يملعلا ثحبلا و يلاعلا ميلعتلا ةرازو
B
ADJIM
OKHTAR-A
NNABAU
NIVERSITYU
NIVERSITEB
ADJIM
OKHTAR-A
NNABAة
ـــــ
بانع
–
راتخم يجاب ةعماج
Faculté des Sciences de L’Ingéniorat Année 2014-2015
Département d’Informatique
THESE
Présentée en vue de l’obtention
du diplôme de Doctorat 3
èmeCycle
Approches Bio-inspirées
pour la Fouille de Données en Bioinformatique
Domaine : Mathématiques et Informatique
Filière : Informatique
Spécialité : STIC
(Sciences et Technologies de l’Information et de la Communication)par
M
elleZEKRI Meriem
Directeur de Thèse
Mme Labiba SOUICI-MESLATI
Professeur Université Badji Mokhtar-Annaba
Co-Directeur de ThèseMme Karima HARBI-ALEM
MCB
Université Badji Mokhtar-Annaba
DEVANT LE JURY
Président :
Mme Hassina SERIDI
Professeur
Université Badji Mokhtar-AnnabaExaminateurs :
Mme Habiba BELLEILI
MCA
Université Badji Mokhtar-AnnabaMr Yacine LAFIFI
MCA
Université 8 Mai 1945- Guelma
Dédicaces
Remerciements
Je souhaite remercier en premier Mme Labiba Souici-Meslati, Professeur à l’université d’Annaba sans qui je ne serais pas là où je suis aujourd’hui, pour ses encouragements et pour avoir été présente à chaque instant au cours de mon parcours de recherche, pour son sérieux et son dévouement.
Je tiens à remercier également Mme Karima Harbi-Alem, Maître de Conférences à l’université d’Annaba pour son aide précieuse durant tout mon travail et pour ses encouragements.
Je remercie vivement Mme Hassina Seridi, Professeur à l’Université d’Annaba pour m’avoir fait l’honneur de présider mon jury de thèse. Je remercie également Mr Abdelouahab Moussaoui, Professeur à l’Université de Setif, Mr Yacine Lafifi, Maître de Conférences à l’Université de Guelma, Mme Habiba Belleili, Maître de Conférences à l’Université d'Annaba et Mme Yamina Mohamed Ben Ali, Professeur à l’Université d'Annaba, pour l’intérêt qu’ils ont bien voulu porter à mon travail en acceptant la lourde tâche d'examinateur.
Je réserve les derniers remerciements à toute ma famille et mes amis, en particulier ma mère pour ses sacrifices, ses encouragements, sa présence et son soutien au quotidien.
T
ABLE DES MATIERES
Introduction Générale ... 1
1. Problématique et objectifs ... 2
2. Contenu du document ... 4
Chapitre 1 :Introduction à la Bioinformatique ... 7
1.1. Introduction ... 7
1.2. Les fondements biologiques de la Bioinformatique ... 8
1.2.1. De la génomique vers la protéomique ... 8
1.2.2. Les protéines ... 10
1.2.3. Les acides aminés ... 11
1.3. Généralités sur la bioinformatique ... 13
1.3.1. Historique de la bioinformatique ... 14
1.3.2. Les objectifs de la bioinformatique ... 15
1.3.3. Les concepts et techniques de la bioinformatique ... 16
1.3.3.1. Les bases de données biologiques ... 17
1.3.3.2. La comparaison de séquences ... 19
1.4. La fouille de données en bioinformatique ... 20
1.4.1. Les tâches de la fouille de données en Bioinformatique ... 21
1.4.1.1. Classification et règles de classification ... 21
1.4.1.2. Clustering ... 22
1.4.2. Synthèse de travaux de la fouille de données en Protéomique ... 23
1.4.2.1. Prédiction de fonctions de protéines ... 24
1.4.2.2. Prédiction de structures de protéines ... 26
1.4.2.3. Prédiction de localisations de protéines ... 28
1.4.2.4. Prédiction des interactions protéine-protéine ... 29
1.5. Conclusion ... 33
Chapitre 2: Systèmes Immunitaires Artificiels et Swarm Intelligence : Concepts et Méthodes ... 35
2.1. Introduction ... 35
2.2.1. Algorithme de sélection clonale ... 38
2.2.1.1. Inspiration biologique ... 38
2.2.1.2. Description ... 39
2.2.1.3. Les variantes de l‘algorithme de la sélection clonale ... 40
2.2.2. Algorithme de sélection négative ... 40
2.2.2.1 . Inspiration biologique ... 41
2.2.2.2 . Description ... 41
2.2.2.3 . Variantes de l‘algorithme de la sélection négative ... 43
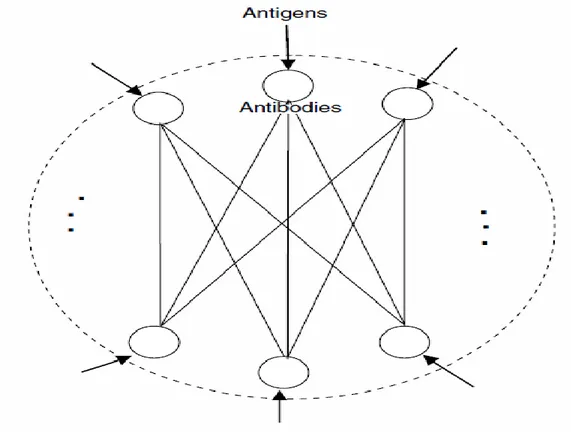
2.2.3. Algorithme des réseaux immunitaires artificiels ... 43
2.2.3.1. Inspiration biologique ... 43
2.2.3.2. Description ... 44
2.2.3.3. Variantes des réseaux immunitaires artificiels ... 45
2.2.4. La théorie du danger ... 46
2.2.4.1. Inspiration biologique ... 46
2.2.4.2. Description ... 47
2.3. Les approches de la Swarm Intelligence ... 48
2.3.1. Optimisation par colonies de fourmis (ACO) ... 49
2.3.1.1. Les fourmis dans la nature ... 49
2.3.1.2. Les colonies de fourmis artificielles ... 51
2.3.1.3. Les variantes de Ant-Miner ... 52
2.3.2. Optimisation par essaim de particules (PSO) ... 52
2.3.2.1. Principe général ... 53
2.3.2.2. Formalisation ... 53
2.3.2.3. Les variantes de PSO ... 55
2.3.3. Les colonies d‘abeilles artificielles (ABC) ... 56
2.3.3.1. Les abeilles dans la nature ... 56
2.3.3.2. Les abeilles artificielles ... 57
2.3.3.3. Les variantes de l‘ABC ... 59
2.4. Conclusion ... 59
Chapitre 3 :Les Récepteurs Couplés aux Protéines G et leur Classification ... 61
