Mélanges de polymères à gouttes composites : application recyclage
Texte intégral
Figure
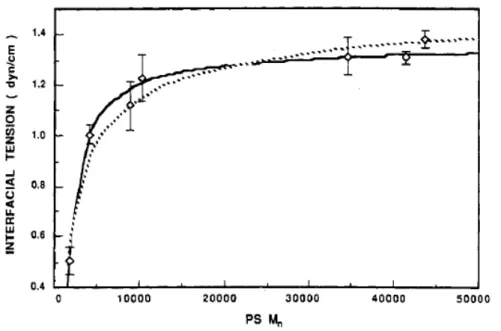
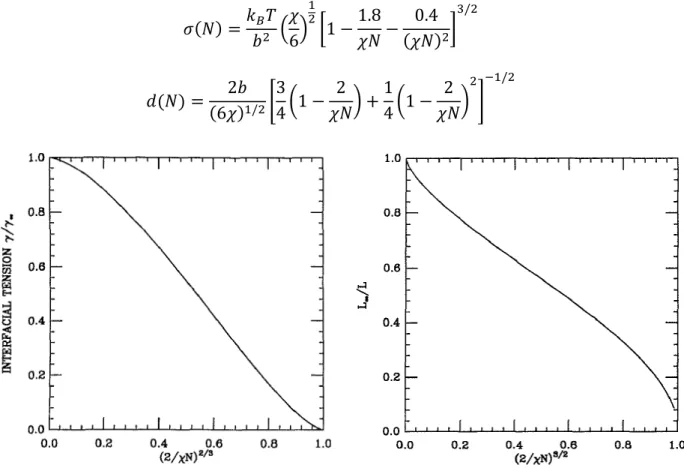

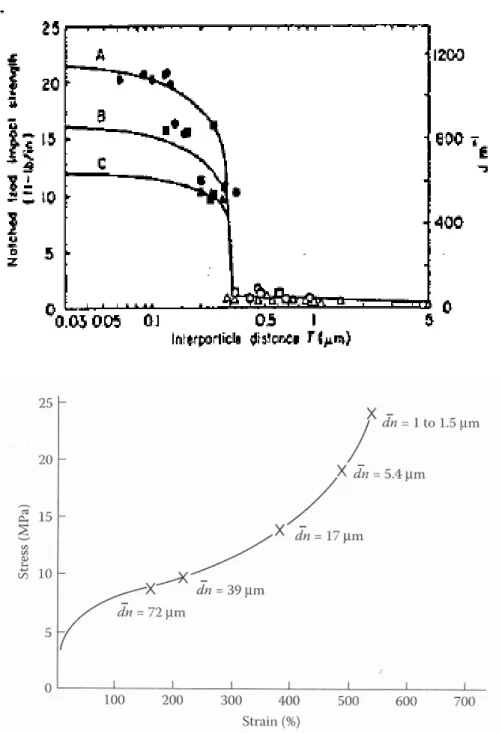

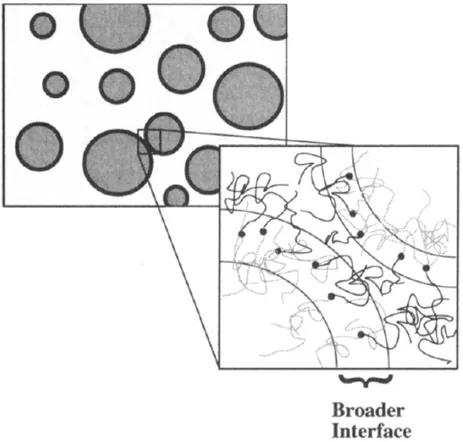


Outline
Documents relatifs
One simple approach for liquid-liquid systems consists in measuring water contact angles on plates of dense polymer making up the membranes (PP or PTFE) when immersed in
Abstract- Investigations were made of the influence of cyclic reversible martensitic transformations on variations in internal friction for a Cu- (14.5%wt.) Zn-(8.5%wt.) A1 al1oy.A
Quantitative data analyzed the performance scores of the pre-cognitive test,pre-procedural test on a Class II composite restoration on tooth # 45(MO) and creation of two Class
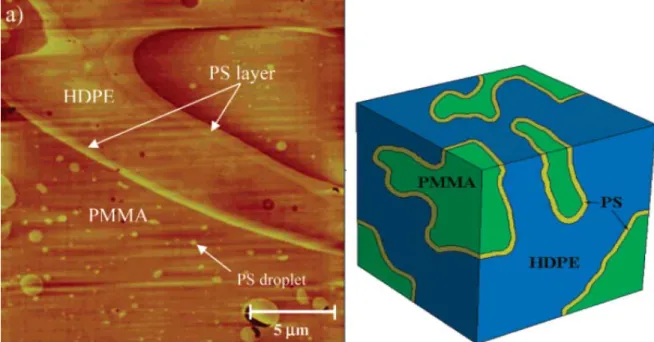
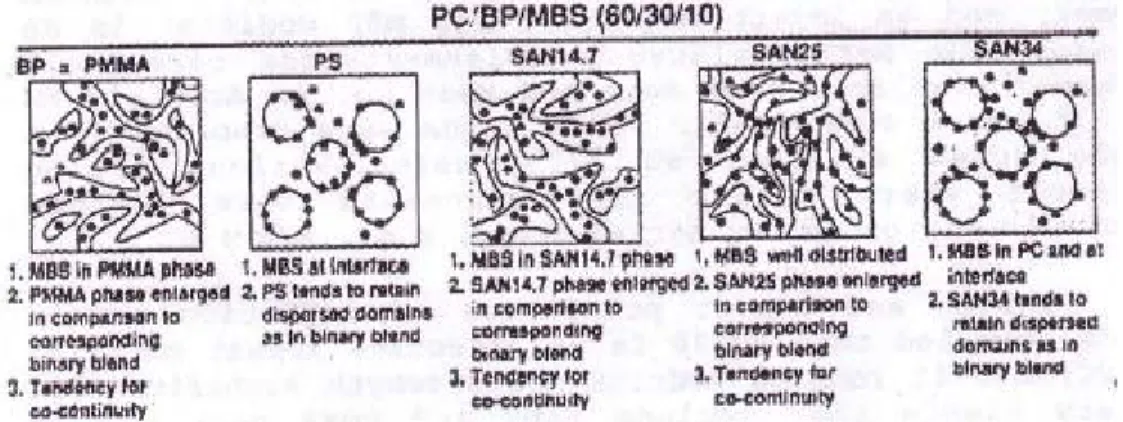
Compatibilization of PMMA/PS blends by nanoparticles and block copolymers: effect on morphology and interfacial relaxation phenomena Abstract In this thesis, the
The degradation tests consist in 4 steps as follows (Figure 4.3): (1) preparation of the polymer films, (2) addition of the hydrolysis media such as the buffered solution pH 9,
Under compression, the Young’s modulus and stress at 50 % deformation of the samples was found to increase with increasing silylation level. Notably, the shape recovery of the NFC
In this section, the starch was plasticized by BMIMCl and glycerol by extrusion in the micro-compounder (section 2.2.1.2). The advantage of this technique is that the
Substantial research has been carried out on the surface modification of wood fibers with coupling agents to improve the strength properties of WPCs (Woodhams et al., 1984; Li
