INTRODUCTION
Depuis plusieurs années, de nombreux travaux ont étayé la fonction endocrine du cœur. L’existence de systèmes hormonaux dans le tissu cardiaque plus particulièrement celui des peptides natriurétiques, a conféré au cœur la propriété de glande endocrine en plus de sa simple fonction de pompe.
Le BNP constitue le second membre d’une famille d’hormones natriurétiques qui comprend également l’ANP (Atrial Natriuretic Peptide, De Bold et al., 1981), le CNP (C type Natriuretic Peptide, Sudoh et al., 1990) et le DNP (D type Natriuretic Peptide, Stein et al., 1998). Le BNP fut initialement isolé du cerveau de porc (Sudoh et al., 1988). Chez l’homme, il est majoritairement produit par les myocytes ventriculaires en réponse à des surcharges de pression ou de volume et relargué en permanence dans la circulation générale. Ce peptide exerce ses effets physiologiques par sa liaison sur ces récepteurs natriurétiques à activité guanylate cyclase, NPRs (NPR-A, -B et –C). Il antagonise le système rénine- angiotensine- aldostérone (SRAA) et agit aussi comme un agent hypotenseur, natriurétique, vasodilatateur, antifibrotique et antihypertrophique. Vu son triple intérêt, diagnostique- marqueur de la décompensation cardiaque, thérapeutique- agent antifibrotique, diurétique, natriurétique et vasodilatateur, et pronostique- facteur prédictif de mortalité chez les insuffisants cardiaques (Scardovi AB., 2004), le BNP semble être une véritable hormone de stress cardiaque. En effet, divers travaux ont souligné une corrélation étroite entre le taux plasmatique du BNP et le dysfonctionnement cardiaque (Redfield MM. et al., 2004).
Toutefois, aussi qu’elles soient nombreuses les mesures du taux plasmatique du BNP dans les conditions physiologiques normales et pathologiques, ces mesures visaient surtout l’aspect macroscopique du rôle de ce peptide, de ses applications, de son potentiel thérapeutique, plutôt que l’aspect microscopique des mécanismes contrôlant la régulation de sa sécrétion et de ses effets modulateurs à l’échelle cellulaire et moléculaire.
Dans le cardiomyocyte ventriculaire, le calcium intracellulaire joue un rôle central dans la régulation de l’expression de nombreux gènes dont celui du BNP (Kudoh S. et al., 2003). A son tour, ce peptide agit à travers des mécanismes GMPc-dépendants (Potter et al., 2006) et semble être impliqué dans la modulation de l’homéostasie calcique, régulant ainsi la fonction contractile du cœur.
Avec comme modèle expérimental, les cellules ventriculaires isolées de cœurs de rats adultes normaux et hypertrophiés par surcharge de pression, nous avons tout d’abord mesuré, à l’aide de la technique ELISA, les concentrations de BNP dans le sang et dans le milieu de culture des cardiomyocytes en présence et en absence d’antagonistes calciques. Nous avons également évalué, en immunohistochimie, le degré d’expression du BNP dans le myocarde normal et hypertrophié ainsi que l’expression génique de ses récepteurs NPR-A, -B et –C par RT-PCR. Parallèlement, nous avons analysé, à l’aide de la technique de patch-clamp, l’effet du BNP sur le potentiel d’action et la densité du courant calcique de type L, ICaL dans les cardiomyocytes normaux.
Ces objectifs visent à rapporter des éléments supplémentaires pour la compréhension des effets bénéfiques paracrines et autocrines du BNP sur la fonction cardiaque normale ainsi que son effet dans le remodelage myocardique en réponse à un stress hémodynamique.
HISTORIQUE
1. Le cardiomyocyte ventriculaire adulte normal
Un événement clé dans le développement cardiaque s’opère à la naissance, où les cardiomyocytes vont perdre leur phénotype d’hyperplasie pour continuer leur développement selon un processus de type hypertrophique uniquement (Figure H1).
Durant leur développement, les cardiomyocytes sous l’action de plusieurs facteurs humoraux (le système nerveux, l’angiotensine II, les peptides natriurétiques, etc..) et hémodynamiques (tension artérielle, étirement..), vont acquérir toutes leurs propriétés morphologiques et électrophysiologiques caractéristiques de la cellule ventriculaire contractile.
Figure H1 : Le développement cardiaque D’après Pignier, 1999.
1.1. Propriétés morphoanatomiques 1.1.1. Morphologie
Les cellules ventriculaires sont organisées d’une façon complexe, à la manière d’une turbine. Elles possèdent une morphologie adaptée à la structure en spirale de la musculature lui permettant d’assurer une puissance nécessaire pour l’ejection de sang lors de la systole (Katz AM, 1992).
Le cœur est constitué de faisceaux musculaires constitués de cardiomyocytes connectés entre eux au niveau des disques intercalaires.
L’observation en microscopie photonique du cardiomyocyte ventriculaire isolé montre une cellule à forme allongée, rectangulaire (ou en bâtonnet « rode shape ») mesurant environ 100 µm de longueur et 20 µm de largeur (Figure H2). Le cardiomyocyte ventriculaire présente des stries qui correspondent aux extrémités des sarcomères et dont le nombre varie de 50 à 90 par myocyte.
Figure H2 : Cardiomyocyte ventriculaire isolé d’un cœur de rat adulte normal
1.1.2. Ultrastucture
Le cardiomyocyte ventriculaire est constitué essentiellement d’un système membranaire, d’un système contractile et d’un cytosquelette.
a) Système membranaire
Le système membranaire (Figure H3) comprend tout d’abord le sarcolemme, épais de 7,5 à 10 nm, de nature phospholipidique, entouré par le glycocalix, épais de 20 à 80 nm, et qui contient des fibrilles constituées notamment de fibronectine et de laminine (Sommer JS. et Jennings RB., 1992). Le sarcolemme contient tous les systèmes (pompes, échangeurs, canaux, enzymes, récepteurs) lui permettant les échanges intercellulaires et avec le milieu externe. Egalement le système membranaire comprend les tubules T (ou système tubulaire transverse) qui sont des invaginations en doigts de gant, qui pénètrent profondément dans la cellule. Ils forment avec le réticulum sarcoplasmique des structures appelées diades ou triades. La surface des tubules T peut représenter jusqu’à 20 % de la surface de la membrane qui assurent la propagation du potentiel d’action. Le réticulum sarcoplasmique, un réseau de tubules membranaires interconnectés, contient le calcium fixé à un ensemble de protéines réticulaires comme la calséquestrine, et dont la fonction est de réguler l’activité calcique intracellulaire contribuant au mécanisme du couplage excitation-contraction. Les cavéoles qui correspondent à de petites invaginations membranaires de 50 à 100 nm de diamètre riches en cholestérol, régulent de façon dynamique des fonctions spécialisées à la surface des cellules. Et finalement les disques intercalaires, localisés aux extrémités de chaque cardiomyocyte qui permettent de connecter la cellule à ses voisines. Ce sont les sites de transmissions ioniques intercellulaires.
b) Système contractile
Le système contractile est constitué d’un ensemble de protéines formant les myofibrilles. Elles présentent une succession de bandes transversales claires et sombres donnant au myocyte son aspect strié (Katz AM., 1992). Elles sont constituées par la répétition de sarcomères, unités contractiles du muscle strié. Chaque sarcomère s’étend entre deux stries Z et est formé de bandes sombres A (anisotropes) où sont localisés les filaments épais, et de zones claires I (isotropes) contenant les filaments fins. Les filaments épais sont composés principalement de molécules de myosine (Figure H4). La myosine est un hexamère composé de deux chaînes lourdes (MHC pour ″Myosin Heavy Chain″) et de deux paires de chaînes légères (MLC pour ″Myosin Light Chain″), l’une dite essentielle (ELC ou LC1) et l’autre régulatrice (RLC ou LC2). ). La chaîne lourde (200 kDa) est formée d’une partie filamenteuse, le ″rod″ et d’une partie globulaire, le sous fragment 1, qui porte l’activité enzymatique de la molécule. La MHC cardiaque est codée par deux gènes α et β (Mahdavi V. et al, 1984) dont l’expression varie au cours du développement ontogénique et dans diverses situations pathologiques. Les filaments minces (Figure H5) sont composés d’actine, de tropomyosine (Tm) et du complexe de troponines. La troponine est un complexe formé de 3 sous-unités : La troponine C qui fixe le calcium, la troponine T qui lie le complexe de la troponine au filament d’actine et la troponine I qui inhibe la contraction. Les filaments d’actine sont essentiellement composés de deux hélices chacune d’elles composée de monomères d’actine globulaire G polymérisées en actine filamenteuse F qui constitue le squelette du filament fin. La tropomyosine est formée de deux sous-unités α et β. Le complexe tropomyosine / troponines permet le réglage fin de la contraction (Solaro RJ. et al, 1998).