3.1. Introduction ... 61
3.2.1. Structure des RCPGs ... 62
3.2.2. Mécanisme des RCPGs ... 64
3.2.2.1. Les protéines G ... 64
3.2.2.2. Transduction du signal par activation des protéines G ... 65
3.2.3. Processus physiologiques des RCPGs ... 67
3.3. Classification des RCPGs ... 67
3.4. Les bases de données RCPGs ... 69
3.5. Les RCPGs comme cibles de médicaments ... 71
3.6. Synthèse de travaux sur l‘identification des RCPGs ... 73
3.6.1. Serveurs web d‘identification de RCPGs ... 74
3.6.2. Méthodes basées sur les machines à vecteurs de support ... 77
3.6.3. Méthodes basées sur les k-plus proches voisins ... 79
3.6.4. Méthodes basées sur les arbres de décision ... 80
3.6.5. Méthodes basées sur les réseaux de neurones artificiels ... 81
3.6.6. Méthodes basées sur les approches de la swarm intelligence ... 82
3.6.7. Méthodes basées sur les algorithmes immunitaires artificiels ... 83
3.7. Conclusion ... 88
Chapitre 4 :Approches Immunologiques pour la Prédiction de Fonctions des RCPGs 89 4.1. Introduction ... 89
4.2. Prédiction de fonctions de protéines par les AIS ... 90
4.3. Algorithme immunitaire artificiel de reconnaissance (AIRS) ... 92
4.3.1. Initialisation ... 94
4.3.2. Identification des cellules mémoires et génération des ARBs ... 94
4.3.3. Compétition pour les ressources et développement des cellules mémoires candidates ... 95
4.3.4. Introduction des cellules mémoires ... 95
4.3.5. Classification ... 95
4.3.6. La version améliorée d‘AIRS : AIRS2 ... 96
4.4. Algorithme de sélection clonale (CLONALG) ... 97
4.5. Algorithme de classification de sélection clonale (CSCA) ... 99
4.6. Ensemble de données et prétraitement ... 101
4.6.1. Ensemble de données GDS ... 102
4.6.2.1. La composition en pseudo acides aminés (PseAAC) ... 103
4.6.2.2. La normalisation moyenne / variance ... 106
4.7. Expérimentations et résultats ... 106
4.7.1. Résultats des classifieurs immunologiques proposés... 106
4.7.2. Comparaison avec d‘autres classifieurs ... 110
4.7.3. Comparaison avec des méthodes publiées ... 111
4.8. Conclusion ... 113
Chapitre 5 : Approches de la Swarm Intelligence pour la Prédiction de Fonction des RCPGs ... 115
5.1. Introduction ... 115
5.2. Approches de Swarm Intelligence pour la prédiction de fonctions de protéines ... 116
5.3. Les colonies de fourmis pour la classification : cAnt-Miner ... 118
5.3.1. Ant-Miner... 118
5.3.2. Ant-Miner pour les attributs continus : cAnt-Miner ... 120
5.3.2.1. Construction du graphe ... 120
5.3.2.2. Mesure d‘entropie ... 120
5.3.2.3. Construction de la règle ... 121
5.3.2.4. Mise à jour des phéromones ... 121
5.4. Les essaims particulaires pour la classification : CPSO ... 122
5.4.1. Représentation de la règle ... 122
5.4.2. Découverte des règles ... 123
5.4.3. Evaluation de la règle ... 124
5.4.4. Algorithme de couverture pour la construction de l‘ensemble de règles ... 125
5.4.5. Elagage d‘une règle et nettoyage de l‘ensemble de règles ... 125
5.5. Classification basée sur l‘hybridation PSO/ACO ... 126
5.5.1. Formalisation ... 126
5.5.2. Version améliorée de l‘hybridation PSO/ACO : PSO/ACO2 ... 127
5.6. Ensemble de données et Prétraitement... 129
5.7. Outils pour les expérimentations... 131
5.7.1. Myra 3.6.1 ... 132
5.7.2. KEEL-GPLv3... 132
5.7.3. PSO/ACO2 ... 134
5.8. Jeux de paramètres et résultats ... 135
5.8.1. Jeux de paramètres... 135
5.8.2. Résultats ... 137
5.8.2.1. Comparaison avec les classifieurs immunitaires ... 139
5.8.2.2. Comparaison avec des méthodes publiées ... 140
5.9. Conclusion ... 140
Conclusion et Perspectives ... 143
1. Résumé des contributions ... 144
2. Perspectives de recherche ... 146 Références Bibliographiques ... 147 Annexe 1 ... 171 Annexe 2 ... 172 Annexe 3 ... 173 Biographie de l'auteur ... 175 Contributions scientifiques ... 176
L
ISTE DES FIGURES
Figure 1.1 Synthèse des protéines 9
Figure 1.2 Les différentes structures d‘une protéine 10 Figure 1.3 Configuration générale d‘un acide aminé [ALB 08] 12 Figure 1.4 Diagramme de Venn des propriétés des acides aminés [SEL 08] 13
Figure 1.5 Analyse des données biologiques 16
Figure 1.6 Processus général d‘une tâche de fouille de données [INZ 10] 20 Figure 1.7 Quelques applications de la fouille de données en protéomique [INZ 10] 23 Figure 2.1 Algorithme générique de la sélection clonale [DAS 09] 39 Figure 2.2 Processus de génération des détecteurs [DAS 09] 42
Figure 2.3 Processus de détection du NSA [DAS 09]. 42
Figure 2.4 Illustration d‘un réseau immunitaire, les nœuds, arêtes et flèches représentent les anticorps, les interactions entre eux, et la simulation de l‘AIN par des antigènes étrangers, respectivement [DAS 09]
45
Figure 2.5 Modèle de la théorie du danger [AIC 14] 47
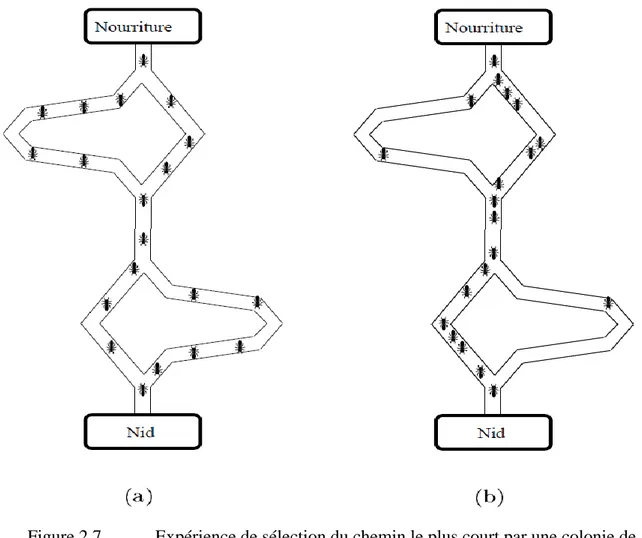
Figure 2.6 Principe de base du comportement collectif 49 Figure 2.7 Expérience de sélection du chemin le plus court par une colonie de
