologie de la Pitié-Salpêtrière… Me le Professeur Brigitte Autran et Mr le Professeur Guy Gorochov, je vous remercie d’avoir rendu cela possible et de m'avoir laissé libre d'arranger mon emploi du temps pour concilier les deux activités ces deux dernières années. Merci à toute l'équipe d'immunochimie. Rue de la Peyronnie, vous m'avez conquise à l'immunologie, sans aucun regret ! Merci Guislaine de m'avoir transmis un peu de ta passion pour le travail hospitalo-universitaire. Merci aussi à toute l'équipe d'immunologie cellulaire, travailler avec vous fut un plaisir, encore plus grand depuis que vous analysez les lymphocytes B !
Un immense merci à mes amis, d'une fidélité et d’une joie de vivre incroyables. Vous participez grandement à ce que je suis et à ce que je fais, j’ai de la chance de vous avoir !
Last but not least, un merci sûrement bien trop faible pour mes parents qui m'ont
toujours incité à prendre la voie d’un métier qui me passionnerait, et à mes frère et sœur. Vous m'avez supportée, écoutée et soutenue tout au long de ce parcours, merci et rassurez-vous, c'est presque terminé.
Table des matières
Abréviations ... 7
1 Introduction ... 8
1.1 Le microbiote intestinal ... 8
1.1.1 Généralités ... 8
1.1.2 Méthodes d’étude du microbiote ... 8
1.1.3 Composition du microbiote intestinal ... 10
1.1.4 Fonctions du microbiote ... 16
1.2 Physiologie des Immunoglobulines A ... 20
1.2.1 Structure et sous-‐classes d’IgA ... 20
1.2.2 Synthèse des IgA intestinales ... 21
1.2.3 Répertoire et spécificité des IgA intestinales ... 27
1.2.4 Les IgA sécrétoires : modulateurs du microbiote ... 33
1.2.5 Interactions des IgA avec ses récepteurs ... 37
1.2.6 Conséquences du déficit en IgA chez l’homme ... 40
1.2.7 Production d’IgA humaines in vitro ... 43
1.3 Place des IgG et IgM dans la réponse anti-‐microbiote ... 46
1.3.1 IgG sériques anti-‐microbiote ... 46
1.3.2 IgM sécrétoires anti-‐microbiote ... 48
2. Problématique et objectifs ... 51
3. Résultats ... 52
3.1 Article I : Human IgA bind a diverse array of commensal bacteria ... 52
3.2 Article II : Systemic IgG and secretory IgA converge toward common gut microbiota targets ... 91
4. Discussion ... 113
5. Conclusion et perspectives ... 121
6. Annexes ... 124
Table des figures et des tableaux
Figure 1 : Variations spatiales de la composition du microbiote intestinal. ... 13
Figure 2 : Schémas représentatifs des interactions réciproques microbiote-système immunitaire. ... 19
Figure 3: Structure des IgA. ... 20
Figure 4 : Représentation schématique de l’induction des réponses IgA dans le GALT. ... 24
Figure 5 : Transcytose des IgA à travers les entérocytes. ... 26
Figure 6 : Proposition de modèle décrivant les réponses IgA vis-à-vis du microbiote. ... 30
Table 1 : Table récapitulative des taxons ciblés par les IgA intestinales. ... 32
Figure 7 : L’agglutination classique et l’agglutination de bactéries en croissance par les IgA interviennent à différentes concentrations de pathogènes. ... 37
Figure 8 : Des variants génétiques dans plusieurs gènes impliqués dans la production des IgA ont été identifiés chez les patients DIgA. ... 41
Figure 11: Schéma récapitulatif de la répartition des deux sous-classes à la surface des bactéries commensales dans l’intestin humain. ... 121
Annexe 1 : Commutation de classe des gènes des immunoglobulines ... 124
Annexe 2 : Analysis of bacterial-surface-specific antibodies in body fluids using bacterial flow cytometry ... 124
Abréviations
AID : Activation-Induced cytidine Deaminase CDR : Complementarity Determining Region CMH : Complexe Majeur d’Histocompatibilité DICV : Déficit immunitaire commun variable DIgA : Déficit en IgA
EBV : Epstein Barr Virus Fab : Fragment antigen binding Fc : Fragment cristallisable
FcαR : Récepteur au fragment Fc des immunoglobulines A FcγR : Récepteur au fragment Fc des immunoglobulines G FWR : Framework Region
GALT : Gut Associated Lymphoid Tissue Ig : Immunoglobuline
IVIG : Intravenous Immunoglobulines G LPS : lipopolysaccharide
LTA : acide lipotechoïque
pIgR : Récepteur aux Immunoglobulines polymériques PCR : Polymerase Chain Reaction
PRR : Pattern Recognition Receptor SFB : Segmented Filamentous Bacterium sIgA : IgA sécrétoire
UFC : Unité Formant Colonie
1 Introduction
1.1 Le microbiote intestinal
1.1.1 Généralités
La lumière intestinale est colonisée par un grand nombre de micro-organismes, jusqu’à 1012 micro-organismes/g dans le colon. Composant notre microbiote, ces micro-organismes incluent des bactéries, des archaea, des parasites, des champignons et des virus (bactériophages et virus eucaryotes). Les bactéries (seuls micro-organismes traités dans ce travail) sont largement majoritaires et représentent plus de 95% des micro-organismes dans l’intestin. La flore bactérienne colique compte ainsi plus de 1014 bactéries pour un poids d’environ 2kg, ce qui permet de la considérer comme un organe à part entière. La diversité de ces espèces commensales est tout aussi étonnante que leur quantité. Evaluée par des techniques de culture in vitro ou des tests d’identification biochimique, une centaine d’espèces ont été caractérisées dans les années 1970. Depuis les années 1980, plus de 1000 espèces capables de coloniser l’intestin humain ont été identifiées avec le développement de techniques de biologie moléculaire s’intéressant à l’ARN 16S spécifique des procaryotes puis à l’ensemble de l’ADN bactérien (1, 2).
L’ensemble des gènes apportés par la communauté bactérienne intestinale est dénommé microbiome. Le projet MetaHit, en utilisant les techniques de séquençage à haut débit, a publié un large catalogue de gènes du microbiome intestinal à partir d’échantillons de selles d’individus sains. Ce catalogue compte à ce jour plus de 9 millions de gènes (3). Comme de nombreux gènes bactériens partagent les mêmes fonctions, les microbiomes présentent moins de variabilité inter-individuelle que la composition du microbiote en tant que telle. Cette redondance fonctionnelle entre les individus a permis de définir un microbiome essentiel c’est à dire un tronc commun de gènes microbiens chez les individus sains incluant des gènes impliqués dans la dégradation des sucres complexes, la synthèse d’acides aminés ou de vitamines et la détoxification de xénobiotiques (4).
1.1.2 Méthodes d’étude du microbiote
Les techniques traditionnelles de culture bactérienne ont été supplantées par deux techniques de biologie moléculaire : d’une part le séquençage des gènes codants pour l’ARN
entier. Ces deux techniques sont réalisables sur des échantillons de selles ou sur bactéries triées en cytométrie de flux après extraction de l’ADN bactérien.
Long d’environ 1500 paires de bases, le gène de l’ARN ribosomal 16S code pour une sous-unité des ribosomes 30S bactériens. Ce gène mosaïque, présent dans tous les génomes bactériens, alterne des régions hautement conservées au sein du règne bactérien et neuf régions variables (V1 à V9). Le pyroséquençage 16S utilise des amorces universelles complémentaires aux régions conservées tandis que les régions variables permettent d’identifier les différentes espèces bactériennes. De grandes bases de données comme la Ribosomal Database Project ont été créées afin de compiler les séquences, identifier les espèces ou encore dessiner des amorces spécifiques (5). Les régions V1-V3 ou V3-V5 sont les plus fréquemment séquencées pour distinguer les espèces bactériennes alors dénommées OTU (Operational Taxonomic Unit). Lors d’un séquençage se limitant à 3 régions hypervariables, certaines espèces bactériennes ne peuvent être distinguées l’une de l’autre : c’est le cas par exemple d’espèces appartenant au genre Streptococcus qui ont moins de 0,5% de divergence entre elles (6). De même, le séquençage de l’ADNr 16S ne permet pas de discriminer les souches au sein d’une même espèce qui diffèrent l’une de l’autre par la présence d’éléments génétiques mobiles contenant des gènes de virulence ou de résistance aux antibiotiques. Il est aussi important à noter qu’une bactérie possède plusieurs copies du gène de l’ARNr 16S (1 à 5) et que ce nombre varie d’une espèce à une autre ainsi qu’au sein d’une même espèce, la quantification des abondances bactériennes par cette technique est donc délicate et nécessite l’utilisation d’algorithmes spécialisés. Deux autres limitations de cette technique sont liées à l’étape de PCR : d’une part, l’utilisation d’amorces universelles qui ne présentent pas la même efficacité d’hybridation entre les espèces peut induire un biais d’amplification ; d’autre part, des chimères comprenant des fragments de gènes de plusieurs espèces peuvent être générées au cours de l’amplification et être interprétées comme une nouvelle espèce (7). Néanmoins, le séquençage d’ADNr 16S est une méthodologie peu onéreuse, rapide et robuste y compris sur des quantités faibles de bactéries (105 UFC).