Figure H4 : La molécule de myosine
c) Cytosquelette
La cellule musculaire striée possède des associations de protéines entre les myofibrilles et avec le sarcolemme qui sont hautement organisées pour former un réseau de connexions appelé cytosquelette. Le cytosquelette du cardiomyocyte (Figure H6) est composé d’un compartiment myofibrillaire et d’un compartiment extramyofibrillaire ou cytoplasmique.
Le compartiment myofibrillaire contient des protéines associées aux sarcomères, qui vont contrôler le mouvement. Ces protéines sont la titine, l’α-actinine et la nébuline (Gregorio CC. et al, 1999). Un certain nombre de protéines associées à la bande Z liées à l’extrémité du filament d’actine, et en relation avec le sarcolemme ont été caractérisées. Il s’agit de la taline, la métavinculine, la MLP (muscle-lim-protein), les intégrines (Chen J. et Chien KR., 1999) et d’un complexe membranaire composé des sarcoglycans et de la dystrophine (Campbell KP., 1995).
Le compartiment extramyofibrillaire ou cytoplasmique composé de filaments intermédiaires, des microtubules et des microfilaments d’actine. Le principal constituant des filaments intermédiaires est la desmine. Ils forment le lien entre deux myofibrilles adjacentes. Ces filaments relient les myofibrilles au sarcolemme. Ils entourent les disques Z et sont très concentrés au niveau des desmosomes des disques intercalaires. Les microtubules sont des assemblages d’hétérodimères de tubulines α et β associés à une collection de protéines, les MAPS (Mitogen-Activated-Proteins) (Wade RH. et al, 1990). Le réseau de microtubules entoure le noyau et les myofibrilles ; il est aussi concentré sous la membrane cytoplasmique, participant ainsi à la rigidité de l’ensemble.
Les microfilaments d’actine permettent la mobilité cellulaire, les changements de forme et le maintien de la viscoélasticité du cytoplasme. La polymérisation, la dépolymérisation et l’assemblage supra-moléculaire sont réglés par des protéines associées.
Les microtubules associés aux filaments d’actine et filaments intermédiaires, établissent et maintiennent l’architecture interne du cytoplasme, et déterminent la forme cellulaire.
Figure H6 : Schéma détaillé du cytosquelette
1.2. Propriétés électrophysiologiques 1.2.1. Potentiel d’action
Chaque cellule se contracte en réponse à une excitation qui se propage le long des fibres sous la forme d’une impulsion électrique ou potentiel d’action. Ce potentiel d’action résulte d’une variation transitoire du potentiel de membrane due à des variations de perméabilités ioniques transmembranaires essentiellement sodique, calcique et potassique. Les premiers enregistrements transmembranaires de l’activité électrique ont été réalisés en 1949 par Coraboeuf et Weidmann sur le tissu conducteur de cœur de chien à l’aide de la technique d’enregistrement par microélectrode.
Le potentiel d’action (PA) du cardiomyocyte ventriculaire se développe ainsi en quatre phases successives (Figure H7) :
- une phase rapide de dépolarisation (phase 0) induite par le courant sodique INa, sensible à la tétrodotoxine,emmène le potentiel de membrane à +30 mV ;
- une courte phase de repolarisation (phase 1) suite à un courant transitoire sortant de nature potassique, sensible à la 4-aminopyridine (4-AP) ;
- une phase plateau prolongée autour de 0 mV (phase 2), dont le calcium en est responsable ;
- une phase de repolarisation (phase 3) qui ramène le potentiel de membrane à la valeur du potentiel diastolique de repos négatif (-80 mV) suite à un courant potassique retardé ;
- une phase stable (la phase 4) qui correspond à la diastole ventriculaire où le potentiel de membrane conserve sa valeur de repos.
Ces courants constituent les principales perméabilités ioniques responsables du décours du PA qui dépend aussi d’autres courants ioniques plus faibles (par exemple les courants chlore), ou résultant de l’activité des mécanismes d’échanges tels que la pompe Na+/K+ ou l’échangeur Na+/Ca2+. Ces courants ioniques transitent à travers la membrane par des structures protéiques bien déterminées.
Figure H7 : Schéma montrant un potentiel d’action ventriculaire avec ses différentes phases (APD50 et APD90 : la durée à 50% et 90% du PA
1.2.2. Courants ioniques transmembranaires
Les courants ioniques impliqués dans la genèse du potentiel d’action sont maintenant assez bien connus tant par leurs propriétés et distributions cellulaires que par leur structure moléculaire et leur codage génétique (Figure H8).
a) Le courant sodique INa
La phase 0 du potentiel d’action de la cellule ventriculaire (dépolarisation rapide de la membrane) est due à l’activation des canaux sodiques voltage-dépendants. L’ouverture des canaux sodiques donne lieu à un courant sodique entrant de courte durée et d’amplitude importante. Il s’active entre -70 et -60 mV, et est maximal pour un potentiel de -30 à -20 mV. Son potentiel d’inversion est de l’ordre de +40 mV en milieu physiologique normal.
b) Le courant transitoire sortant (Ito)
Ce courant est formé d’une composante potassique (Ito1) et d’une composante portée par des ions chlorures activés par le calcium (Ito2). Dans la composante potassium, bloquée par la 4-aminopyridine, on distingue un courant d’inactivation rapide (Ito, fast) et un autre d’inactivation lente, (Ito, slow). La composante chlorure est non voltage-dépendante, de faible amplitude, s’active en présence de calcium très rapidement à -25 mV et s’inactive rapidement. Le courant (Ito) est responsable de la repolarisation précoce du ventricule (phase 1) et contrôle la durée du potentiel d’action.
c) Le courant sortant potassique retardé (IK)
Ce courant sortant est le facteur déterminant de la durée du potentiel d’action et de sa configuration. Le courant IK est formé de trois composantes différentes : l’une rapide IKr, une autre lente IKs et une composante ultra-rapide IKur. La proportion de ces trois courants varie selon la préparation cardiaque utilisée. IKur est présent dans les cellules d’oreillette de rat et humaine alors que IKr est la seule composante présente dans les cardiomyocytes ventriculaires de rat (Pond AL. et al, 2000) et de souris (Zhou J. et al, 1998).
d) Les courants sortants chlorures (ICl)
Les courants chlorures sont dus à un mouvement entrant d’anions, donc de nature repolarisante. Le courant chlorure est principalement sortant et participe au raccourcissement du potentiel d’action. Trois classes différentes de conductances d’anions chlorure (Cl-) ont été distinguées: (1) une conductance chlorure Ca2+- dépendante (ClCa), déclenchée par la stimulation des cellules avec l'endothéline-1 ou un Ca2+-ionophore, (2) une conductance AMPc / protéine kinase A dépendante, activée par l'ajout de forskoline, (3) une classe distincte de canaux Cl- dépendants du volume cellulaire, potentialisée en présence de l'endothéline-1 ou de l'inhibiteur de la phosphatase phosphotyrosine, le pervanadate (Tilly BC. et al., 1996).
Les canaux ClCa sont ligand-dépendants activés par l’augmentation de la concentration du Ca2+ intracellulaire dans divers types cellulaires, y compris les cellules cardiaques. Parce que les canaux ClCa sont normalement fermés au repos, lorsque la concentration de Ca2+ libre intracellulaire est d’environ 100 nmol/L, ils ont généralement été considéré de nature excitables, et déclenchent le mécanisme de transduction du signal pour l’excitabilité membranaire, l’équilibre osmotique, les mouvements transépithéliaux de chlorure, ou la sécrétion de liquide (Leblanc N. et al., 2005). Au cours de l’hypertrophie, le courant chlorure calcium-dépendant (Ito2) semble contribuer aux altérations du potentiel d’action suite à une augmentation significative de sa densité (Guo D., 2008).
e) Le courant calcique entrant
L’ion calcium est le signal de transduction le plus commun des cellules vivantes allant des bactéries aux neurones spécialisées. Dans la cellule, le calcium joue le rôle de second messager indispensable dans la plupart des processus physiologiques : contraction musculaire, croissance et différentiation cellulaire, sécrétion, transmission nerveuse. Il intervient donc dans la régulation de nombreuses protéines. Ainsi toute altération de l’homéostasie calcique en particulier l’élévation durable de la concentration de Ca2+ intracellulaire [Ca2+]i va engendrer des situations pathologiques pouvant aller jusqu'à la mort cellulaire. Cette homéostasie est maintenue par l’influx d’ions calcium dans la cellule par des canaux voltage dépendants.
Les canaux calciques transmembranaires voltage dépendants, jouent un rôle central dans le développement et le contrôle de la contractilité cardiaque qui est modulée par la concentration des ions calcium (Ca2+) libres cytosoliques.