fourmis
43
Figure 2.8 Eléments du comportement des particules d‘un essaim 53
Figure 2.9 Les différentes topologies de voisinages 55
Figure 2.10 Diagramme de l‘algorithme des colonies d‘abeilles [AKA 12] 58 Figure 3.1 Structure de la majorité des récepteurs couplés aux protéines G [ALE
11]
63 Figure 3.2 Transmission des signaux par les récepteurs couplés aux protéines G
[ALE 11]
66
Figure 3.3 Vue simplifiée de l‘arbre des familles des récepteurs couplés aux protéines G (classification du système d‘information GPCRDB) [HUA 04]
68
Figure 3.4 Les médicaments ciblant les RCPGs [LUN 06] 72 Figure 3.5 Schéma représentant un récepteur couplé aux protéines G et les
possibles sites de liaison du ligand [ZHA 13]
73
Figure 4.1 Organigramme du système immunitaire artificiel de reconnaissance [BRO 05b]
Figure 4.2 Déroulement des étapes de l‘algorithme CLONALG [BRO 05b] 99
Figure 4.3 Les étapes de l‘algorithme CSCA [OLI 12] 100
Figure 4.4 Prédiction des fonctions de RCPGs à quatre niveaux 107 Figure 5.1 Corrélation séquence-ordre entre les résidus les plus contiguës le long
d‘une chaîne de protéine à différents niveaux [CHO 05b]
L
ISTE DES TABLEAUX
Tableau 1.1 Les vingt acides aminés natifs et leur code officiel 11
Tableau 1.2 Historique de la bioinformatique 14
Tableau 1.3 Synthèse de travaux en protéomique utilisant les tâches de la fouille de données
30
Tableau 2.1 Immunité basée sur des modèles de calcul et de concepts immunologiques spécifiques [DAS 09]
37
Tableau 3.1 Les différentes bases de données RCPGs disponibles 70 Tableau 3.2 Serveurs web de prédiction et d‘identification des RCPGs 75 Tableau 3.3 Synthèse de travaux sur l‘identification des RCPGs 84 Tableau 4.1 Correspondance entre le système immunitaire biologique et le système
immunitaire artificiel de reconnaissance [WAT 04]
92
Tableau 4.2 Paramètres de l‘algorithme AIRS2 [BRO 05a] 96
Tableau 4.3 Paramètres de l‘algorithme CLONALG 97
Tableau 4.4 Paramètres de l‘algorithme CSCA [BRO 05b] 101 Tableau 4.5 Valeurs des paramètres knn et ressources totales à chaque niveau 108 Tableau 4.6 Précision prédictive de l‘algorithme AIRS2 aux quatre niveaux 109 Tableau 4.7 Précision prédictive des trois classifieurs aux quatre niveaux 109 Tableau 4.8 Comparaison de la précision prédictive des trois classifieurs
immunitaires avec d‘autres classifieurs
110
Tableau 4.9 Comparaison de la prédiction prédictive d‘AIRS2 avec des méthodes publiées
111
Tableau 5.1 Paramètres de l‘algorithme cAnt-Miner dans la plateforme myra 132 Tableau 5.2 Paramètres de l‘algorithme CPSO sous KEEL 133
Tableau 5.3 Paramètres de l‘algorithme PSO/ACO2 134
Tableau 5.4 Valeurs des paramètres nombre d‘itération et nombre de fourmis à chaque niveau
135
Tableau 5.6 Précision prédictive du classifieur cAnt-Miner avec les paramètres avec défaut et les paramètres modifiés
137
Tableau 5.7 Précision prédictive du classifieur CPSO avec les paramètres avec défaut et les paramètres modifiés
138
Tableau 5.8 Comparaison de la précision prédictive des trois classifieurs basés swarm intelligence
138
Tableau 5.9 Tableau 5.9. Comparaison de la précision prédictive des meilleurs classifieurs bio-inspirées PSO/ACO2 et AIRS2
139
Tableau 5.10 Comparaison de la précision prédictive de PSO/ACO2 avec des méthodes publiées
L
ISTE DES ABREVIATIONS
AAC Amino Acid Compisition (Composition en acides aminés) ABC Artificial Bee Colony (Colonie d‘abeilles artificielles)
ACO Ant Colony Optimization (Optimisation par colonies de fourmis) ADN Acide Désoxyribonucléique
AIN (aiNET) Artificial Imunne Network (Réseau immunitaire artificiel) AIS Artificial Immune System (Système immunitaire artificiel)
AIRS Artificial Immune Recognition System (Système immunitaire artificiel de reconnaissance)
ARB Artificial Recognition Balls (Balles de reconnaissance artificielles) ARN Acide Ribonucléique
BA Bat Algorithm (Algorithme de chauves-souris)
BLAST Basic Local Alignment Search Tool (Outil de recherche d‘alignement local basique)
CD-HIT Cluster Database at High Identity with Tolerance (Base de données de clusters à haute identité avec tolérance)
CLONALG CLOnal selection ALGorithm (Algorithme de sélection clonale) CSA Clonal Selection Algorithm (Algorithme de sélection clonale) CPSO Constricted Particle Swarm Optimization
CSCA Clonal Selection Classification Algorithm (Algorithme de classification de sélection clonale)
DCA Dendritic Cell Algorithm (Algorithme des cellules dendritiques) FA Firefly Algorithm (Algorithme de lucioles)
FPA Flower Pollination Algorithm (Algorithme de la pollenisation des fleurs) GDS G protein coupled receptor Data Set (ensemble de données de RCPGs) GPCR G-Protein Coupled Receptors (Traduction de RCPGs)
GPCRDB G-Protein Coupled Receptor Data Base (Base de données de RCPGs) GRK G protein coupled Receptor Kinase