Le séquençage aléatoire haut débit (métagénomique) consiste à diviser la totalité de l’ADN bactérien en petits fragments de taille constante et à les séquencer. Les génomes bactériens sont ensuite reconstitués par une analyse bioinformatique basée sur la co-abondance et la co-variance des amplicons : au sein d’un même échantillon, tous les gènes d’une même espèce doivent être présents en quantités égales. Cette méthode a conduit à la description de nouvelles espèces, notamment des espèces extrêmement sensibles à l’oxygène
unités fonctionnelles ainsi générées par l’analyse bioinformatique sont appelées CAG (Co-Abundant Genes) ou MGS (MetaGenomics Species) si elles comprennent plus de 700 gènes. Toutes les espèces constituant le microbiote peuvent aussi être quantifiées de manière plus précise que par l’approche 16S à condition qu’elles soient présentes à une fréquence de 10-7 -10-8 (8). En donnant accès à tout le génome bactérien, cette nouvelle approche permet d’étudier les voies métaboliques portées par l’ensemble du microbiote (métabolome) ou d’analyser la présence et la diversité des gènes de résistance aux antibiotiques encore appelé résistome (2).
Parallèlement à l’émergence de ces deux techniques, de nombreux algorithmes bioinformatiques ont été développés afin de manipuler l’ensemble des données brutes générées (9). L’analyse des données commence par un assemblage des séquences sens et anti-sens en une séquence conanti-sensus, un filtre des séquences de mauvaise qualité ou des séquences chimériques puis une correction des erreurs de séquençage avant de grouper les séquences obtenues en OTU ou CAG. Plusieurs algorithmes existent pour effectuer ces mêmes tâches et de récentes études mettent en lumière une variabilité des résultats obtenus à partir d’un même échantillon. Ces différences ont été observées au niveau du genre bactérien, point important à retenir dans l’interprétation des données issues du séquençage de l’ADNr 16S (10).
1.1.3 Composition du microbiote intestinal
1.1.3.1 Structure du microbiote
Le microbiote intestinal est un écosystème complexe et divers à deux niveaux : on distingue la diversité alpha au sein d’un individu et la diversité bêta entre les individus. Plus de 1000 espèces ont été décrites à l’échelon de la population globale néanmoins un individu n’abrite qu’en moyenne 200 espèces. Malgré cette grande diversité, la majorité des espèces bactériennes colonisant l’intestin appartiennent à quatre phylums principaux : les Firmicutes et Bacteroidetes qui comptent pour 90% de la population bactérienne colique et dans une moindre mesure les Actinobacteria et Proteobacteria. D’autres espèces appartenant aux phylums Verrucomicrobia, Spirochaetes, Tenericutes et Fusobacteria ont été décrites dans l’intestin mais à des taux très faibles ou de façon inconstante parmi les individus (11). Au-delà de la variabilité inter-individuelle, a émergé la notion de « core microbiote » ou microbiote essentiel à savoir un consortium bactérien partagé par la majorité des individus sains. Dans une étude portant sur 4000 individus sains, ce consortium bactérien comptait 17
genres différents qui étaient partagés par 95% des individus. A l’échelle individuelle, le microbiote essentiel représente environ 72% du microbiote total (12).
Ci-dessous, nous détaillerons les quatre phylums majoritaires afin de mieux comprendre la structure du microbiote.
• Phylum des Bacteroidetes
Les Bacteroidetes incluent des bactéries Gram-négatives, allongées, aérobies ou anaérobies, incapables de former des spores mais capables de coloniser différentes régions du tractus intestinal. Elles se caractérisent par un large panel d’enzymes de dégradation des polysaccharides complexes d’origine végétale ou animale. Ainsi le genre Prevotella, associé au microbiote essentiel des individus avec une alimentation riche en fibre, est spécialisé dans la digestion de la cellulose et du xylane (13). Au sein du genre Bacteroides, prédominant dans l’intestin, plusieurs espèces sont capables de dégrader des polysaccharides complexes dérivés de l’hôte, comme les sucres associés à la mucine ce qui favorise leur implantation parmi la flore intestinale ; c’est le cas de Bacteroides thetaiotaomicron. Bien que le genre Bacteroides entretienne dans son ensemble une relation bénéfique avec l’hôte en participant à son métabolisme, certaines de ses espèces peuvent être considérées comme pathobiontes c’est à dire qu’elles peuvent se révéler pathologiques pour l’hôte dans certaines circonstances. Par exemple, Bacteroides fragilis se trouve habituellement dans la lumière intestinale colique où il stimule de façon positive le développement du système immunitaire local. Cependant, cette espèce est responsable d’abcès abdominaux et de sepsis lorsque la muqueuse est altérée (14).
• Phylum des Firmicutes
Le phylum des Firmicutes comprend des bactéries anaérobies strictes ou facultatives. La plupart de ces membres sont Gram-positifs et sont capables de former des endospores, ce qui leur confère un avantage de survie important. Au sein de ce phylum, la classe très hétérogène des Clostridia est subdivisée en groupes parmi lesquels les XIVa et IV représentent une part conséquente du microbiote total (10-40%). Ces bactéries résident préférentiellement dans les cryptes, proches des entérocytes dont elles contribuent à l’homéostasie. Certaines espèces des groupes XIVa et IV sont en effet reconnues pour libérer du butyrate à la suite du processus de fermentation, butyrate qui favorise le maintien des entérocytes et de l’homéostasie intestinale (15). D’autres membres de ces deux groupes majoritaires jouent un rôle dans l’immunité de
groupes XIVa et IV, d’autres groupes des Clostridia contiennent des pathobiontes pour l’homme comme C. difficile (groupe I) voire même des pathogènes stricts comme C. tetani ou
C. perfringens (groupe XI) (16). Enfin, le phylum des Firmicutes compte la classe des Bacilli
qui comprend les genres Enterococcus et Streptococcus. Leurs membres, tolérants à l’oxygène, sont présents en faible abondance en situation physiologique mais peuvent être associés à des dysbioses (17, 18).
• Phylum des Actinobacteria
Comme les Firmicutes, ce phylum est composé de bactéries Gram-positives mais qui se caractérisent par un génome riche en GC (guanine et cytosine). Le genre Bifidobacteria, genre majoritaire de ce phylum dans le tractus intestinal, est bien adapté au milieu intestinal : il exprime des pilis qui facilitent l’adhésion aux entérocytes et possède des transporteurs qui lui confèrent une résistance aux acides biliaires. Nombre de ses espèces considérées comme pro-biotiques sont impliquées dans la résistance aux pathogènes en entrant en compétition avec eux ou en sécrétant des substances antimicrobiennes (19).
• Phylum des Proteobacteria
Le phylum des Proteobacteria comprend une grande diversité de bactéries Gram-négatives. Tandis que la majorité des bactéries intestinales sont anaérobies strictes, les membres de ce phylum sont des bactéries aéro-anaérobies. Minoritaires au sein d’un microbiote sain, les Proteobacteria semblent signer un état de dysbiose quand leur prévalence augmente (20). Ainsi la famille des Enterobacteriaceae (classe des Gammaproteobacteria) contient des pathobiontes tels qu’Escherichia coli et Klebsiella spp. qui peuvent envahir le microbiote intestinal au cours de dysbioses et de contextes pathologiques (immunodépression, maladies métaboliques, inflammation) (18, 20, 21).