Figure H8 : Bases ioniques du potentiel d’action cardiaque « global » reprenant différentes caractéristiques de différents types cellulaires
D’après Snyders, 1999.
Le décours du PA est caractéristique du PA de cellules ventriculaires mis à part les courants ICaT et If qui ne
sont rencontrés que dans les cellules cardiaques à activité autorythmique (cellules nodales et cellules du système de conduction). Le sens entrant ou sortant des courants ioniques est figuré sous le PA. La localisation des différentes composantes ioniques intervenant dans les PA cardiaques est indiquée dans
Les canaux calciques sont fermés au potentiel de repos membranaire normal des cellules cardiaques. Pendant le potentiel d’action, ils sont en état ouvert suite à la dépolarisation membranaire et transforment alors le signal électrique en signal chimique. En plus de leur contribution au plateau du potentiel d’action (PA), l’influx de calcium Ca2+ à travers les canaux calciques de type L induit une libération d’ions calcium du réticulum sarcoplasmique (RS) ce qui initie la contraction. Grâce à leur rôle central dans le phénomène de couplage excitation contraction, les canaux calciques de type L régulent l’inotropie. Le rôle des canaux calciques de type T est plus obscur. Ils sont impliqués d’un côté dans l’activité rythmique du coeur, de même que dans les stades précoces du développement et dans les pathologies des tissus contractiles (Richard S et al., 1997).
Contrairement aux neurones qui expriment six types de canaux calciques, les types L (Long Lasting), N (Neuronal), P (Purkinje), Q (Q après P), R (R après Q) et T (Transient), les cellules cardiaques expriment deux types, les canaux de type T et les canaux de type L. Ces deux types de canaux calciques sont retrouvés sur la plupart des cellules excitables où ils sont coexprimés.
En résumé, on peut distinguer deux principales catégories de canaux calciques en fonction de leur seuil d’activation. Les canaux à bas seuil d’activation (LVA, « low voltage activated ») qui s’activent pour de faibles dépolarisations et s’inactivent rapidement d’une manière voltage dépendante, il s’agit du type T ; les canaux à haut seuil d’activation (HVA, « high volatage activated ») qui s’activent par des dépolarisations plus élevées et engendrent des courants qui s’inactivent lentement, il s’agit des types L, N, P, Q, et R.
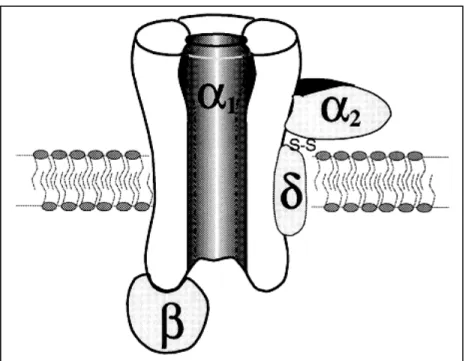
e-1) Les canaux calciques de type L (ICaL )
(i) Structure, distribution et rôle de ICaLdans le cœur
Le canal calcique de type L est une protéine multimérique. Une sous unité α1 organisée en 4 domaines répétés forme le pore. Elle est associée à une sous unité transmembranaire α2-δ et une sous unité β cytoplasmique (Figure H9). La sous unité α1 est codée par le gène de la classe C (sous unité α1c) (Richard S. et al., 1998). La diversité est due à la variété de combinaisons avec différentes sous unités β (4 gènes codent pour la sous unité β ; β1, β2, β3 et β4). Toutes les sous unités β augmentent le niveau
d’expression de α1c dirigeant Ica dans les systèmes d’expression. La sous unité α1c en elle seule induit seulement des courants calcium faibles, mais quand elle est co-exprimée avec les sous unités β, le courant est augmenté, les cinétiques d’activation et d’inactivation sont accélérées (Nargeot J. et al., 1997). C’est pourquoi la sous unité β est considérée comme un vrai modulateur endogène qui influence l’activité électrique de même que les propriétés pharmacologiques des canaux calciques. Les études au niveau ARNm suggèrent que les canaux calciques contiennent les sous unités α1c et β2a.
Contrairement à ICaT, ICaL a été trouvé dans toutes les cellules cardiaques étudiées jusqu’à présent : sinus, nœud sinusal, fibres de Purkinje, oreillete et ventricule. Soit il coexiste avec le courant de type T soit il est exprimé seul comme dans le cas des myocytes auriculaires de lapin, des myocytes auriculaires et ventriculaires de veau (Bean B.P., 1989), des myocytes auriculaires humains (Ouadid H. et al., 1991) et des myocytes ventriculaires de grenouille (Argibay JA. et al., 1988) et de rat adulte (Hering S. et al., 1989 ;Richard S. et al., 1990).
Dans le cœur, ICaL constitue la voie d’entrée principale du calcium lors d’une dépolarisation membranaire. L’influx de calcium par ce canal déclenche la libération de calcium des sites de stockage intracellulaire, le réticulum sarcoplasmique et joue également un rôle dans le couplage excitation contraction (calcium induced calcium release, CICR) (Fabiato A., 1989).
(ii) Caractéristiques électrophysiologiques et modulation pharmacologique de ICaL
ICaL s’écoule par des canaux voltages dépendants. Ces canaux s’ouvrent en réponse à la dépolarisation membranaire. Le seuil d’activation d’ICaL est plus élevé que celui d’ICaT, il est de -40 mV et son intensité maximale est atteinte pour des potentiels voisins de 0 mV et son potentiel d’inversion pour des potentiels de ≥ +50 mV (Figure H10). La voltage dépendance se produit par une gamme de potentiel d’environ 50mV, entre -40 et +10mV. Celle de son inactivation se produit entre -50 et +10 mV. L’activation de ce courant est très rapide, de 2 à 4 ms pour une dépolarisation à 0 mV (Isenberg G. et Klockner U., 1980, 1982). Sa cinétique d’inactivation est décrite selon un processus mono exponentiel (Cohen NM. et Lederer WJ., 1988; sur le rat nouveau-né)
ou bi exponentiel (Isenberg G. et Klocker U., 1982). L’inactivation de ICaL est principalement calcium dépendante pour un courant calcique induit par une dépolarisation modérée alors que pour un courant induit par une forte dépolarisation l’inactivation est gouvernée principalement par le voltage. Ce canal peut transporter d’autres ions divalents tels que SR2+ et Ba2+ (Hess P. et al., 1986) et est aussi perméable aux cations monovalents tels le Li+, Na+, K+, Cs+.
Une grande variété de molécules chimiques module les canaux calciques de type L. Des inhibiteurs inorganiques tels le La3+, le Cd2+, le Ni+, Co+, ou Mn2+ et Mg2+ bloquent le canal sans variation de l’amplitude du courant unitaire (Lansman JB. et al., 1986), des inhibiteurs organiques qui ont des effets sur la contraction cardiaque, le potentiel d’action et le courant calcique et qui agissent sur un site défini au niveau du canal, qui sont les dihidropyridines (DHPs) dont la première substance connue est la nifédipine, les phénylalkylamines dont le vérapamil et les benzothiazépines dont principalement le diltiazem. Certaines dihydropyridines peuvent avoir un effet agoniste et augmenter ICaL, tel la nitrendipine et le BayK 8644.
Des études biochimiques ont démontré que plusieurs kinases, inclus la PKA, PKC, PKG et la calmoduline kinase II peuvent phosphoryler les canaux calciques cardiaques. La meilleure régulation de ICaL dans les cellules cardiaques est la régulation par la stimulation du récepteur β-adrénergique qui active la cascade AMPc intracellulaire résultant en une augmentation de la probabilité d’ouverture des canaux calciques.
e-2) Les canaux calciques de type T ou ICaT
(i) Structure, distribution et rôle de ICaTdans le cœur
Peu de données récentes décrivent la structure moléculaire des canaux calciques de type T. Les résultats expérimentaux suggèrent que les canux calciques de type T constituent une famille hétérogène de canaux calciques qui pourraient être constitués de plusieurs isoformes structurales et fonctionnelles (Chen CF. et Hess P., 1990 ; Soong TW. et al, 1993). Les canaux calciques de type T ont été trouvés dans une grande variété de cellules excitables et non excitables; leur présence dans le coeur est bien établie.
Figure H9 : Représentation schématique de la structure en 4 sous unités du canal calcique de type L
D’après Richard S., 1998.