KEEL Knowledge Extraction based on Evolutionnary Learning (Extraction des connaissances basée sur l‘apprentissage évolutionnaire)
KNN k-Nearest Neighbors (k-plus proches voisins) LVQ neural Learning Vector Quantization algorithm
multicouches)
NSA Negative Selection Algorithm (Algorithme de sélection négative) Opt-aiNET Optimization aiNET
PDB Protein Data Bank (banque de données de protéines)
PIO Pigeon Inspired Optimization (Optimisation inspirée des pigeons) PIR Protein Information Resource (ressource d‘information sur les protéines) PRF Protein Research Foundation (fondation de recherche sur les protéines) PseAAC Pseudo Amino Acid Composition (Composition en pseudo acides aminés) PSO Particle Swarm Optimization (Optimisation par essaims particulaires) RCPGs Récepteurs Couplés aux Protéines G
ROC Receiver Operating Characteristic
SCOP Structural Classification of Proteins databases (classification structurelle des bases de données de protéines)
SI Swarm Intelligence
SVM Support Vector Machine (Machine à vecteurs de support)
I
NTRODUCTION
G
ENERALE
Dans le cadre de leurs travaux, les biologistes ont à collecter et à interpréter un grand nombre de données. Ces dernières années, ce nombre a augmenté de façon exponentielle grâce aux nouvelles technologies expérimentales complexes qui permettent une collecte de données beaucoup plus rapide que leur interprétation. Afin d‘aider les biologistes dans la collecte, le stockage et l‘analyse de ces données, une nouvelle discipline a vu le jour, appelée
Bioinformatique. La bioinformatique est une discipline en plein essor, elle concerne le
développement de modèles, de méthodes et d‘outils afin d‘analyser des données biologiques (génomes, protéomes, etc.), et produire de nouvelles connaissances pour mieux comprendre et résoudre les problèmes posés par la biologie. Elle représente aussi un domaine interdisciplinaire qui s‘appuie largement sur l‘informatique, les statistiques, les mathématiques et l‘étude du vivant (la biologie).
La bioinformatique se développe au fur et à mesure que nous continuons à générer et à intégrer de grandes quantités de données biologiques. Dans ce domaine, il est essentiel de comprendre les problèmes biologiques tout autant que les solutions informatiques à mettre en œuvre pour produire des outils utiles à leur résolution. Il est donc nécessaire de savoir ce que représente un gène ou une protéine et dans quel but les étudier. La bioinformatique peut être divisée en deux grands axes de recherche, la génomique et la protéomique. La génomique représente l‘étude, à l‘échelle du génome, de tous les organismes, organes, maladies, etc. tandis que la protéomique, qui découle de la génomique, représente l‘étude du protéome, c‘est-à-dire, l‘étude, à grande échelle, des fonctions de toutes les protéines exprimées dans un organisme, ainsi que de leurs modifications, localisation et les interactions entre elles.
Notre travail se place dans le cadre de la protéomique, plus précisément, celui de la prédiction de fonctions de protéines, qui représente une tâche de première importance dans ce domaine. La prédiction de fonctions de protéines consiste à assigner à la protéine, la classe fonctionnelle correspondante, afin de déterminer le rôle biologique et biochimique qu‘elle joue dans l‘organisme. Cet axe de recherche s‘appelle également la protéomique in silico, il concerne l‘étude des séquences protéiques par simulation sur ordinateur. A l‘inverse, la protéomique in vivo, qui, pour annoter une protéine au laboratoire, nécessite de réaliser de nombreuses expériences dans diverses conditions expérimentales qui peuvent être longues, très coûteuses et dont les résultats ne sont pas garantis. L‘identification des protéines par des méthodes computationnelles peut réduire le champ de recherche en indiquant au biologiste le
point de départ pour orienter ses investigations, ce qui réduit le temps des expérimentations et garantit ainsi un certain résultat.
Le champ le plus important dans le domaine de la bioinformatique est l‘application de la fouille de données pour la résolution de problèmes biologiques. La fouille de données joue un rôle fondamental dans la compréhension des problèmes bioinformatiques émergents tels que l‘annotation des gènes et des protéines. Les tâches de la fouille de données les plus couramment utilisées sont la classification et le clustering. La tâche de classification, incluant la découverte des règles de classification permet de construire des modèles de prédiction et de classification performants pour l‘annotation des séquences, alors que la tâche de clustering est le plus souvent utilisée, en combinaison avec les méthodes de sélection de caractéristiques, pour représenter les séquences nucléiques et protéiques.