1.1.3.2 Distribution anatomique du microbiote
Le tractus intestinal n’est pas un environnement homogène : les conditions de pH, de taux d’oxygène, les nutriments disponibles, la concentration en acides biliaires, la durée du transit ou encore l’épaisseur de mucus varient de l’estomac jusqu’au rectum mais aussi de l’épithélium jusqu’à la lumière intestinale. Ces modifications environnementales façonnent la composition du microbiote tant sur le plan longitudinal que sur le plan transversal.
De l’estomac (milieu acide et partiellement aérobie) au colon (milieu anaérobie), la concentration bactérienne augmente de 105 bactéries à 1014. Dans l’iléon, où le transit est rapide et le métabolisme des acides aminés ou sucres simples est privilégié, la communauté bactérienne est dominée par les Lactobacillales et les Proteobacteria. Avec un transit plus lent, la fermentation des polysaccharides complexes devient un élément clé du métabolisme colique ce qui favorise les ordres des Bacteroidales et Clostridiales (Figure 1). La diversité des espèces est également supérieure à celle retrouvée dans l’iléon (22).
Figure 1 : Variations spatiales de la composition du microbiote intestinal.
Représentation schématique adapté d’après (23, 24).
Au sein d’un même segment colique, les bactéries s’organisent aussi sur un plan transversal. Dans la couche de mucus vont subsister des bactéries capables de dégrader la mucine comme Akkermansia muciniphila ou certaines espèces de Bacteroides tandis que les bactéries installées dans les cryptes intestinales se distinguent par leur capacité d’adhésion (Figure 1). Au plus proche de l’épithélium, ces bactéries sont aussi aéro-anaérobies, elles appartiennent principalement aux Proteobacteria (23, 25).
Pour des raisons pratiques, la composition du microbiote en contexte physiologique ou pathologique a été largement étudiée à partir des selles. Néanmoins, ce type d’échantillon ne rend pas compte des populations microbiennes associées au mucus ni même des populations iléales. L’intérêt de la « biogéographie » du microbiote a été notamment démontré dans le
Estomac Duodenum Jejunum Ileum Colon
Densité bactérienne pH Oxygène Lactobacillus Veillonella Helicobacter Bacilli Streptococcaceae Actinobacteria Actinomycineae Corynebacteriaceae Lachnospiraceae Bacteroidetes
Epithélium Mucus Lumen
Clostridium Lactobacillus Enterococcus Bacteroidetes Bifidobacterium Streptococcus Enterobacteriaceae Enterococcus Clostridium Lactobacillus Ruminococcus 1012 CFU/g
colique, les auteurs ont démontré une sur-représentation des genres Enterococcus, Veillonella et Bifidobacterium chez les patients. L’abondance de ces espèces a aussi été corrélée à l’inflammation ainsi qu’à un score de sévérité clinique. De façon intéressante, ces différences n’étaient pas décelables sur l’analyse des selles (26). La conception des études les plus récentes prend en compte ces bactéries associées à la muqueuse, ce qui permet aussi de comparer la composition du microbiote dans deux lieux différents chez le même individu, au niveau d’une tumeur et de la muqueuse normale par exemple (27).
1.1.3.3 Evolution au cours de la vie
Longtemps considéré comme stérile, l’environnement utérin a été l’objet d’études récentes qui ont démontré la présence d’un microbiote spécifique au placenta et au liquide amniotique (28, 29). La colonisation bactérienne du tractus digestif commence donc in utero et se poursuit dans les trois premières années de vie au terme desquelles la composition du microbiote est considérée comme « adulte » (30). Au cours du temps, la diversité alpha augmente ainsi que l’abondance globale. Le mode d’accouchement est le premier facteur influençant la composition du microbiote : suite à un accouchement par voie basse, les nouveau-nés présentent une flore proche de la flore vaginale maternelle tandis qu’après césarienne, les nouveau-nés développent une flore plus hétérogène mêlant des bactéries de la flore maternelle cutanée mais aussi des bactéries de l’environnement hospitalier (31). Preuve de leur mauvaise adaptation au milieu intestinal, ces premiers colonisateurs ont tendance à disparaître avec le temps pour laisser place à des bactéries sélectionnées notamment pour leurs capacités métaboliques. Ainsi, dans les quatre premiers mois de vie, les bactéries capables de dégrader les protéines du lait sont majoritaires (Bifidobacterium spp.) même s’il existe une différence de composition de microbiote entre les nouveau-nés allaités et ceux nourris avec du lait industriel (32, 33). Ces derniers présentent une maturité de microbiote plus précoce. Avec la diversification alimentaire, les bactéries métabolisant les sucres complexes et les acides aminés prennent le dessus (Bacteroides, Bilophila, Roseburia,
Clostridium, Anaerostipes). Plus les enfants grandissent, plus leur microbiome acquiert des
voies métaboliques de carbohydrates. En parallèle, des gènes codant pour la synthèse d’hormones comme la mélatonine apparaissent aussi dans les premiers mois ce qui va de pair avec l’établissement du rythme circadien chez le nouveau-né (34).
Cette phase de colonisation et de diversification du microbiote coïncide avec la maturation du système immunitaire, ces deux éléments étant liés l’un à l’autre. Des perturbations précoces du microbiote ont ainsi été associées à la survenue de manifestations
allergiques (atopie ou asthme). Par exemple, une moindre diversité du microbiote néonatal a été corrélée à un risque accru de rhinite allergique avant l’âge de 6 ans. De même, une étude canadienne a pu mettre en lien une faible diversité microbienne dans les 100 premiers jours de vie avec la survenue d’asthme dans l’enfance (35–37).
Une fois installé, le microbiote intestinal démontre une grande stabilité tant en composition qu’en diversité chez l’adulte. Une analyse longitudinale du microbiote de 37 adultes suivis pendant 5 ans a illustré cette stabilité : plus de 60% des espèces appartenant majoritairement aux phylums des Bacteroides et Actinobacteria étaient retrouvées à chaque échantillonnage (38). Ce n’est qu’après 65 ans que la structure du microbiote tend à changer. Avec l’âge, la diversité microbienne décline, les bactéries glycolytiques décroissent au profit des bactéries protéolytiques, le microbiote essentiel à savoir les espèces dominantes (appartenant aux familles Ruminococcaceae, Lachnospiraceae ou encore Bacteroidaceae) laisse la place à des espèces jusque-là minoritaires. Cette tendance se confirme chez les centenaires (39). Certaines de ces modifications du microbiote ont été associées à une espérance de vie plus longue que la normale. En effet, un maintien de Faecalibacterium spp. et de faible taux de LPS (lipopolysaccharide) corrèle avec un nombre accru de centenaires. De même, Biagi et ses collaborateurs ont pu décrire une signature de longévité en regroupant certains genres bactériens (40–42).
1.1.3.4 Facteurs influençant la composition du microbiote
Outre l’âge qui vient d’être discuté, la diversité et la composition du microbiote est influencée par un grand nombre de facteurs dont la liste ne cesse d’augmenter : une récente étude hollandaise a décompté plus d’une centaine de facteurs intrinsèques (sexe, fond génétique) et extrinsèques (alimentation, survenue d’une infection) (12, 43). Seuls les déterminants majeurs seront détaillés ci-dessous.
Parmi les facteurs intrinsèques, plusieurs études tendent à montrer l’importance du terrain génétique : (i) association entre certains genres bactériens et des gènes impliqués dans le métabolisme des sucres, l’adhésion aux cellules épithéliales ou le système immunitaire chez l’homme (44), (ii) association entre le Complexe Majeur d’Histocompatibilité (CMH) et la composition microbienne chez des souris congéniques exprimant des molécules de CMH différentes (45), (iii) similarité des microbiotes entre jumeaux monozygotes comparativement à des jumeaux dizygotes ou des enfants non apparentés (33, 46). L’origine géographique (cohorte comparative entre enfants et adultes d’Amérique du Nord ou du Sud, (47)) et la
favorisées en cas de transit rapide ; (48)). Dernier facteur intrinsèque que nous citerons, le cycle circadien: les bactéries « diurnes » expriment plus de gènes participant à la production de protéines que les bactéries « nocturnes » exprimant plus de gènes assurant des voies de détoxification (49).