Par exemple dans les cellules sinusales de grenouille ( Bois P. et Lenfant J., 1991) ou de lapin (Hagiwara et coll, 1988), dans les cellules auriculaires de grenouille (Bonvallet R., 1987), de chien (Bean BP. , 1985) ou de rat (Xu XP. et Best PM., 1990), dans les cellules de Purkinje de chien (Hirano Y. et al, 1989) et dans les cellules ventriculaires de chien (Tseng GN.et Boyden PA.,1989) et de cobaye (Nilius B. et al, 1985 ; Mitra R. et Morad M., 1986). ICaT a été premièrement enregistré dans des cellules non pathologiques de ventricule de porc et récemment dans des myocytes de différentes espèces et tissus. L’ICaT est rare dans les cellules ventriculaires de mammifères, y compris les rats et lapins, sauf peut être chez le cobaye (Richard S. et al., 1998).
Dans les conditions normales, ICaT ne joue pas un rôle important dans le développement de l’activité électrique des cellules auriculaires et ventriculaires (Bean BP., 1985; Bonvallet R. et Rougier O., 1989) alors que dans les tissus partiellement dépolarisés ICaT facilite le déclenchement d’une activité spontanée et pourrait être impliqué dans
différents types d’arythmies, tels que les post-dépolarisations précoces et retardées. L’influx de calcium via les canaux de type T cardiaques semble insuffisant pour induire la libération de calcium du réticulum sarcoplasmique alors que dans certaines conditions pathologiques, telles que l’ischémie et l’hypoxie, ICaT pourrait participer à l’accumulation du calcium a l’intérieur de la cellule (Vassort G. et Alvarez J.,1994).
(ii) Caractéristiques électrophysiologiques et modulation pharmacologique de ICaT
Le courant calcique de type T pour « transient » ou « tiny » a un bas seuil d’activation et est rapide. Ses principales propriétés sont : un seuil d’activation entre -70 et -60 mV, un pic autour de -30 mV et un potentiel d’inversion entre +10 et +60 mV. (Vassort G. et Alvarez J., 1994) (Figure H10). Sa cinétique d’activation est rapide puisque son pic est atteint en moins de 20 ms. Son inactivation est mono exponentielle, avec une constante d’inactivation, de l’ordre de 5 à 50 ms quel que soit l’ion qui transite ; elle est dépendante du voltage et indépendante de l’influx de calcium. Sa gamme d’activation, négative suggère son implication dans la dépolarisation diastolique des cellules à activité pacemaker.
Le courant peut transporter d’autres ions divalents que le calcium, tel le Ba2+ ou Sr2+ (Hagiwara N. et al, 1988 ; Bois P. et Lenfant J., 1991). Sa conductance élémentaire est faible, de l’ordre de 5 à 10 pS en milieu 110 mM Ba2+. Son temps d’ouverture est court.
Les canaux calciques se caractérisent par leur faible sensibilité aux agents pharmacologiques et aux neurotransmetteurs. Il n’existe pas de ligands spécifiques pour inhiber ICaT, les seuls agents dont on a pu montrer un effet sont les dihydropyridines (DHPs, le Benidipine hydrochlorure, nouveau dérivé DHPs inhibiteur des canaux calciques L, N et T (Alvarez JL. et Vassort G., 1992), les agents anti-arythmiques et les neurotransmetteurs (Vassort G. et Alvarez J., 1994). D’autres composés pharmacologiques tel les alcools à haut poids moleculaires (octanol, menthol), le neuroleptique chloropromazine, l’amiloride, l’acide retinoique le U-92032, la tétrametrine, peuvent bloquer le courant calcique de type T (Hagiwara N. et al., 1988).
Parmi les substances inorganiques, l’efficacité inhibitrice des ions est la suivante : La3+>Ni2+>Cd2+≥Co2+.
Les canaux calciques cardiaques de type T sont insensibles à la stimulation β-adrénergique (Tytgat J. et al., 1988). Cependant, il a été montré dans les cellules ventriculaires de cobaye, que l’isoprénaline augmente ICaT par un mécanisme de couplage direct avec les récepteurs β2-adrenergiques (Mitra R. et Morad M., 1986). Un effet agoniste a été mis en évidence, via la protéine kinase C (PKC), avec des agonistes α1 -adrénergiques tels que l’angiotensine II, l’endothéline et l’ATP (Vassort G. et Alvarez J., 1994).
Figure H10 : Enregistrements de deux types de courants ICaT et ICaL par la technique de patch clamp en configuration cellule entiere dans un myocyte ventriculaire de cobaye (Les courants sont induits par des dépolarisation à 0mV (A) et à -40 mV (B) à
partir de potentiel de maintien (HP) de -90 mV et -50 mV ; (C) Courbes courant-potentiel pour des courants induits à partir d’un HP de -90 mV (ronds noirs) où les deux types de courant s’activent, à partir d’un HP de -50 mV (ronds blancs) où le seul courant de type L est présent. La courbe courant-potentiel du
courant calcique de type T (triangles noirs) s’obtient par soustraction des deux précédentes) D’après Balke CW., 1992.
2. Le cardiomyocyte ventriculaire hypertrophié
L’hypertrophie cardiaque est définie par une augmentation de la masse totale du coeur, relative à la surface corporelle. Au niveau histologique, elle est caractérisée par une augmentation de la taille des myocytes, sans prolifération (sans hyperplasie). Cependant, certaines autres cellules présentes dans le myocarde, en particulier les cellules endothéliales des vaisseaux coronariens et les fibroblastes des espaces interstitiels, augmentent en taille et prolifèrent, avec pour conséquence une production excessive de matrice extracellulaire et de collagène, entraînant des lésions irréversibles de fibrose myocardique (Choukroun G. et al., 2002).
La réponse hypertrophique de la cellule se manifeste tout d’abord par un processus adaptatif puis un processus pathologique.
2.1. L’hypertrophie cardiaque 2.1.1. Processus adaptatif
La mise en jeu d’un processus adaptatif est d’origine mécanique et fait l’objet d’une cascade d’évènements. L’étirement de la cellule est un facteur universel d’activation de la transcription mais l’activité mécanique joue un rôle supplémentaire aussi bien dans l’activation de la synthèse protéique que dans celle d’ARN messagers spécifiques.
a) Les récepteurs à l’étirement
Il existe très probablement des récepteurs à l’étirement. L’étirement active des mécanismes autocrines/paracrines locaux tel que la production d’angiotensine II ainsi que la synthèse et la production d’endothéline. Les facteurs humoraux (angiotensine II et endothéline) vont se lier sur les récepteurs couplés aux protéines G et activer la phospholipase C (PLC) qui va générer le 1,4,5-inositol-trisphosphate (IP3) et le diacylglycérol (DAG) suivi d’une libération de calcium du réticulum sarcoplasmique (RS). L’expression des protéines induites par la libération de Ca2+ peut s’opérer à travers différentes voies Ca2+- dépendantes. La calcineurine, une protéine Ca2+ et calmoduline dépendante, est impliquée dans la voie de signalisation calcique (Molkentin J.D. et al.,
1998 ; Vega R.B. et al., 2003). Elle va phosphoryler le substrat NFAT (Nuclear Factor of Activated T cells) qui sera transloqué du cytoplasme au noyau où il va engager une multitude de facteurs de transcriptions qui vont activer la réponse génique. Plusieurs données suggèrent que la libération du Ca2+ des stocks du RS résulte en un influx de Ca2+ extracellulaire dans le cytosol ce qui va augmenter le calcium intracellulaire.
Une relation entre les stocks Ca2+, l’activation de l’influx de Ca2+ et la modification génétique via la voie calcineurine/NFAT a été démontrée dans les myocytes (Rosenberg P., 2004). Cette entrée calcique provient de différents canaux perméables au Ca2+ dont principalement le canal calcique de type L (Figure H11) ainsi que d’autres canaux perméables au Ca2+ tels les canaux SOCs (store operated Ca2+-entry channels), les SACs (stretch activated channels) et probablement d’autres canaux cationiques non séléctifs, les canaux TRPs (Transient Receptor Potential) qui eux aussi semblent également impliqués dans le développement de l’hypertrophie cardiaque (TRPC1, TRPC3, TRPC6) (Guinamard R et Bois P., 2007).
Figure H11 : Schéma montrant la regulation de l’expression génique suite à l’entrée de calcium à travers les canaux calciques de type L
b) Les voies de transduction
La cascade utilisée par la transduction mécanogénique détermine tout un jeu de phosphorylations/dephosphorylations dominé par la MAP kinase (Figure H12). La régulation est à la fois transcriptionelle et post-transcriptionelle, pouvant utiliser le cycle phosphoinositol et l’AMP cyclique. De nombreuses expériences transgéniques démontrent le rôle de la filière MAP kinase : la surexpression de ras, de raf, de la MAPKK3/6 et de p38β entraînent une hypertrophie ; la coéxpression de MKK7 et MKK6 inhibe l’hypertrophie. La MEKK inhibe l’hypertrophie induite par la phényléphrine.