Etant donnée l‘importance des problèmes protéomiques posés, différentes méthodes ont été développées et exploitées dans le but d‘y apporter des solutions fiables, efficaces et peu coûteuses. Parmi les méthodes les plus utilisées, nous pouvons citer les machines à vecteurs de support, les k-plus proches voisins, les arbres de décision, etc. Bien que ces méthodes aient fait leurs preuves, les chercheurs se sont tournés, ces dernières années vers des paradigmes particuliers de l‘intelligence artificielle, ceux des approches bio-inspirées. Ces approches s‘inspirent de la nature et connaissent un grand succès dans de nombreux domaines d‘application. En effet, elles se basent sur la biologie, comme source d'inspiration, pour résoudre des problèmes computationnels et des expériences du monde naturel (insectes, animaux,…) pour résoudre les problèmes du monde réel. Le fait que des organisations et des structures simples dans la nature soient capables de faire face à des problèmes complexes avec aisance a inspiré les chercheurs à concevoir des modèles intéressants pour la résolution de problèmes dans de nombreux domaines.
Les deux familles d‘approches bio-inspirées les plus récentes sont les systèmes immunitaires artificiels (Artificial Immune Systems – AIS) qui, comme leur nom l‘indique, s‘inspirent des processus et des mécanismes de défense des systèmes immunitaires biologiques, et la swarm intelligence (SI). Cette dernière s‘inspire du comportement collectif d‘insectes et d‘animaux tels que les colonies de fourmis et d‘abeilles, les bancs de poissons, les volées d‘oiseaux. Ces groupes font preuve de comportements fascinants qui combinent, à la fois, l‘efficacité à la flexibilité et la robustesse. Les approches bio-inspirées sont connues pour apporter des solutions optimales à coût réduit, ce qui a motivé leur application, entre autres, dans le domaine de la prédiction de fonctions de protéines.
1.
Problématique et objectifs
Dans le cadre de notre travail, nous nous intéressons à la famille de récepteurs couplés aux protéines G (RCPGs), l‘une des familles les plus larges et les plus importantes des protéines membranaires, responsables de la communication cellulaire, jouant un rôle clé dans divers processus physiologiques (la vision, l'olfaction, la contraction du muscle cardiaque, la
régulation de la pression artérielle,... etc). Leur diversité et les nombreuses fonctions qu'ils contrôlent les font intervenir dans de nombreuses pathologies. Bien que des milliers de séquences de récepteurs couplés aux protéines G soient connues, nombre d‘entres-elles restent orphelines (leur fonction est inconnue) à ce jour, malgré les efforts mis en œuvre par les chercheurs et l‘industrie pharmaceutique. Il est donc primordial de trouver des techniques et des méthodes efficaces pour remédier à ce problème. La prédiction de fonction de protéines, qui est une tâche de première importance en bioinformatique, a pour objectif d‘identifier le rôle central biologique qu‘elle occupe dans l‘organisme et pouvoir ainsi, si nécessaire, inhiber ou activer son fonctionnement. La prédiction de fonctions des protéines est un domaine de recherche très actif pour plusieurs raisons, telles que la nécessité cruciale d‘une meilleure compréhension des protéines responsables de maladies, le développement de médicaments plus efficaces, la médecine préventive, etc. Cette prédiction est généralement effectuée à partir de la séquence primaire d'acides aminés des protéines, mais elle peut également être réalisée à partir de leurs structures secondaires et tertiaires, ainsi que leurs localisations et leurs interactions avec les autres protéines, qui apportent des informations pertinentes.
Au cours des dernières années, de nombreuses méthodes ont été développées et exploitées pour apporter des solutions prometteuses à ce problème, cependant, beaucoup d‘entre-elles atteignent rapidement leur limites. De ce fait, des chercheurs se sont tournés vers de nouveaux paradigmes, comme les approches bio-inspirées récentes, utilisées avec succès dans de nombreux domaines. L‘application de ces approches en bioinformatique et, en particulier, pour la prédiction de fonctions des récepteurs couplés aux protéines G, représente un axe de recherche prometteur. En effet, ces approches bénéficient des caractéristiques des processus et organismes dont elles s‘inspirent comme l‘auto-organisation, la mémorisation, l‘apprentissage, la robustesse et l‘efficacité à résoudre des problèmes complexes à coût réduit. L‘objectif de notre travail est d‘étudier l‘apport des approches bio-inspirées récentes, issues de l‘immunologie artificielle et de l‘intelligence en essaims (swarm intelligence), au problème de prédiction de fonctions des récepteurs couplés aux protéines G (RCPGs). Nous avons commencé notre travail par une étude bibliographique approfondie des travaux dans le domaine de la fouille de données en protéomique et, en particulier, celui de la prédiction de fonctions des RCPGs. Nous avons ensuite effectué deux études expérimentales, la première en utilisant des classifieurs basés sur l‘approche immunologique et la seconde, des classifieurs basés sur les techniques de la swarm intelligence.
Les algorithmes immunitaires sélectionnés sont le système immunitaire artificiel de reconnaissance (Artificial Immune Recognition System – AIRS), l‘algorithme de sélection clonale (CLONal Selection ALGorithm – CLONALG) et l‘algorithme de classification de sélection clonale (Clonal Selection Classification Algorithm – CSCA). Ces trois algorithmes ont la particularité d‘être basés sur la théorie de la sélection clonale, connue pour ses caractéristiques de mémorisation et d‘apprentissage. L‘algorithme AIRS introduit par Watkins en 2001 [WAT 01, 04] est l‘un des premiers algorithmes immunitaires conçus pour les problèmes de classification. CLONALG est l‘un des algorithmes immunitaires les plus populaires du fait qu‘il soit de faible complexité et qu‘il dispose de peu de paramètres
utilisateurs et l‘algorithme CSCA est une version améliorée de CLONALG, conçue spécialement pour la tâche de classification.