Premier facteur extrinsèque étroitement lié à l’âge (cf 1.1.3.3) et à l’origine géographique, l’alimentation joue un rôle fondamental dans l’établissement et la composition du microbiote (50). Ainsi, les entérotypes décrits par Arumugam en 2011 (trois clusters regroupant l’ensemble des individus en fonction de l’abondance des genres Bacteroides,
Prevotella et Ruminococcus) ont été associés à des régimes alimentaires particuliers
(entérotype Prevotella et alimentation végétarienne vs entérotype Bacteroides et alimentation riche en protéines animales) (11, 51). Les médicaments, au premier rang desquels les antibiotiques, sont aussi à même de moduler voire d’altérer la composition du microbiote, favorisant la survenue d’infections comme celle à Clostridium difficile ou à Salmonella (52, 53)). L’administration d’antibiotiques s’accompagne le plus souvent d’une diminution de la diversité microbienne qui peut être durable dans le temps (plus de 2 ans après une cure d’antibiotiques à large-spectre) (54, 55). Les inhibiteurs de la pompe à proton, en diminuant l’acidité gastrique, ont également un impact sur la composition du microbiote (56). Enfin, les infections gastro-intestinales, qu’elles soient bactériennes, parasitaires ou virales, peuvent altérer la flore intestinale dite normale (57, 58).
1.1.4 Fonctions du microbiote
Le microbiote intestinal entretient une relation symbiotique avec son hôte en lui assurant certaines fonctions vitales. Organe à part entière, il supporte des voies métaboliques qui lui sont propres permettant de dégrader, fermenter et transformer les substrats provenant de l’alimentation (glucides, protéines et lipides). Son rôle métabolique s’étend au métabolisme de certains xénobiotiques comme la digoxine (59). Il joue également un rôle dans le développement et le maintien de l’intégrité de la barrière épithéliale : Lactobacillus rhamnosus et survie des entérocytes (60), Bacteroides thetaiotaomicron et angiogenèse (61).
Le microbiote est un écosystème très diversifié. Sa composition du microbiote varie entre les individus et au sein d’un même individu le long de l’intestin, sur le plan transversal et aux âges extrêmes de la vie.
Nous détaillerons ci-dessous deux autres rôles du microbiote qui ont pour objectif commun de maintenir le mutualisme entre l’hôte et la flore microbienne : d’une part, un rôle « écologique » de protection contre les pathogènes, d’autre part, un rôle immunologique.
1.1.4.1 Protection contre les pathogènes
Les bactéries commensales forment une communauté stable qui résiste à l’invasion de bactéries extérieures ou à l’expansion de pathobiontes. Reconnue depuis les années 1950, ce phénomène inclut la résistance à l’infection initiale mais aussi la clairance du pathogène (62). On peut distinguer deux types de mécanismes de résistance : directs et indirects.
De façon directe, les bactéries entrent en compétition pour les nutriments et l’espace : en consommant les ressources énergétiques, le microbiote empêche la croissance des pathogènes ; en couvrant les sites d’adhésion aux entérocytes, le microbiote ne permet pas aux pathogènes d’envahir l’épithélium (S. Typhimurium, certains E. coli pathogènes) (63, 64). Les bactéries commensales ont aussi développé des molécules bactéricides ou inhibitrices. Par exemple, certaines souches d’E. coli sont capables de sécréter des bactériocines actives sur
Salmonella sp ou E. coli entéro-hémorragique (65).
De façon indirecte, la flore intestinale interagit avec l’hôte pour prévenir l’invasion par des pathogènes. Le microbiote est ainsi capable de stimuler la production de peptides antimicrobiens par les cellules de Paneth et les entérocytes (les plus connus d’entre eux étant les défensines et RegIIIγ, Figure 2) ou de réguler l’épaisseur du mucus qui permet de maintenir les bactéries à distance des cellules épithéliales (22, 66, 67). En marge du système immunitaire inné de l’hôte, le microbiote peut utiliser certaines molécules de l’hôte à des fins anti-microbiennes, c’est le cas des acides biliaires qui peuvent être transformés par
Clostridium scindens en composés toxiques pour Clostridium difficile (68). 1.1.4.2 Interactions avec le système immunitaire
L’étude de souris axéniques, (sans microbiote/germ-free), a rapidement mis en évidence l’impact du microbiote sur le développement des réponses immunes de l’hôte, qu’elles soient locales ou systémiques. Au niveau local, le tissu lymphoïde associé à l’intestin, le GALT (Gut Associated Lymphoid Tissue = plaques de Peyer, follicules lymphoïdes isolés, cellules T et B dispersées dans la lamina propria) est sous-développé dans ce modèle de souris, observation qui s’accompagne d’une diminution de lymphocytes T CD4+ de type Th17 et T régulateurs, de lymphocytes T CD8+ intra-épithéliaux et de plasmocytes sécréteurs d’IgA. Outre les lignées lymphoïdes, l’immunité innée est elle aussi modulée par l’absence de microbiote e.g
réversibles lorsque les souris axéniques sont colonisées par un microbiote (1, 69). Ces modèle de souris colonisées par un microbiote de composition contrôlée ou une espèce bactérienne particulière (souris gnotobiotiques) ont permis d’étudier plus spécifiquement l’impact d’une espèce sur le système immunitaire et les mécanismes sous-jacents. Des travaux princeps ont ainsi identifié le rôle d’un consortium de Clostridia dans la différenciation et la survie des lymphocytes T régulateurs ou encore celui de SFB (Segmented Filamentous bacterium) dans la maturation des lymphocytes de type Th17 (Figure 2 ; (70–72)). Récemment, Geva-Zatorsky et ses collaborateurs ont largement développé ce système de souris gnotobiotiques en étudiant l’impact de 53 espèces bactériennes fréquemment retrouvées dans le microbiote humain. En plus d’une vision détaillée des relations bactéries-hôte, les auteurs aboutissent à trois conclusions intéressantes : (i) il existe une redondance des effets immunologiques induits par différentes bactéries, (ii) la modulation du système immunitaire par une bactérie semble indépendante de sa phylogénie, (iii) des différences d’effets immunologiques sont observées au sein même d’une espèce, au niveau souche (73). Nombre de ces interactions microbiote-immunité semblent liées à l’activité métabolique des bactéries telles que la production d’acides gras à chaînes courtes qui interviennent dans l’homéostasie des lymphocytes T régulateurs (74).
De façon intéressante, les effets du microbiote sur le système immunitaire ne se limitent pas à la muqueuse intestinale mais sont aussi visibles au niveau central ou en périphérie. Ainsi, chez des souris axéniques colonisées par Bacteroides fragilis, le nombre de lymphocytes T CD4 circulants varie selon le sous-type de B. fragilis, produisant ou non du polysaccharide A (75). Ou encore, la moelle osseuse de souris axéniques présente un défaut de cellules souches hématopoïétiques et une dysgranulopoïèse (76).