La surcharge mécanique entraîne rapidement l’apparition d’un certain nombre de changements transitoires dans l’expression génétique. Plusieurs proteines apparaissent, certaines oncoproteines (c-myc, c-fos), des proteines de stress (HSP 90), et plusieurs facteurs de croissance. La plupart de ces marqueurs transitoires sont des MAPK (par exemple JNK) ou dépendant de l’activité des MAPK (c-jun).
2.1.2. Processus pathologique
Dans les diverses formes d’insuffisance cardiaque, un travail excessif conduit à un élargissement du cœur de façon à pouvoir gérer l'augmentation de la demande hémodynamique. Ce processus, connu sous le nom d'hypertrophie, est classé comme "physiologique" (lorsque l'hypertrophie se produit chez des individus sains après un exercice physique ou une grossesse et qui n'est pas liée à des dommages cardiaques), ou bien "pathologique" (Figure H13). Dans le cas d’hypertrophie pathologique, bien que l'augmentation de la taille du coeur est d'abord un mécanisme de compensation soutenu, l’hypertrophie peut conduire à une diminution de la fonction ventriculaire gauche et représente alors un facteur de risque pour l'insuffisance cardiaque (Levy D et al., 1990).
Figure H12 : La cascade d’évènements à l’origine du processus adaptatif D’après Swynghedauw, 2002.
Les principales causes de l'hypertrophie pathologique sont l'hypertension, les polymorphismes génétiques, et la perte de myocytes suite à l’ischémie. L’altération du métabolisme cardiaque peut aussi être un élément important conduisant à l'hypertrophie (Rajabi M. et al., 2007).
Figure H13 : L’hypertrophie pathologique et physiologique D’après Barry, 2008.
La croissance de l’hypertrophie s’opère de deux façons : L’hypertrophie concentrique, induite par la surcharge chronique de pression et conduit à une réduction du volume ventriculaire gauche et une augmentation de l’épaisseur de la paroi ventriculaire ; alors que l'hypertrophie excentrique est induite par une surcharge de volume et entraîne une dilatation et un épaississement de la paroi du myocarde (Wakatsuki T. et al., 2004). Mécaniquement, l'expansion excentrique se produit par l'ajout de sarcomères en série provoquant une élongation des cellules; d'autre part l’expansion concentrique est causée par l'ajout de sarcomères en parallèle, entraînant une augmentation de l'épaisseur des cellules. L'augmentation de la taille des myocytes est accompagnée par une augmentation du nombre de fibroblastes cardiaques ce qui va provoquer une fibrose et une augmentation de la rigidité du myocarde (Wakatsuki T. et al., 2004). Cela en retour va conduire à une surcharge et favoriser alors l'hypertrophie et l’apoptose cellulaire, résultant en un cycle cardiaque de croissance et de perte myocytaire.
Une surcharge de pression ou de volume conduit à une hypertrophie concentrique et excentrique via l’activation de voies de signalisation intracellulaires spécifiques, qui vont altérer la forme du myocyte de manière à s’adapter le mieux à la surcharge.
2.2. Mécanismes moléculaires
Diverses voies moléculaires sont responsables du contrôle du programme hypertrophique. Suite à l'augmentation du stress mécanique ou d’une surcharge de pression, des changements moléculaires vont réactiver le programme génétique fœtal qui va permettre de coordiner la synthèse des protéines nécessaires pour apporter l’augmentation de la taille des myocytes cardiaques et l’ajustement à la modification des demandes énergétiques de ces grandes cellules.
La réinduction du programme génétique fœtal est associée à l’hypertrophie excentrique et concentrique qui provoque la réexpression des gènes foetaux, ce qui n’est pas le cas dans l'hypertrophie physiologique. Bien qu'il existe une réexpression d’une multitude de gènes fœtaux au cours de l'hypertrophie, les marqueurs moléculaires les plus communément utilisés sont les taux d’expression de la chaîne lourde de myosine et les peptides natriurétiques.
2.2.1. La chaîne lourde de myosine (Myosin Heavy Chain, MHC)
Une des caractéristiques de l'hypertrophie cardiaque chez les patients et les rongeurs, est l’augmentation de l’expression de la β-MHC et la diminution de l’expression de l’α-MHC (Izumo S. et al., 1987; Mahdavi V. et al., 1984). Leur rapport influence la fonction cardiaque. Une augmentation de la β-MHC diminue l’activité de l'enzyme myosine ATPase ce qui diminue le taux de contraction myocytaire, d’où une adaptation à la surcharge de travail (Lowes BD. et al., 1997).
En cas de disparition de l'hypertrophie on assiste à un retour du taux de l’isoforme MHC à la normale. Chez les patients atteints de cardiomyopathie dilatée et qui subissent une thérapie par des β-bloqueurs, leur récupération est associée à l’augmentation de l’expression de l’α-MHC et à la diminution de celle du β-MHC (Lowes BD. et al., 2002). Dans les cardiomyopathies dilatées, 90% de la quantité totale de MHC est représentée par l'isoforme β-MHC chez l'homme adulte, alors que chez les rongeurs adultes l’α-MHC est la forme dominante (LeWinter MM., 2005). Par conséquent, alors que l’expression de la β-MHC est augmentée chez l'homme dans le cas d'insuffisance cardiaque, elle n'est pas aussi prononcée que l'augmentation perçue chez les rongeurs.
Malgré le lien génétique entre les protéines MHC et l'hypertrophie, des travaux récents ont suggéré que l'augmentation de l’expression de la β-MHC dans une cellule individuelle n’est pas nécessairement associée à une augmentation de la taille du coeur (Pandya K. et al., 2006).
2.2.2. Les peptides natriurétiques
Les peptides natriurétiques (NPs) sont une famille d’hormones qui agissent sur le système cardiovasculaire et endocrinien par l'intermédiaire de leurs actions diurétiques, natriurétiques, vasorelaxatrices et inhibitrices d’aldostérone et de rénine (Nishikimi T. et al., 2006). Les NPs sont des inhibiteurs endogènes de l'hypertrophie. Il existe trois types: le Peptide Natriurétique de type A (ANP), également appelé facteur natriurétique auriculaire (ANF), le Peptide Natriurétique de type B (BNP) et le Peptide Natriurétique de type C (CNP).
Dans le cœur, l’expression de l’ANP est limitée aux oreillettes, alors que le BNP est exprimé dans les oreillettes et les ventricules mais la mesure de l’expression du CNP
dans le cœur n'est pas encore bien élucidée. Après leur sécrétion, les NPs sont clivés pour libérer les dérivés peptidiques biologiquement actifs et exercer leurs effets en se liant aux récepteurs peptidiques natriurétiques (NPRs), également connus sous le nom de récepteurs à guanylyl cyclase.
L’ANP et le BNP se trouvent à des taux élevés au cours du développement embryonnaire et chez les nouveaux nés, mais sont absents chez les adultes sains (Gardner DG., 2003). Le stimulus hypertrophique augmente de manière spectaculaire l'expression de l’ANP et du BNP par le facteur de transcription GATA-4 (Gardner DG. et al., 2007; Richards AM., 2007).
La suppression de l'ANP chez les souris provoque l'hypertension et l'hypertrophie malgré les conditions de repos (Mori T. et al., 2004;Wang D. et al., 2003).
L’effet anti-hypertrophique de l’ANP est indépendant de l'augmentation de la pression artérielle (Feng JA. et al., 2003). Les souris déficientes en ANP ont une plus grande expression des protéines de la matrice extracellulaire (ECM) suite à l’hypertrophie, ce qui suggère que l’ANP peut réduire le remodelage hypertrophique en inhibant le dépôt de l’ECM (Wang D. et al., 2003). Alors que la suppression du BNP cause une augmentation de la fibrose interstitielle dans les ventricules en réponse à la surcharge ventriculaire (Tamura N. et al., 2000).
Un autre argument en faveur de l'activité anti-hypertrophique de l’ANP et du BNP provient des études faites sur des souris déficientes en NPR-A. Le NPR-A est le seul récepteur de l'ANP et du BNP, et sa délétion supprime leur fonctionnalité dans le système cardiovasculaire (Lopez MJ. et al., 1997). Les souris NPR-A-/ - souffrent d'hypertension, de fibrose interstitielle, d’hypertrophie ventriculaire et de symptômes similaires à l’insuffisance cardiaque (Kishimoto I et al., 2001; Knowles JW. et al., 2001 ;Oliver PM. et al., 1997). La suppression spécifique du NPR-A dans le cœur montre que les peptides natriurétiques ont un effet direct sur le myocarde (Holtwick et al., 2003).