Plusieurs approches de swarm intelligence ont été développées ces dernières années, comme l‘algorithme des chauves-souris, l‘algorithme des lucioles, l‘algorithme de la pollenisation des fleurs, … Dans notre étude, nous nous sommes intéressés aux deux approches les plus populaires et les plus couramment utilisées qui sont : l‘optimisation par colonies de fourmis (Ant Colony Optimization – ACO) et l‘optimisation par essaims particulaires (Particle Swarm Optimization – PSO). Nous avons choisi d‘utiliser cAnt-Miner, une version pour les attributs continus d‘Ant-Miner qui représente le premier algorithme basé ACO proposé pour la découverte de règles de classification, ainsi que CPSO (Constricted Particle Swarm Optimization) qui représente une version améliorée de l‘algorithme original PSO, adaptée pour la tâche de classification. Enfin, nous avons évalué la combinaison des deux approches à travers l‘hybridation PSO/ACO afin de déterminer le modèle qui correspond le mieux à la problématique posée.
Afin de construire, évaluer et comparer les performances des classifieurs que nous avons proposés, il est important de choisir une base de données adéquate. Nous avons opté pour l‘ensemble de données GDS parce qu‘il représente l‘ensemble de données le plus utilisé dans le domaine d'identification des RCPGs, ce qui nous permettra de comparer nos résultats avec les différents travaux utilisant ce même ensemble de données.
Afin de maximiser les chances d‘obtenir de bons résultats, une phase de prétraitement est réalisée sur cet ensemble de données pour les deux études effectuées. Cette phase concerne l‘utilisation de méthodes de sélection des caractéristiques pour représenter les séquences des RCPGs. Les séquences primaires des RCPGs, comme celles de toutes les protéines se présentent sous forme d‘enchaînement d‘acides aminés variant, en longueur (de 250 à 1200 acides aminées, voir Annexe 1) et qui se composent de différentes lettres correspondant aux 20 acides aminés natifs, ce qui représente un nombre trop important d‘attributs, il est donc nécessaire de réaliser une sélection des caractéristiques, en ne retenant que les attributs les plus pertinents. Il y a plusieurs méthodes pour la représentation des séquences protéiques mais la plus appropriée et la plus utilisée reste la composition en pseudo acides aminés (Pseudo Amino Acid Compoisition – PseAAC) introduite par Chou en 2001 [CHO 01, 09], elle se base sur la sélection des propriétés physico-chimiques de la protéine.
2.
Contenu du document
Ce document est organisé en cinq chapitres dont les trois premiers sont théoriques et comportent, respectivement, une introduction à la bioinformatique, une description des méthodes et concepts des approches bio-inspirées utilisées : les systèmes immunitaires artificiels et la swarm intelligence ainsi qu‘une étude de la famille de protéines considérées : les récepteurs couplés aux protéines G. Les deux derniers chapitres concernent l‘étude expérimentale effectuée, qui consiste en la prédiction de fonctions de protéines en utilisant
des systèmes immunitaires artificiels dans le chapitre quatre et des méthodes issues de la swarm intelligence dans le cinquième et dernier chapitre.
Chapitre 1. Introduction à la bioinformatique
Dans ce chapitre, nous introduisons la bioinformatique qui représente un domaine en pleine émergence et qui consiste en l‘application de diverses disciplines pour la résolution de problèmes biologiques. En premier lieu, nous présentons les fondements biologiques de la bioinformatique, plus spécifiquement, la protéomique qui découle de la génomique et dont les principaux fondements sont les protéines et leurs composantes, les acides aminés. Par la suite, nous abordons des généralités sur la bioinformatique, son historique, ses objectifs, ses concepts et ses techniques. Nous discutons ensuite de l‘importance de la fouille de données en bioinformatique, de ses tâches les plus exploitées qui sont la classification et le clustering. Nous terminons le chapitre par une synthèse de travaux portant sur l‘application des tâches de la fouille de données aux problèmes protéomiques.
Chapitre 2. Systèmes immunitaires artificiels et swarm intelligence : Concepts et méthodes
Nous abordons dans ce chapitre les deux familles bio-inspirées choisies pour notre étude, les systèmes immunitaires artificiels et la swarm intelligence. Nous présentons, en premier lieu, les systèmes immunitaires artificiels dont le fonctionnement est inspiré des systèmes immunitaires naturels, ensuite, nous abordons les différentes méthodes immunitaires connues à ce jour dans la littérature et qui sont, la sélection clonale, la sélection négative, les réseaux immunitaires et la théorie du danger. En second lieu, nous abordons la swarm intelligence, qui s‘inspire de divers comportements animaliers tels que ceux des volées d‘oiseaux, des bancs de poissons ou des colonies d‘insectes. Nous nous intéressons particulièrement aux approches de la swarm intelligence les plus populaires et les plus utilisées comme l‘optimisation par les colonies de fourmis, l‘optimisation par les essaims particulaires et les colonies d‘abeilles artificielles.
Chapitre 3. Les récepteurs couplés aux protéines G et leur classification
Ce chapitre consiste en une étude de la famille protéique des récepteurs couplés aux protéines G (RCPGs). Les RCPGs représentent l‘une des familles les plus larges et les plus importantes des protéines membranaires. Nous abordons leur structure et leur mécanisme qui consiste en la transduction du signal et l‘activation de la protéine G, ainsi que les processus physiologiques dont ils sont responsables dans l‘organisme. Nous présentons, par la suite, la classification la plus connue des RCPGs en plusieurs niveaux hiérarchiques où chaque niveau a son importance. Ensuite, nous présentons les différentes bases de données disponibles et nous discutons du fait que les RCPGs représentent des cibles de médicaments en raison du rôle clé qu‘ils jouent dans plusieurs processus physiologiques. Nous terminons ce chapitre par une synthèse de méthodes, proposées dans la littérature, pour l‘identification des RCPGs.