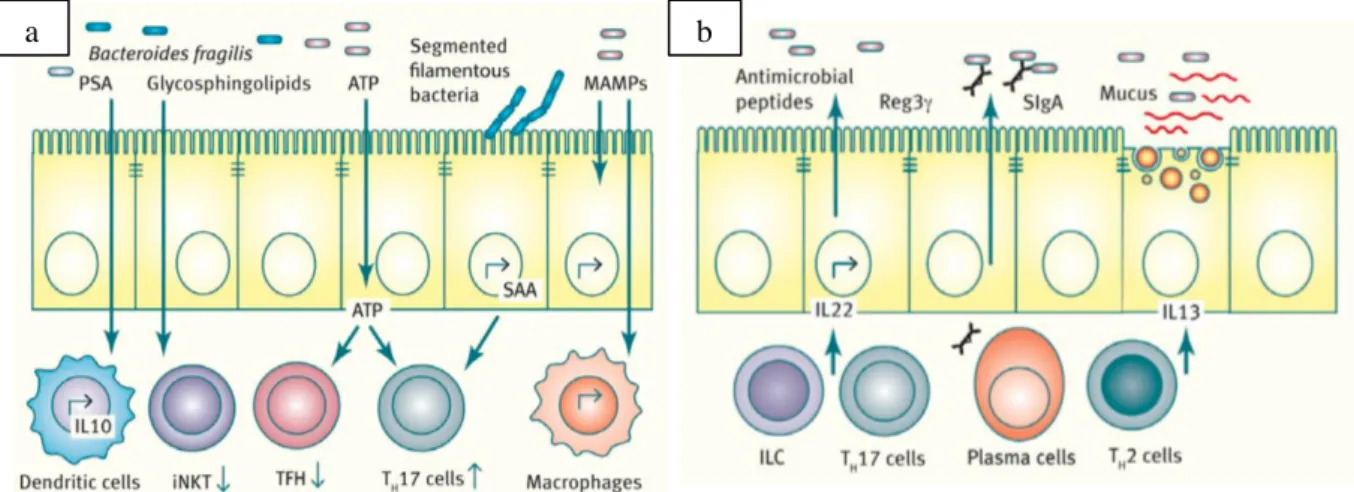
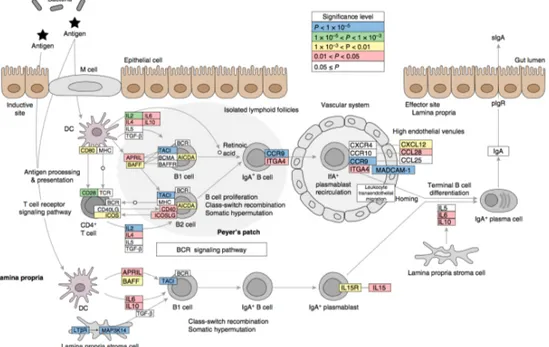
Figure 2 : Schémas représentatifs des interactions réciproques microbiote-système immunitaire.
(a) Les constituants bactériens (comme le polysaccharide A, PSA) ou les métabolites synthétisés de novo par les bactéries ont un impact sur le système immunitaire. Par exemple, les glycosphingolipides de Bacteroides fragilis module l’homéostasie des cellules iNKT. L’ATP libéré par le microbiote limite la différenciation des T folliculaires helper mais favorise celle des Th17. En adhérant directement aux cellules épithéliales, le SFB induit la production de SAA (Serum Amyloid A) par les entérocytes qui promeut la différenciation Th17. Enfin, les MAMPs (Microbe-associated molecular patterns) sont à même d’être reconnus par de nombreux récepteurs à la surface des cellules épithéliales ou immunes, notamment les macrophages. (b) En réponse au microbiote, le système immunitaire le module. Les ILC2 (Innate Lymphoid Cells type 2) et les lymphocytes Th17 sécrètent de l’IL-22 qui déclenche la production de peptides antimicrobiens dont RegIIIγ par les entérocytes. Les plasmocytes de la lamina propria sécrètent de l’IgA sous forme dimérique, transporté dans la lumière intestinale via les cellules épithéliales. Enfin, la production de cytokines par les cellules immunitaires présentes dans la lamina propria peut moduler la production de mucus par les cellules de Paneth et ainsi réguler l’accès du microbiote à la barrière épithéliale. D’après (1).
Si le microbiote contribue au développement du système immunitaire, ce dernier s’est adapté au cours de milliers d’années de co-évolution afin de favoriser les relations symbiotiques tout en diminuant le risque d’invasion opportuniste ou d’inflammation. Les mécanismes de protection de l’hôte sont multiples et ne sont pas encore tous élucidés : (i) production de mucus, (ii) sécrétion d’IgA et de peptides antimicrobiens dans la lumière intestinale, (iii) épithélium intestinal avec des jonctions serrées empêchant la pénétration, (iv) immunité innée avec un rôle important des TLR (77), (v) développement du GALT (Figure 2). L’ensemble de ces acteurs agit de manière flexible et complémentaire, l’un pouvant suppléer l’autre dans le but de maintenir l’homéostasie intestinale. Il a en effet été montré chez les souris que l’absence d’anticorps au niveau intestinal provoquait une activation accrue de l’immunité innée (78). Au sein de cette immunité muqueuse, les IgA ont une place centrale et seront détaillées dans le chapitre suivant.
Le microbiote est la première barrière contre les pathogènes. Il stimule le système immunitaire de l’hôte et favorise un environnement tolérogène.
1.2 Physiologie des Immunoglobulines A
1.2.1 Structure et sous-classes d’IgA
La production quotidienne d’IgA, 66mg/kg/j, dépasse celle de toutes les autres classes d’immunoglobulines réunies (79). Contrairement à la plupart des mammifères (dont la souris), il existe deux isotypes d’IgA chez l’Homme : IgA1 et IgA2. Les IgA1 se caractérisent par une région charnière –région flexible qui sépare les domaines de fixation de l’antigène Fab des domaines effecteurs Fc – allongée de 13 acides aminés (Figure 3). Cette particularité des IgA1 est apparue tardivement dans l’évolution et n’est pas présente dans les IgA murines (80, 81). Elle confère aux IgA1 une forme singulière, en « T » et non en « Y » comme les autres immunoglobulines et l’expose à la protéolyse bactérienne (82). Aucune protéase n’a été décrite à ce jour pour les IgA2 (83).
Figure 3: Structure des IgA.
(a) IgA1 ; (b) IgA2 ; (c) IgA1 dimérique ; les domaines des chaînes lourdes sont représentées en rose,
ceux des chaînes légères en bleu clair, la chaîne J en jaune. Les sites de N- et O- glycosylation sont indiqués en rouge et en vert respectivement. D’après (83).
La répartition des sous-classes d’IgA dépend des compartiments : l’IgA1 est majoritaire dans le sérum (90-95% des IgA totales, soit en moyenne 1,7g/l ; (84)) alors que l’IgA2 est plus abondante au niveau des muqueuses. Il est à noter que l’IgA1 n’est pas absente des muqueuses : elle représente 80% des IgA au niveau nasal et 60% au niveau de la salive et de l’iléon (85, 86). Dans le lait maternel, les deux isotypes cohabitent en proportions égales (87).
L’IgA existe sous forme de monomères prédominants dans le sérum, ou de dimères prédominants au niveau des muqueuses. Bien qu’à faible concentration, des polymères de plus grande taille (trimères ou tétramères) sont aussi présents dans les muqueuses (83, 88).
Chaîne J dIgA1 Région charnière Fc Fa b Cα2 Cα3 a b c
monomères d’IgA sont liés entre eux de manière covalente via leur fragment Fc et une chaîne J synthétisée par les plasmocytes. La chaîne J est un peptide très conservé qui possède 8 cystéines, lui permettant d’établir des ponts disulfures avec les résidus terminaux des Fc. Dans le cas de polymères de plus grande taille (IgA ou IgM), une seule chaîne J suffit pour stabiliser la structure (89).
Enfin, les IgA sont des molécules hautement glycosylées. Le degré de glycosylation varie en fonction de l’isotype et du degré de polymérisation (80, 90). En effet, l’IgA1 présente en plus de sites de N-glycosylation (glycosylation sur les groupements amines d’asparagine ou d’arginine) communs avec l’IgA2, des sites de O-glycosylation sur la région charnière (glycosylation sur des groupements hydroxyles présents par exemple sur la sérine ou la thréonine, Figure 3). La chaîne J possède quant à elle un site de N-glycosylation qui contribue à la stabilisation des dimères (89). La large diversité d’oligosaccharides branchés sur les IgA leur confère un degré d’hétérogénéité supplémentaire (plusieurs glycoformes d’IgA détectables dans le lait maternel) et des propriétés intéressantes d’interaction avec les surfaces bactériennes, épithéliales ou avec certains récepteurs comme la Dectin-1 (90, 91).