Contrairement à l’ANP et au BNP, le rôle du CNP dans l'hypertrophie est moins connu. Le CNP est principalement produit dans les cellules vasculaires et endothéliales où il joue un rôle important dans la régulation du tonus vasculaire, du débit sanguin et de la pression (Ahluwalia A et al., 2005). Il a également été démontré que le CNP
et le BNP (Rosenkranz AC. et al., 2003; Tokudome T. et al., 2004). En outre, l’administration de CNP inhibe in vivo la fibrose et l'hypertrophie suite à la ligature de l’artère coronaire (Soeki T. et al., 2005). Ce résultat a été confirmé chez les souris transgéniques surexprimant le CNP et dans des rats transgéniques surexprimant le NPR-B, seul récepteur au CNP (Langenickel TH. et al., 2006;Wang D. et al., 2007).
2.3. Modifications d’expression des courants ioniques
Des altérations des courants ioniques cardiaques sont associés au processus d'hypertrophie. Un changement caractéristique dès les premiers stades de l’hypertrophie est la prolongation du potentiel d’action (PA). Cette prolongation est liée à une surexpression du ICa et une sous expression du Ito. Une fois l’hypertrophie cellulaire est établie, seul la réduction de Ito persiste alors que les valeurs de ICa retrouvent leur niveau contrôle. La diminution du Ito repolarisant résulte en la prolongation du PA. Le ralentissement du taux de repolarisation augmente l’activation du ICa, ce qui va moduler la durée du PA (Sah R. et al., 2002). Cette hypothèse n’exclut pas le fait que des changements au sein d’autres courants (augmentation du INa et IK) (Li X. et Jiang W., 2000) ou l’augmentation d’expression d’autres courants tel l’ICaT (Romain G. et Bois P., 2007) peuvent aussi être impliqués dans la prolongation du PA. Ces changements dans l’ICa et Ito sont controversés. Il est généralement rapporté une diminution dans Ito alors qu’une augmentation, une diminution ou pas de changement dans ICa ont été décrits (Armoundas A. et al., 2001 ; Benitah JP. et al., 2002, 2003). Ces divergences peuvent refléter différentes étiologies, espèces, conditions expérimentales et plus particulièrement différents stades d’hypertrophie.
3. Le peptide natriurétique de type B
3.1. Mise en évidence
Dès les premiers jours de la microscopie électronique, il a été décrit des “granules atriales spécifiques” dans l’atrium cardiaque, possédant plusieurs dispositifs morphologiques des granules sécrétrices dans les cellules endocrines (Baxter GF., 2004).La fonction physiologique de ces granules et la nature de leur produit de sécrétion sont restés mystère jusqu’à sa révélation par le travail d’Adolfo de Bold et ses collègues travaillant dans Ontario vers les années 1970. En 1979, De Bold décrit comment les granules atriales étaient altérées par le système hémodynamique et la balance électrolytique. L’année suivante De Bold et ses collègues décrivent comment les extraits de tissu atrial favorisent une diurèse et natriurèse intense chez les rats; cette observation démontre l’activité endocrine de l’atrium caradiaque et décrit l’existence d’un “facteur atrial natriurétique”. En 1982, de Bold confirme la relation entre la granulité atriale et le facteur natriurétique. La structure de l’hormone polypeptidique qu’on connaît sous le nom de peptide natriurétique atrial (ANP) a été confirmée par Flynn et al. en 1983.
En 1988, Sudoh et al isolent du cerveau de porc un peptide avec une activité biologique similaire à celle de l’ANP. Le peptide a été nommé peptide natriurétique de type B (BNP) malgré le fait que les ventricules cardiaques ont été trouvés comme la source majeure de BNP circulatoire. En 1990, Sudoh et al. isolent du cerveau de porc un autre peptide structuralement similaire au BNP, qu’ils nomment peptide natriurétique de type C, CNP.
Ces trois peptides, l’ANP, BNP et CNP, sont codés par des gènes différents, mais leur forme mature possède un caractère commun qui est la structure en anneau de 17 acides aminés (Figure H14).
D’autres membres de la famille des peptides natriurétiques méritent d’être mentionnés. Le peptide natriurétique de type D, le DNP, isolé pour la première fois du venin du Mamba vert (Dendroaspis angusticeps; Schweitz H. et al., 1992). L’urodilatin qui est un peptide de 32 acides aminés produit par le clivage alternatif du proANP dans la partie distale des tubules rénaux et qui peut être considéré comme un dérivé rénal du peptide natriurétique (Schulz-Knappe P. et al., 1988). Le guanylin et l’uroguanylin qui
sont exprimés dans l'épithélium intestinal où ils sont impliqués dans l'absorption d’eau dans l'intestin, et sont également présents dans la circulation (Beltowski J., 2001).
Figure H14 : Schéma représentant la forme mature des peptides natriurétiques partageant l’anneau de 17 acides aminés.
D’après D’Souza, 2004.
3.2. Peptide natriurétique de type B, BNP
Le BNP constitue le second membre de la famille d’hormones polypéptidiques. Chez l’homme, le BNP est majoritairement produit par les myocytes ventriculaires, relargué dans la circulation de façon permanente, et semble être une véritable hormone de stress cardiaque (Valli N. et al., 2000).
3.2.1 Biochimie du BNP
Le gène Nppb code pour le BNP. Chez l’homme, ce gène est situé sur la partie distale du bras court du chromosome 1, alors que chez le rat il est situé sur le chromosome 5. La structure du gène codant pour le BNP est formé de 3 exons séparés par
2 introns (McGrath MF et al., 2005;Sudoh et al.,1988). La séquence de signalisation est située au niveau de l'exon 1, alors que la majorité des séquences codantes pour la protéine, qui contiennent aussi la partie bioactive de la molécule, se trouvent au niveau de l'exon 2.
3.2.2 Biosynthèse du BNP
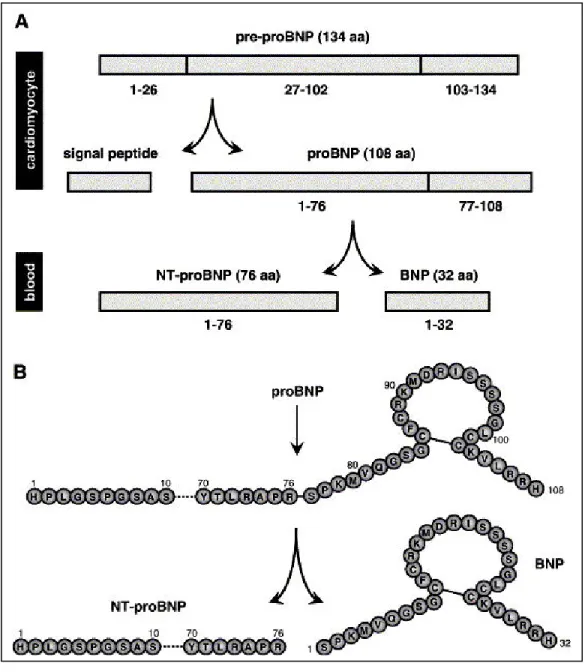
Chez l’homme, le BNP est synthétisé comme un prépropeptide de 134 acides aminés, qui est clivé par une endopeptidase en propetide de 108 acides aminés (le proBNP). Le propeptide est à son tour clivé en fragment carboxy-terminal de 32 acides aminés, biologiquement actif, et en un fragment N-terminal de 76 acides aminés (NT-proBNP) (Figure H15).
A la différence de l’ANP qui est stocké sous forme de propeptide, la forme la plus abondante de BNP dans le myocarde humain est le peptide mature de 32 acides aminés. Dans les cardiomyocytes atriaux, ce peptide est stocké dans des granules atriales spécifiques (McGrath MF et al., 2005).
3.2.3. Expression et sécrétion du BNP
L’expression du gène du BNP est trouvée en grande partie dans les oreillettes cardiaques (1-3 % du total des ARNm). D'autres sites, tels que les ventricules et l’arc aortique, ainsi que les tissus extracardiaques, expriment également le BNP, mais à des niveaux beaucoup plus bas que dans les oreillettes (Ogawa T. et al., 1996 ; Ruskoaho H., 2003).
Le BNP mature est secrété par les oreillettes et les ventricules mais le rapport de préproBNP ventriculaire est plus élevé que celui atriale. Le ventricule cardiaque reste la source principale de sécrétion de BNP. Il est relargué en permanence à des concentrations de picoMolaires dans le sang des sujets sains.
Figure H15 : La biosynthèse du BNP et NT-proBNP D’après Jarolim, 2006.
3.2.4. BNP dans la circulation
La demi-vie plasmatique du BNP est de 22 minutes, alors que la demi-vie du NT-proBNP, plus stable, est de 120 minutes. Le niveau normal de BNP et de NT-BNP varie en fonction de l’âge et du sexe. Pour le BNP, la concentration varie de quelques pg/ml chez les jeunes hommes, à plus de 100 pg/ml chez les femmes saines de plus de 70 ans. Le NT-proBNP est principalement éliminé par clairance rénale, alors que le BNP est en majorité éliminé suite à sa liaison aux récepteurs peptidiques; Cependant dans l’insuffisance rénale on assiste une augmentation des niveaux sanguins de BNP et de NT-proBNP ( Jarolim P., 2006).