Chapitre 4. Approches immunologiques pour la prédiction de fonctions des RCPGs
Ce chapitre est consacré à notre première contribution principale, qui consiste en la proposition de trois classifieurs immunitaires pour déterminer les fonctions des récepteurs couplés aux protéines G. Nous commençons ce chapitre par un bref état de l‘art sur
l‘utilisation des approches immunologiques pour la prédiction de fonctions de protéines afin d‘avoir une idée générale sur ce qui a été réalisé jusqu‘à présent dans ce domaine. Ensuite, nous présentons les trois algorithmes immunitaires choisis pour la proposition de nos classifieurs : le système immunitaire artificiel de reconnaissance (Artificial immune Recognition System – AIRS), l‘algorithme de sélection clonale (Clonal Selection Algorithm – CLONALG) et l‘algorithme de classification de sélection clonale (Clonal Selection Classification Algorithm – CSCA). Dans le cadre de notre première contribution principale, et afin d‘évaluer les performances des trois classifieurs proposés, un ensemble de données de séquences RCPGs est utilisé et des prétraitements sont effectués pour rendre ces données exploitables et maximiser les chances d‘obtenir de bons résultats. Les expérimentations concernant ces trois classifieurs avec l‘ensemble de données sélectionné sont présentées à la fin de ce chapitre ainsi que la comparaison de nos propositions avec d‘autres méthodes publiées utilisant le même ensemble de données.
Chapitre 5. Approches de la swarm intelligence pour la prédiction de fonctions des RCPGs
Ce chapitre concerne notre seconde contribution principale, portant sur la prédiction de fonctions des RCPGs en proposant trois classifieurs basés sur des approches issues de la swarm intelligence. Le premier se base sur cAnt-Miner (version pour attributs continus d‘Ant-Miner) fondé sur l‘optimisation par colonies de fourmis (Ant Colony Optimization – ACO). Le deuxième classifieur se base sur l‘algorithme CPSO (Constricted PSO) fondé sur l‘optimisation par essaims particulaires (Particle Swarm Optimization – PSO) et le troisième classifieur se base sur l‘algorithme hybride PSO/ACO. Ces trois classifieurs sont testés en utilisant le même ensemble de données que pour les approches immunologiques mais avec des prétraitements différents, plus adéquats aux algorithmes sur lesquels se basent les classifieurs proposés car ces algorithmes ne peuvent faire face à la grande dimensionnalité de l‘ensemble de données obtenu précédemment. Différentes expérimentations ont été réalisées avec divers jeux de paramètres afin d‘adapter, aux mieux, les classifieurs proposés à la problématique abordée.
C
HAPITRE
1
I
NTRODUCTION A LA
B
IOINFORMATIQUE
La bioinformatique est l‘application des techniques de traitement de l‘information à la gestion de données biologiques ou, en d‘autres termes, c‘est la discipline de l‘analyse « in silico » de l‘information biologique. Elle est apparue avec la création des premières banques de données biologiques. Ce chapitre consiste en une introduction au domaine de la bioinformatique, il est organisé comme suit : dans la deuxième section, nous aborderons les fondements biologiques de la bioinformatique, tels que les protéines et les acides aminés, nous ne détaillerons pas les aspects relatifs aux gènes et aux acides nucléiques car notre étude se focalise sur les protéines. Dans la troisième section, nous présenterons des généralités sur la bioinformatique, son historique, ses objectifs, ses concepts et ses techniques. Dans la quatrième section, nous discuterons de l‘application de la fouille de données en bioinformatique, qui représente un domaine émergent, ainsi que les tâches de la fouille de données les plus exploitées en bioinformatique et qui sont, la classification et le clustering. Nous terminerons la section avec une synthèse de travaux sur la fouille de données dans le domaine de la protéomique qui représente l‘étude de l‘ensemble des protéines.
1.1.
Introduction
Ces dernières années, une croissance massive de l‘information biologique recueillie par les communautés scientifiques a vu le jour. Le déluge de ce type d‘informations sous la forme de génomes, de séquences protéiques, de données d‘expressions génétiques, …a conduit à la nécessité de concevoir des outils informatiques performants pour stocker, analyser et interpréter ces données, ce qui a engendré une science nouvelle, appelée Bioinformatique… Le terme bioinformatique signifie littéralement la science de l‘informatique appliquée à la recherche biologique. D‘autre part, l‘informatique est la gestion et l‘analyse de données en utilisant diverses techniques computationnelles de pointe. Ainsi, en d‘autres termes, la bioinformatique peut être décrite comme l‘application de méthodes computationnelles pour la découverte de connaissances biologiques. Elle représente une symbiose de plusieurs domaines différents de la science, notamment, l‘informatique, la biologie, les mathématiques et les statistiques [DAS 08a].
Les tâches de la bioinformatique ne cessent de s‘accroître avec les données biologiques. Les premiers temps, elles consistaient en une simple analyse des séquences à travers des comparaisons, aujourd‘hui, une séquence est analysée sous toutes les coutures. A partir d‘un gène, il est possible de construire une puce ADN, de réaliser une transcription de ce gène et d‘obtenir la protéine correspondante ou de chercher des séquences similaires afin d‘étudier
l‘homologie1
d‘une espèce donnée. Cependant, la tâche majeure en bioinformatique demeure la prédiction de fonction de protéine, qui consiste en l‘identification de son rôle cellulaire et biologique dans l‘organisme afin de mieux comprendre son comportement.