1.2.2 Synthèse des IgA intestinales
1.2.2.1 Induction des réponses IgA dans la muqueuse intestinale
Le GALT est le principal lieu de production d’IgA, en témoigne le grand nombre de plasmocytes IgA+ présents dans la lamina propria intestinale (92). Trois de ses structures interviennent dans l’induction de la réponse IgA : (i) les ganglions mésentériques localisés sous la lamina propria (dans le mésentère), (ii) les plaques de Peyer organisées en follicules contenant des centres germinatifs avec un dôme recouvert de cellules M (microfold cells, cellules épithéliales spécialisées), les plaques de Peyer sont plus fréquentes dans l’iléon et se situent sous l’épithélium, (iii) les îlots lymphoïdes isolés ressemblant à des follicules dispersés dans la lamina propria. Le développement des plaques de Peyer et des îlots lymphoïdes isolés est dépendant du microbiote. En effet, bien que les plaques de Peyer apparaissent in utero elles s’hypertrophient lors de la colonisation bactérienne. Les îlots lymphoïdes isolés sont absents chez les souris axéniques (93).
Il existe deux sous-classes d’IgA chez l’Homme qui sont présentes au niveau des muqueuses sous forme de dimères hautement glycosylés.
Plusieurs mécanismes de capture et transit des antigènes depuis la lumière intestinale vers les sites inducteurs ont été décrits. La transcytose des antigènes (recouverts ou non par des IgA, cf 1.2.4.2) via les cellules M semble être la voie la plus importante : chez des souris dépourvues de cellules M, le nombre de plasmocytes IgA+ est drastiquement réduit (94). Ces antigènes sont ensuite délivrés aux cellules dendritiques présentes dans les plaques de Peyer. Certains pathogènes comme Salmonella sont à même d’utiliser cette transcytose à leur profit afin de traverser l’épithélium et envahir la lamina propria (95). D’autre part, les cellules dendritiques de la lamina propria peuvent directement capter des antigènes en prolongeant leurs dendrites à travers les jonctions serrées des cellules épithéliales, ces cellules dendritiques rejoignent ensuite les sites inducteurs. A l’occasion d’une brèche de l’épithélium, des antigènes peuvent aussi atteindre directement la lamina propria : ils sont alors soit captés par les cellules dendritiques soit drainés par le circuit lymphatique et conduits vers les ganglions mésentériques. Enfin, des PAMP (pathogen-associated molecular patterns) tels que la flagelline ou des métabolites bactériens peuvent être reconnus par des PRR (pattern recognition receptors) exprimés par les cellules épithéliales et les cellules dendritiques et ainsi induire une réponse IgA (96). L’ensemble de ces mécanismes intervient lors d’infection par des pathogènes mais aussi dans la régulation « normale » de la flore commensale.
On distingue classiquement les réponses IgA T-dépendantes et T-indépendantes : les premières sont générées dans des centres germinatifs à la suite d’une stimulation antigénique et aboutissent à la synthèse d’anticorps de haute affinité ; les deuxièmes sont indépendantes des centres germinatifs et se caractérisent par des anticorps de faible affinité, faiblement mutés. Bien que ces deux voies soient impliquées dans l’induction des réponses IgA anti-microbiote, les réponses T-indépendantes semblent prépondérantes selon des études chez des souris CD40 ou TCR-knock out (97, 98). Les réponses T-indépendantes ont lieu principalement dans les îlots lymphoïdes isolés et les ganglions mésentériques contrairement aux réponses T-dépendantes qui sont majoritairement localisées dans les centres germinatifs des plaques de Peyer (Figure 4, (99)). De plus, les réponses T-dépendantes à un antigène étranger au microbiote semblent être synchronisées entre plusieurs plaques de Peyer (profil oligoclonal) tandis que les réponses aux bactéries commensales sont largement multi-centriques et polyclonales (100). Chez l’Homme, l’importance relative et les mécanismes de ces deux types de réponse sont encore à élucider. Néanmoins, la présence de plasmocytes IgA+ dans la muqueuse intestinale de patients infectés par le Virus de l’Immunodéficience Humaine (donc avec un défaut de lymphocytes T muqueux) et de lymphocytes B CD27-
IgA+ intestinaux chez des patients présentant une mutation de CD40L sont en faveur de l’existence d’une réponse IgA T-indépendante chez l’homme (101, 102).
Plusieurs facteurs solubles, principalement sécrétés par les cellules dendritiques, influencent la commutation isotypique vers l’IgA dans la muqueuse intestinale. La liste ci-dessous n’est pas exhaustive mais reprend les éléments les plus importants issus d’études murines (Figure 4):
- le TGFβ et son récepteur exprimé à la surface des lymphocytes B. En effet, les souris
knock-out pour le récepteur au TGFβ sont déficitaires en IgA (103, 104). L’expression du TGFβ-R est régulée positivement par le NO (monoxyde d’azote) produit par les cellules dendritiques via iNOS (inducible Nitric Oxyde Synthase). Ces acteurs sont particulièrement importants dans le cas des réponses T-dépendantes (105).
- APRIL (Proliferation Inducing Ligand) : les souris déficitaires pour APRIL présentent une
absence d’IgA, même après vaccination muqueuse (106).
- BAFF (B-cell activating factor) qui renforce l’effet d’APRIL : la sur-expression de BAFF
chez des souris transgéniques induit un syndrome hyper-IgA (107).
- l’IL-10, l’IL-4 et l’IL-21 : l’ajout de ces cytokines à une culture de lymphocytes B de
patients déficitaires en IgA permet de restaurer la sécrétion d’IgA (108).
- l’IL-33 agit en synergie avec le TGFβ : les souris IL-33 ko présentent un défaut de réponse
IgA qui s’accompagne d’une dysbiose et d’une colite inflammatoire (109).
Si les cellules dendritiques jouent un rôle clé dans la commutation isotypique vers l’IgA en sécrétant des facteurs solubles cités ci-dessus, elles entrent aussi directement en contact avec les lymphocytes B sous le dôme des plaques de Peyer. Pour atteindre cette zone, les lymphocytes B activés sur-expriment le CCR6 et répondent à un gradient de CCL20, chimiokine sécrétée notamment par les cellules mésenchymateuses présentes dans le dôme sub-épithélial (110, 111). Les lymphocytes B entrant dans les plaques de Peyer sont des lymphocytes B naïfs provenant de la rate via la circulation sanguine (112).
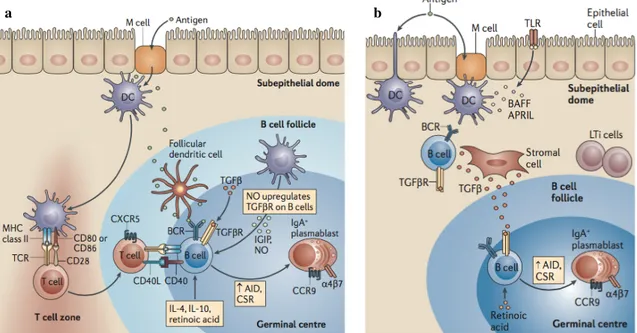
Figure 4 : Représentation schématique de l’induction des réponses IgA dans le GALT.
(a) L’induction des réponses IgA T-dépendantes a lieu principalement dans les plaques de Peyer. Les antigènes
sont capturés par les cellules M et délivrés aux cellules dendritiques du dôme sub-épithélial. Les cellules dendritiques entre dans la zone T pour y activer la différenciation de lymphocytes T naïfs en T effecteurs ; les cellules dendritiques rejoignent également la zone B où elles sécrètent des cytokines inductrices d’IgA. La coopération T-B aboutit à l’expression d’AID et à la commutation isotypique vers l’IgA. (b) L’induction des réponses IgA T-indépendantes a lieu principalement dans les follicules lymphoïdes isolés comme représenté sur le schéma ou dans la lamina propria. Les cellules dendritiques et les TLR exprimés par les cellules épithéliales coopèrent pour sécréter BAFF, APRIL et ainsi activer les lymphocytes B. D’après (113).
Une fois induites, les réponses IgA T-dépendantes sont finement régulées dans les centres germinatifs par la présence des lymphocytes T folliculaires helpers (Tfh). Ces lymphocytes T se caractérisent par une forte expression de molécules de co-stimulation à leur surface telles que CD40L, PD-1 (Programmed cell Death-1), OX-40 et la production de cytokines (IL-21 et IL-4) impliquées dans le contrôle des lymphocytes B (114). Ils contribuent à la sélection d’IgA spécifiques. Ainsi chez des souris PD1-/-, Kawamoto et ses collaborateurs ont constaté une diminution de la proportion de bactéries commensales reconnues par des IgA parallèlement à une augmentation d’IgA libre dans les eaux fécales, démontrant un manque de spécificité des anticorps générés. Dans ce même modèle murin, les plasmocytes IgA+ présentent moins de mutations somatiques (115). D’une autre façon, les lymphocytes T régulateurs (CD4+FoxP3+, Treg), participent à la sélection des IgA : en présence de Treg, les IgA générées reconnaissent plus spécifiquement (index de maturation d’affinité plus élevé) une large diversité d’espèces bactériennes ce qui contribue au maintien d’un microbiote sain et diversifié (116).