3.2.5. Facteurs influençant le taux de sécrétion du BNP
Chez l'homme, les taux plasmatiques de BNP sont significativement corrélés avec l'âge et le sexe (Cowie M.R. et al., 2003). Chez les sujets ne souffrant pas de pathologies cardiovasculaire, rénale ou pulmonaire connus, les niveaux de BNP sont plus élevés chez les femmes que chez les hommes, et ces niveaux augmentent avec l'âge au sein de chaque sexe (Redfield M.M. et al., 2002).
Des stimuli mécaniques peuvent entraîner l’augmentation de la quantité de BNP libérée avec ou sans augmentation de sa synthèse. En effet ce peptide est sécrété en réponse à une surcharge de pression ou de volume observée dans l’hypertrophie ventriculaire et dans l’hypertension artérielle pulmonaire (Scardovi AB., 2004).
En plus des stimuli mécaniques, des agents neuroendocriniens peuvent agir sur les cardiomyocytes pour moduler la synthèse et la sécrétion du BNP. Il s'agit notamment de l'endothéline-1 (ET-1) (Uusimaa P.A et al, 1992), l’hydroxyvitamine D3 (Li Q. et al., 1994), les glucocorticoïdes, les hormones thyroïdiennes, les facteurs de croissance, la thrombine, l'angiotensine II (Ang II), les prostaglandines et les agonistes α1 -adrénergiques (de Bold A.J. et al., 2001) . Certaines de ces substances contribuent activement à la modulation de la synthèse et la sécrétion du BNP dans les conditions normales et physiopathologiques (Bianciotti L.G. et al., 2001 ; Bianciotti L.G. et al., 2002).
3.3. Récepteurs peptidiques natriurétiques (NPRs)
Les effets biologiques divers des peptides natriurétiques sont médiés par leur liaison à des récepteurs membranaires à domaine guanylate cyclase (GCs), connus sous le nom de récepteurs des peptides natriurétiques (NPRs) (Baxter GF., 2004).
Il existe deux sous type de récepteurs, le NPR-A connu aussi comme GC-A et le NPR-B connu comme GC-B, qui sont majoritairement responsable des effets physiologiques des peptides via la guanylate cyclase (GC) qui va médier la conversion du guanosine triphosphate (GTP) en guanosine monophosphate (GMPc).
Sur le NPR-A se lie l’ANP et le BNP alors que sur le NPR-B se lie sélectivement le CNP. Un troisième récepteur, le NPR-C, sur lequel se lient tous les peptides, joue un rôle primaire de récepteur de clairance et sert d’internalisation cellulaire et de dégradation. Le domaine extracellulaire partage une homologie de séquences des acides aminés avec le GC-A et le GC-B alors que ce récepteur n’a que 37 acides aminés intracellulaires et ne produit pas de GMPc(Kuhn M., 2004)(Figure H16).
3.3.1. Récepteur peptidique natriurétique de type A, NPR-A
L’ARNm du NPR-A humain et celui de rat sont fortement exprimés dans le rein, le cerveau, les poumons, le tissu adipeux, et le coeur. Ce récepteur à guanylyl cyclase est composé de 3 domaines : un domaine de liaison extracellulaire qui se lie aux peptides natriurétiques d'environ 450 acides aminés, une région transmembranaire hydrophobe de 20 à 25 résidus unique et un domaine intracellulaire d'environ 570 acides aminés (Figure H16). Ce dernier est composé d'un domaine d’homologie kinases de 250 acides aminés, d’un domaine de dimérisation de 40 résidus enroulés en bobine et d’un domaine catalytique carboxyl terminal guanylyl cyclase d’approximativement 250 acides aminés. L’affinité relative des peptides natriurétiques pour le NPR-A est dans l’ordre suivant : ANP ≥ BNP ≥ CNP.
Figure H16 : Les récepteurs peptidiques NPR-A, B et C D’après Potter, 2006.
3.3.2. Récepteur peptidique natriurétique de type B, NPR-B
L’ARNm du NPR-B est surtout exprimé dans les poumons, le cerveau, l’utérus, les ovaires et les chondrocytes. Le NPR-B est le principal récepteur peptidique natriurétique dans le cerveau. La protéine NPR-B a été trouvé à des concentrations relativement élevées dans les fibroblastes. Le NPR-B a la même topologie que le NPR-A. L’affinité relative des peptides natriurétiques pour le NPR-B est dans l’ordre suivant : CNP >> ANP ≥ BNP.
3.3.3. Récepteur peptidique de clairance, NPR-C
Le NPR-C est exprimé dans la plupart des tissus, comme le placenta, les poumons, les reins, le tissu veineux, le muscle lisse de l’aorte et les cellules endothéliales, le cœur, le cortex cérébral, et le tissu cérébral. (Wilcox JN. et al., 1991). Le domaine extracellulaire du NPR-C a environ 30% d’homologie avec le NPR-A et NPR-B.
Ainsi, contrairement aux récepteurs liés à une guanylyl cyclase, il contient seulement 37 acides aminés intracellulaires et aucune activité guanylyl cyclase. Le domaine extracellulaire du NPR-C est glycosylé et contient deux séries de ponts disulfures intramoléculaires.
La fonction majeure du NPR-C est la clairance des peptides natriurétiques de la circulation ou du milieu extracellulaire par internalisation ou dégradation médiée par le récepteur, et cette internalisation est indépendante de la liaison d’un ligand sur le récepteur. Il a été également suggéré que le NPR-C pourrait médier des effets biologiques des peptides natriurétiques à travers des seconds messagers autres que le GMPc, comme par example via un couplage aux protéines Gi ou à travers le système adénylate cyclase/AMPc. L'affinité des peptides natriurétiques pour le NPR-C est dans l’ordre suivant : ANP ≥ CNP > BNP à la fois chez les humains et les rats(Potter LR. et al., 2006).
3.4. Antagonistes des récepteurs peptidiques natriurétiques
Trois inhibiteurs des récepteurs peptidiques NPR-A et NPR-B ont jusqu'ici été les plus largement appliqués. Le HS-142-1, un polysaccharide naturel de l’Aureobasidium spp., est relativement un inhibiteur spécifique de l’activité guanylate cyclase, qui inhibe la liaison des ligands et l'activation des deux récepteurs NPR-A et NPR-B par le biais d’un mécanisme allostérique (Morishita Y. et al., 1991). Cet inhibiteur n’antagonise pas les réponses médiées par le NPR-C. Bien que HS-142-1 a été largement utilisé dans la recherche des peptides natriurétiques, sa production a cessé actuellement, probablement pour des raisons commerciales. Le A71915 [Arg6, H-cyclohexyl-Ala8, D-Tic16, Arg17, Cys18)-ANP (6-18) amide] est un peptide synthétique dérivé de l’ANP, présentant des propriétés compétitives antagonistes au récepteur NPR-A (Delporte C. et al., 1992). Un troisième composé, l’anantin, est un peptide cyclique du Streptomyces coerulescens, qui agit comme un antagoniste compétitif au récepteur NPR-A (Weber W. et al., 1991). Bien que l’anantin soit sans doute le premier antagoniste des NPRs à être décrit, il a été moins largement utilisé que le HS-142-1 malgré sa disponibilité.
3.5. Clairance métabolique des peptides natriurétiques
En plus du NPR-C, la clairance des peptides natriurétiques de la circulation peut avoir lieu par dégradation enzymatique par l’endopeptidase neutre (NEP) qui est principalement exprimée dans les tubules proximaux des reins, dans l’endothélium vasculaire, dans les poumons et le cœur. Dans les conditions physiopathologiques, lorsque le NPR-C est saturé, la NEP contribue significativement à l’élimination des peptides natriurétiques (Hashimoto Y. et al., 1994). Le BNP a une forte résistance à la dégradation grâce à son affinité réduite au NPR-C ainsi que son faible taux d’hydrolyse par la NEP.
3.6. Voie de signalisation BNP/GMPc
Le BNP exerce son rôle cardioprotecteur en se fixant sur son récepteur NPR-A (GC-A). Parmi les effets biologiques du BNP via GC-A est la formation du GMPc, un second messager qui est important dans la maintien du débit cardiaque, des réponses cardioprotectrices et de la prolifération cellulaire. Les effets du GMPc sont médiés par sa liaison à trois classes de protéines : les protéines kinases GMPc-dépendantes (PKGs), les canaux ioniques GMPc-dépendants (CNG), et les phosphodiestérases (PDEs) (Figure H17).