Peu d’études ont exploré les facteurs déterminant la commutation isotypique vers l’IgA1 ou l’IgA2. Les deux isotypes ne semblent pas liés à un type de réponse en particulier (T-dépendant ou T-indépendant) puisque des plasmocytes IgA1+ et IgA2+ sont visibles dans la lamina propria de patients mutés pour CD40L (102). Ces mêmes auteurs soulignent que la commutation isotypique de l’IgM vers l’IgA1 a lieu de façon directe tandis que la commutation de l’IgM vers l’IgA2 est plus souvent séquentielle : IgG2-IgA2 ou IgM-IgA1-IgA2 (cf annexe 1, structure des gènes d’immunoglobulines). Cependant ces données n’ont pas été confirmées par une étude plus récente (117).
1.2.2.2 Sécrétion des IgA dans la lumière intestinale
Les IgA sont synthétisées par les plasmocytes de la lamina propria. Ces lymphocytes B spécialisés dans la synthèse d’Ig sont des cellules à longue durée de vie, résidentes du GALT grâce à l’expression de molécules de homing à leur surface (CCR9 et α4β7). Chez l’homme, deux autres populations de lymphocytes B mémoires se trouvent dans la lamina propria : les lymphocytes B CD27+IgA+ et CD27-IgA+ (86, 118, 119).
Une fois synthétisées, les IgA sons transportées activement dans la lumière intestinale à travers les cellules épithéliales via le récepteur aux Immunoglobulines polymériques (pIgR). Ce récepteur est exprimé constitutivement au niveau du pôle basal des entérocytes. Son expression est augmentée par des cytokines pro-inflammatoires comme l’IL-17, les hormones stéroïdiennes ou l’activation des PRR à la surface des cellules épithéliales (120, 121). Le pIgR est une glycoprotéine qui comprend 5 domaines extracellulaires Immunoglobulin-like, un domaine transmembranaire et un domaine intracellulaire. Le cycle de transit des IgA commence par la fixation des IgA dimériques au pIgR. Une fois internalisé, le complexe traverse la cellule épithéliale puis est excrété dans la lumière intestinale après clivage par des protéases du domaine N-terminal du pIgR constituant alors la pièce sécrétoire (Figure 5). Au cours de la transcytose, les 5 domaines Ig-like se lient de façon covalente au Fc des IgA et se replient comme un doigt autour de l’IgA dimérique la protégeant de la protéolyse (122, 123). Ce complexe IgA dimérique – pièce sécrétoire est désigné sous le terme d’IgA sécrétoire (sIgA).
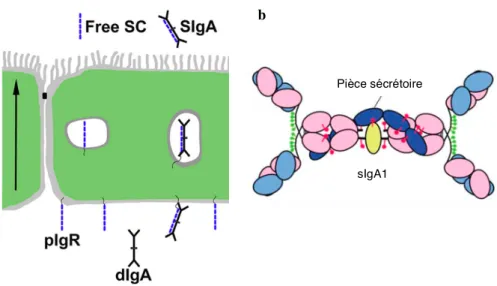
Figure 5 : Transcytose des IgA à travers les entérocytes.
(a) Les dimères d’IgA se lient par la chaîne J au pIgR (en bleu marine) situé au pôle basal de l’entérocyte. Le
complexe formé est alors endocyté et traverse le cytoplasme de la cellule épithéliale. Enfin, le pIgR est clivé au pôle apical et libère l’IgA sécrétoire dans la lumière intestinale. Le trafic intracellulaire du pIgR libère de la pièce sécrétoire libre (SC free). (b) représentation schématique d’une IgA1 sécrétoire. Les domaines du pIgR sont représentés en bleu. Les sites de N-glycosylation sont indiqués en rouge. D’après(83, 124).
Bien que la fixation d’Ig polymériques stimule la transcytose du pIgR, environ 50% du trafic intracellulaire de pIgR libère de la pièce sécrétoire non fixée à des Ig. Cette pièce sécrétoire libre, riche en groupements carbohydrates tout comme la pièce sécrétoire liée, possède des fonctions « immunes » : elle est capable d’inhiber l’adhésion des bactéries Gram négatives à la surface des entérocytes ou de contribuer à l’homéostasie du microbiote par des interactions non spécifiques protéines-protéines ou sucres-protéines (124, 125).
Ce mécanisme de transcytose des Ig polymériques est valable pour les IgM et les IgA, quel que soit la sous-classe d’IgA (IgA1 et IgA2 ont des affinités similaires pour le pIgR) ou le type de muqueuses, les IgA du lait maternel ont en effet subi le même transport (120, 126). A contrario, les IgA monomériques et les IgG synthétisées dans la lamina propria ne sont pas transportées par le pIgR. Elles peuvent néanmoins rejoindre la lumière intestinale par diffusion passive entre les entérocytes ou via le récepteur Fc néonatal (exprimé au pôle basal des entérocytes, uniquement pour les IgG ; (127, 128).
Le microbiote est le principal inducteur et régulateur de la synthèse d’IgA dans l’intestin. Deux types de réponses IgA coexistent : T-dépendante et T-indépendante.
Seules les IgM et les IgA sont transportées de manière active dans la lumière intestinale.
Pièce sécrétoire
sIgA1
1.2.3 Répertoire et spécificité des IgA intestinales
1.2.3.1 Un répertoire diversifié
En raison d’obstacles techniques, la description du répertoire des IgA intestinales s’est longtemps limitée à de grandes tendances : (i) oligoclonalité des plasmocytes IgA+ sur un fond polyclonal, (ii) existence d’une parenté entre IgM et IgA, (iii) dispersion de clones identiques tout au long du colon, (iv) présence de mutations somatiques évoquant une maturation d’affinité et une sélection dirigée par l’antigène (129). Cette vision s’est étoffée et éclaircie avec le développement des techniques de séquençage haut-débit. On distingue désormais deux contingents au sein du répertoire IgA : un contingent principal composé de clonotypes minoritaires (représentés moins de 20 fois parmi 5000 séquences analysées) et un deuxième contingent composé de clonotypes étendus (plusieurs séquences affiliées, retrouvées en grand nombre). La diversité du répertoire IgA est directement liée à la présence du premier contingent à savoir les nombreux clonotypes minoritaires. Il se diversifie avec l’âge selon deux mécanismes : l’accumulation de mutations somatiques et l’augmentation du nombre de clonotypes minoritaires (130). La diversité du répertoire est aussi associée à la diversité du microbiote : le répertoire de souris monocolonisées avec E. coli ont un répertoire restreint comparativement aux souris colonisées avec un microbiote complexe (131). Un dernier niveau de diversité du répertoire IgA chez l’homme concerne les sous-classes IgA1 et IgA2, néanmoins ce point n’a pas encore été étudié.
Le phénomène de dispersion spatiale des clones au sein du colon ou de l’iléon a aussi été confirmé par ces études de Lindner et al., néanmoins le répertoire IgA de ces deux compartiments ne se chevauche que partiellement. De façon étonnante, il a été montré une plus grande similitude entre les répertoires iléon-glandes mammaires qu’entre les répertoires iléon-colon (131). Quant à la relation entre les IgA sériques et les IgA intestinales, Holtmeier
et al. ne trouvent pas de chevauchement entre ces deux répertoires. A l’inverse, en combinant
de façon originale la spectrométrie de masse et le séquençage haut-débit, il a été récemment démontré une relation clonale entre les IgA sériques et intestinales évoquant une origine intestinale des lymphocytes B IgA+ du sang. Cependant, cette étude ne portait que sur des IgA spécifiques de la transglutaminase 2, anticorps impliqués dans la maladie cœliaque (132). L’existence de deux types de lymphocytes B mémoires IgA+ (CD27+ et CD27-) pourrait expliquer ces résultats divergents : les lymphocytes B CD27-IgA+ seraient issus de l’intestin tandis que les lymphocytes B CD27+IgA+ seraient issus des ganglions périphériques (101).
Des investigations supplémentaires sont nécessaires afin d’approfondir le lien entre IgA sériques et IgA intestinales.
Enfin, le répertoire IgA est un répertoire hautement privé : des souris de même fond génétique vivant dans le même environnement, ou colonisées par la même espèce bactérienne, expriment des répertoires IgA génétiquement différents (130, 131). L’importance du fond génétique joue néanmoins un rôle dans la mise en place du répertoire IgA, puisque des souris BALB/c et C57BL/6 élevées dans les mêmes conditions diffèrent quant à leur diversité de répertoire IgA (133).
1.2.3.2 Un répertoire stable mais dynamique
En plus d’être diversifié, le répertoire des IgA se montre à la fois stable et dynamique. Hapfelmeier et al. ont appréhendé ces deux particularités grâce à un système murin de colonisation réversible par une souche d’E. coli mutante (qui ne peut pas se multiplier dans l’intestin murin par manque de nutriments). Ils ont démontré la mise en place rapide et la persistance des réponses IgA dans le temps : les IgA anti-E. coli perduraient malgré l’absence de la souche ou même après recolonisation avec un microbiote complexe et donc diversification du répertoire (134). Ci-dessous, nous envisagerons ces deux caractéristiques lors de la mise en place des réponses IgA au début de la vie et lors d’une infection par un pathogène.
Les réponses IgA suivent la colonisation bactérienne : elles apparaissent progressivement après 3 à 4 semaines chez la souris ou à partir du 3ème mois chez le nouveau-né (33). Au cours de cette première période, l’apport d’IgA par le lait maternel est un élément clé de développement des réponses IgA néonatales. En leur absence, le nombre de plasmocytes IgA+ augmente plus précocement et plus rapidement dans la lamina propria (135). L’allaitement a aussi des conséquences sur l’établissement du microbiote. En croisant des souris pIgR-/- et pIgR+/+, Rogier et al. ont montré en l’absence d’IgA maternelles (i) une translocation bactérienne majoritairement de protéobactéries vers les ganglions mésentériques (ii) une différence de composition microbienne avec une expansion des γ-protéobactéries qui persiste jusqu’à l’âge adulte et (iii) un profil d’expression génique des entérocytes semblable à celui retrouvé dans les colites inflammatoires (136). Ce travail chez la souris a trouvé un parallèle chez l’homme dans une étude longitudinale incluant 40 paires de jumeaux mono- et dizygotes. Dès le 3ème mois, les IgA des nouveau-nés reconnaissent une portion de la flore commensale, les premières cibles étant Clostridium nexile et Bifidobacterium bifidum. Une fois reconnues par les IgA, les cibles bactériennes le sont encore à la fin de l’étude, à 3 ans ce
qui est en faveur d’une stabilité du répertoire IgA. Par ailleurs, l’âge et la famille sont des facteurs plus importants que le caractère mono/dizygote pour prédire la concordance entre les réponses IgA et la composition microbienne (33). La composition microbienne des premiers stades de vie, riche en protéobactéries, influence directement les réponses IgA : les antigènes obtenus à partir d’un microbiote de souris âgées d’une semaine semblent plus immunogènes que ceux obtenus à partir d’un microbiote adulte (137). La mise en place du répertoire IgA dans cette période de l’enfance a des conséquences à long terme : un biais quantitatif ou qualitatif a été associé au risque allergique (138).
Si le répertoire IgA évolue rapidement au début de la vie puis se stabilise, il reste extrêmement flexible en cas de survenue d’un agent pathogène. Lors d’une infection aigüe à
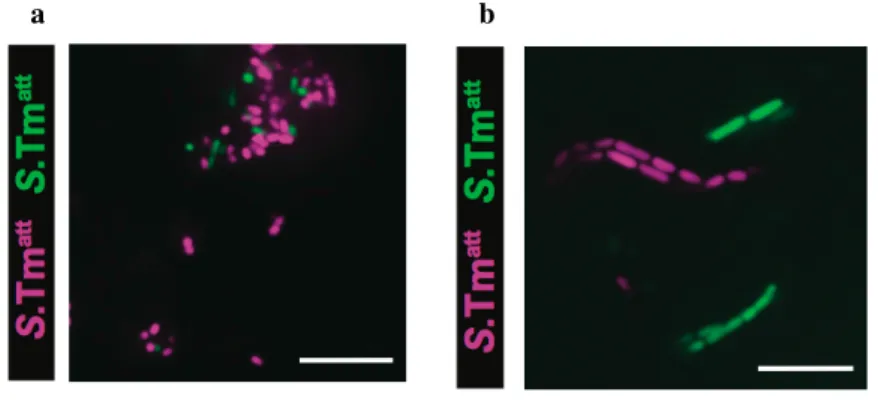
Salmonella typhimurium, Lindner et al. ont observé un changement drastique du répertoire
avec l’apparition de nouveaux clones dominants permettant de prendre en charge l’agent infectieux puis une restauration du répertoire de base, les nouveaux clones se diluant dans le répertoire initial. Cette flexibilité et cette résilience du répertoire sont liées à l’agent pathogène et non au traitement antibiotique. En effet, une antibiothérapie brève n’induit pas de modification majeure du répertoire IgA (131).
1.2.3.3 Les IgA : affinité et réactivité croisée
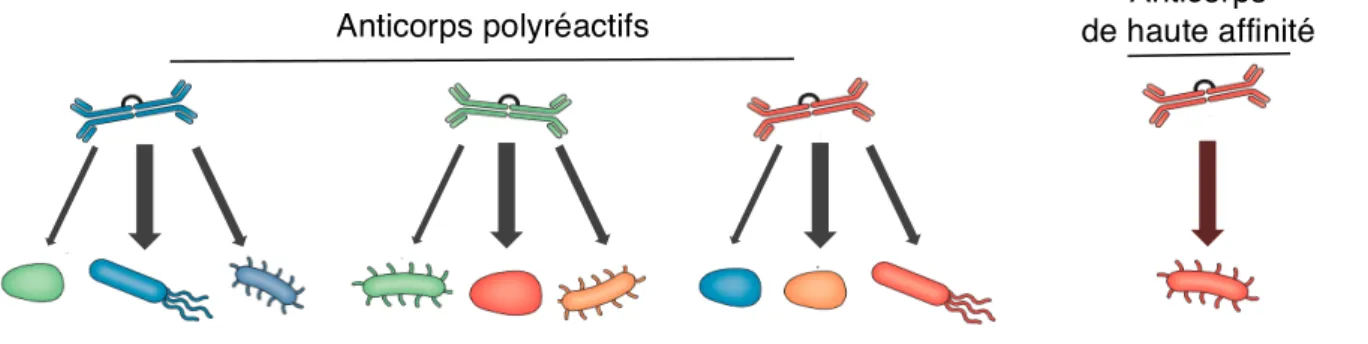
Une vision classique des réponses humorales distingue les anticorps de haute affinité spécifiques d’un pathogène, des anticorps de faible affinité polyréactifs. Les premiers, issus de réponses T-dépendantes, ont accumulé des mutations somatiques et cibleraient les pathogènes tandis que les derniers, T-indépendants, cibleraient les bactéries inoffensives à savoir les commensales.
Les dernières avancées sur les IgA viennent bousculer cette conception. En effet, la majorité des plasmocytes IgA+ de la lamina propria portent des mutations somatiques, ils produisent donc a priori des anticorps de haute affinité (130, 139). En parallèle, une fraction importante du microbiote est reconnue par les IgA (environ 60% dans l’iléon et 30% dans le colon) (97). Ces deux observations supportent donc l’existence d’anticorps de haute affinité vis-à-vis de la flore commensale. Au sein de la flore commensale, certaines bactéries (comme SFB seraient plus à même d’induire ces réponses IgA de haute affinité ce qui est suggéré dans les modèles murins AID-/- (sans mutations somatiques). Ainsi, le microbiote de ces souris incapables de monter des réponses affines est déséquilibré avec une hyper-prolifération de SFB (140). Particularité supplémentaire aux IgA intestinales : elles sont à la fois de haute