Les effets les mieux étudiés du cGMP sont induit par les PKGs, conduisant à des résidus sérines et thréonines (Lohmann SM. et al., 1997 ; Schlossmann J. et al., 2005). Deux différents gènes pour la PKG ont été identifiés. Le gène PKG-I qui est alternativement épissé pour produire deux isoformes α et β qui diffèrent par leur domaine amino terminal. Ces deux isoformes de la PKG-I sont surtout cytosoliques et la PKG-Iα est fortement exprimée dans les reins, le muscle lisse, les poumons, les cardiomyocytes et le cerveau alors que la PKG-Iβ est uniquement présente dans l’utérus.. La délétion fonctionnelle de la PKG-I chez la souris se traduit par une perte de la vasodilatation GMPc-dépendante et par une hypertension juvénile (Pfeifer A. et al., 1998), mais pas chez les adultes (Schlossmann J. et al., 2005).
Figure H17 : Liaison du GMPc à trois classes de protéines pour médier ses effets D’après Potter, 2006.
La PKG-II qui est liée à la membrane et absente du système cardiovasculaire, est exprimée à des concentrations élevées dans l'intestin, les reins, le cerveau, les poumons et les os (Smolenski A. et al., 1998). La délétion fonctionnelle de la PKG-II chez la souris (Pfeifer A. et al., 1996) ou les rats (Chikuda H et al., 2004) se traduit par des animaux normotendus avec un nanisme et une résistance à l'infection par l’Escherichia coli.
L’augmentation du niveau de GMPc active la PKG-I qui est responsable de la phosphorylation de plusieurs cibles dont le phospholambane et les canaux KATP mitochondriaux, de la régulation de la croissance et de l’activation de la pompe calcique du réticulum sarcoplasmique (Pigott LA. et al., 2006).
La protéine kinase AMPc-dependante (PKA) contient des sites de liaison homologues à ceux de la PKG impliquant une activation possible par le GMPc. Toutefois, les cascades de signalisation de l’AMPc et GMPc permettent d’obtenir indépendamment divers effets physiologiques importants (Lucas K.A. et al., 2000).
Les PDEs sont des régulateurs indispensables de la signalisation des nucléotides cycliques, car elles dégradent le GMPc en GMP. Par conséquent, les PDEs règulent les concentrations des seconds messagers intracellulaires. Il existe 11 différentes familles de PDEs contenant au moins 25 protéines différentes. Les familles sont organisées en fonction de la spécificité du substrat (si elles dégradent l'AMPc, le GMPc, ou les deux) et la façon dont elles sont activées ou inhibées. Par exemple les PDE1, -2, -3, -10 et -11 dégradent l’AMPc et le GMPc; Les PDE4, -7 et -8 hydrolysent spécifiquement l'AMPc et alors que les PDE5, -6, -9 dégradent uniquement le GMPc (Figure H18).
La liaison du GMPc à la PDE5 augmente son activité et accélère la dégradation du GMPc. L'activité allostérique des PDEs peut faciliter l’interférence des voies AMPc / GMPc. Par exemple, le GMPc active la PDE2, qui entraîne une diminution de la concentration intracellulaire d’AMPc, alors que la liaison du GMPc inhibe l'activité de la PDE3 entraînant une augmentation de l'AMPc (Potter LR. et al., 2006).
Le GMPc induit aussi des réponses cellulaires par la régulation de canaux ioniques GMPc-dépendants (CNG). Les CNG (cyclic nucleotide-gated channel) sont une famille de canaux cationiques non sélectifs, identifiés pour la première fois dans les photorécepteurs de la rétine et les neurones sensoriels olfactifs, qui laissent passer des cations divalents, en particulier le Ca2+ (Kaupp UB. et Seifert R., 2002). La liaison des nucléotides cycliques, AMPc et le GMPc aux canaux CNG provoque leur ouverture, l'entrée de Ca2+ dans la cellule et l'induction de processus Ca2+-dépendants (Biel M. et al., 1998).
Donc le BNP, en générant du GMPc, induit l’activation des canaux CNG (Castro LR. et al., 2006) et le courant ICNG est augmenté suite à l’inhibition des PDEs. La densité et la conductance du courant ICNG sont plus élevés dans les myocytes ventriculaires hypertrophiés que dans ceux normaux (Fernandez-Velasco M. et al., 2003) et il est
généralement surexprimé dans les myocytes hypertrophiés où il semble jouer un rôle dans l’arhythmogenèse (Stillitano F. et al., 2008).
Figure H18 : Représentation schématique de la structure des PDEs dans le cardiomyocyte
3.7. Effets physiologiques du BNP
Le BNP possède des effets variés sur de multiples sites observés aussi bien chez l’homme que chez l’animal à des doses pharmacologiques (Figure H19). Grâce à ses propriétés vasodilatatrices, natriurétiques, diurétiques, antiprolifératives et modulatrices neurohormonales, le BNP est l’hormone de la décompensation cardiaque (Boerrigter G. et al., 2004).
Le BNP est connu être un antagoniste du système Rénine-Angiotensine-Aldostérone (RAAS) d’où son implication dans la régulation de la pression artérielle et de l’équilibre hydrosodique. Dans l'insuffisance cardiaque, l'activation du système rénine-angiotensine-aldostérone est supprimée par le BNP suite à une inhibition directe de la synthèse et de la sécrétion de l'aldostérone et une suppression de la libération de rénine (Brenner B.M. et al., 1990 ; Burger A.J., 2005).
Le BNP possède également des propriétés antifibrotique et antiremodelage in vitro dans des cultures humaines de fibroblastes cardiaques (Kapoun A.M. et al., 2004). En effet, ces auteurs ont démontré que le BNP inhibe le facteur de croissance TGF-bêta qui induit la fibrose. Des expériences sur des souris génétiquement modifiées suggèrent que l’effet antiproliferatif du BNP est un de ces rôles les plus important physiologiquement (Kuhn M., 2004). Chen HH et al., 2004, ont demontré que l’infusion de BNP pendant l’hospitalisation d’un infarctus myocardique antérieur peut avoir des effets favorable sur le remodelage. Le BNP exerce d’autre part un effet inotrope négatif, anti-mitotique et anti-coagulateur (Valli N. et al., 2000).
De plus, il joue un rôle dans la diminution des lésions tissulaires observées durant l’ischémie et la reperfusion. En effet, dans un modèle de rat à infarctus myocardique, l’administration éxogène de BNP limite la taille de l’infarctus de manière proportionnelle à la concentration (Lucas K.A. et al., 2000 ; Takagi G. et al., 2000).
Enfin, comme le monoxyde d’azote, le BNP faciliterait la neurotransmission vagale via le GMPc couplé aux récepteurs natriurétiques en phosphorylant les canaux calciques de type N présynaptiques augmentant ainsi la libération de l’acétylcholine bradycardisante (Herring N. et al., 2001).
Figure H19 : Effets biologiques généraux du BNP dans différents tissus D’après De Sa DD, 2008.
4. Le BNP dans le cardiomyocyte ventriculaire normal et pathologique
Le BNP est exprimé dans les oreillettes et les ventricules, mais il est principalement sécrété par les ventricules. La cause principale conduisant à sa sécrétion est le stress mécanique, où les niveaux d’ARNm du BNP sont fortement augmentés en réponse à la surcharge cardiaque.
Le BNP peut alors être considéré comme un marqueur spécifique du myocyte permettant d’identifier les altérations dans l’expression des gènes cardiaques couplés à une surcharge mécanique.
4.1. Localisation et sécrétion du BNP dans le cardiomyocyte normal et pathologique
Dans les conditions normales, la concentration myocytaire de BNP est prédominante dans les cardiomyocytes auriculaires. Suite à une dysfonction ventriculaire, ce peptide augmente alors dans les myocytes auriculaires et ventriculaires avec une augmentation de sa concentration plasmatique (Luchner A. et al., 1998).
Le niveau plasmatique du BNP est faible chez les sujets sains et la sécrétion de ce peptide augmente proportionnellement en fonction de la sévérité de la dysfonction ventriculaire. Cela suggère que la sécrétion du BNP est principalement régulée par le stretch mécanique du ventricule et que son taux plasmatique est un marqueur du degré de la dysfonction ventriculaire gauche (Yasue H. et al., 1994).
4.2. Expression du BNP dans le cardiomyocyte normal et pathologique
A l’exception des rats et des souris où l’expression du gène du BNP est présente dans le ventricule gauche normal, ce peptide, dans les conditions normales, n’est exprimé que dans les oreillettes chez les félins, les lapins et les chiens (Biondo AW. et al., 2003). Une nouvelle expression ventriculaire du gène du BNP est décelée dans l’hypertrophie ventriculaire.
Dans un cœur sain, l’expression du gène du BNP dans le tissu cardiaque est limité aux oreillettes et augmente en cas d’une dysfonction ventriculaire accompagnée d’une augmentation dans l’expression du gène du BNP au niveau du ventricule gauche, associée