Dans notre étude, notre intérêt s‘est porté sur cette tâche de bioinformatique et plus précisément de protéomique. Cette dernière représente la science d‘étude des protéines, leur structure, leur localisation ainsi que leur interactions avec d‘autres protéines et leurs fonctions. La prédiction de fonction est une tâche que l‘on peut attribuer au domaine de la fouille de données en bioinformatique [BIC 10], ceci en raison du fait que l‘on doit réaliser une classification pour pouvoir assigner la fonction correspondante à chaque séquence. En effet, grâce aux tâches de la fouille de données telles que la classification et le clustering, il est plus facile d‘apporter des solutions prometteuses aux problèmes bioinformatiques en association avec des méthodes computationnelles.
1.2.
Les fondements biologiques de la Bioinformatique
Il est indispensable d‘avoir des pré-requis biologiques nécessaires pour le développement et l‘évaluation de modèles ou de techniques en bioinformatique. Ceci est effectué en ayant les connaissances basiques sur les principes fondamentaux de la biologie moléculaire tels que les structures des gènes et des protéines...
Dans notre étude, notre intérêt s‘est porté sur la branche de la protéomique qui découle de la génomique. De ce fait, nous n‘allons aborder que les notions fondamentales de la protéomique qui sont les protéines et les vingt acides aminées natifs qui composent leurs séquences.
1.2.1.
De la génomique vers la protéomique
La compréhension du fonctionnement d‘une cellule vivante suppose celle des mécanismes moléculaires complexes qui sous-entendent les diverses activités cellulaires. Tous les gènes d‘un organisme, ou son génome, constituent une base de données statique et spécifique de l‘être vivant. A partir d‘un génome unique, chaque type cellulaire d‘un organisme exprimera un ensemble de protéines (voir Figure 1.1), ou protéome, qui variera en fonction de l‘environnement des cellules.
La synthèse des protéines comprend deux étapes
- La transcription permet de copier l‘ADN en ARN messager (ARNm), elle se déroule dans le noyau
- La traduction correspond au décodage de l‘information portée par l‘ARNm en polypeptidesreliésen protéines
1
Une homologie désigne un même caractère observé chez deux espèces différentes, qui a été hérité d'un ancêtre commun mais dont la fonction chez chacune des espèces n'est pas forcément la même.
La génomique et la protéomique sont intrinsèquement « globales », dans le sens où des centaines, si ce n‘est des milliers de bases de données, de bases de connaissances, de programmes informatiques et de bibliothèques de documents sont disponibles via Internet et sont utilisés par des chercheurs et des développeurs à travers le monde dans le cadre de leurs travaux.
Figure 1.1. Synthèse des protéines.
Comme les protéines sont les principaux acteurs finaux des processus biologiques, leurs études peuvent offrir la vision la plus pertinente du fonctionnement d‘une cellule vivante [DZI 10].
La protéomique désigne la science qui étudie les protéomes, c'est-à-dire l'ensemble des protéines d'une cellule, organite, tissu, organe ou organisme à un moment donné et sous des conditions données. Dans la pratique, la protéomique s'attache à identifier les protéines extraites d'une culture cellulaire, d'un tissu ou d'un fluide biologique, leur localisation dans les compartiments cellulaires, leurs modifications post-traductionnelles ainsi que leur quantité. Elle peut également permettre de quantifier les variations de leur taux d'expression en fonction du temps, de leur environnement, de leur état de développement, de leur état physiologique et pathologique, de l'espèce d'origine. Elle étudie aussi les interactions que les protéines ont avec d'autres protéines, avec l'ADN ou l'ARN ainsi que les fonctions de chaque protéine [BER 07].
1.2.2.
Les protéines
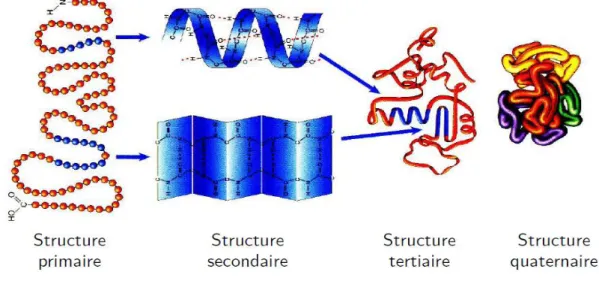
Les protéines représentent l‘une des classes moléculaires les plus importantes dans les organismes vivants. Leurs fonctions incluent la catalyse de processus métaboliques sous la forme d'enzymes, elles jouent un rôle important dans la transmission du signal, les mécanismes de défense, et de transport de molécules, et elles sont utilisées comme matériaux de construction, par exemple dans les cheveux (la protéine de la kératine). Chaque protéine est une macromolécule produite par un organisme vivant. Les protéines sont formées de chaînes d‘acides aminés liées par des liaisons peptidiques. Elles sont généralement représentées sous forme de séquences. Elles se replient en structures tridimensionnelles plus ou moins stables (voir Figure 1.2). Les protéines ont des tailles de plusieurs centaines d‘acides aminés. Plus spécifiquement, les petites chaînes sont appelées peptides, les protéines étant des polypeptides pouvant être réunies par des ponts disulfures.
Figure 1.2. Les différentes structures d‘une protéine.
Les protéines se répartissent en trois classes générales, sur la base de leur structure tridimensionnelle globale et sur la base de leur rôle fonctionnel, en fibreuse, membranaire, et globulaire [DZI 10].
Les protéines fibreuses ont tendance à être de longues molécules étroites. Elles sont utilisées pour construire les structures macroscopiques, en particulier les structures à l'extérieur des cellules. Elles ont également tendance à avoir un rôle structurel, même si certaines ont ainsi des fonctions plus actives.
Les protéines membranaires comprennent une classe unique de protéines. Pour les protéines membranaires, une zone importante de la protéine doit être stable dans un environnement hydrophobe. Ceci est généralement réalisé en ayant des chaînes latérales non polaires sur des zones de surface spécifiques de la protéine. En raison de cette surface hydrophobe exposée, et parce que de nombreuses protéines membranaires sont déstabilisés par l'élimination de la membrane, la plupart des protéines membranaires sont difficiles à manipuler. Par conséquent, l'information structurelle n‘est disponible que pour un nombre relativement faible de ces
protéines, bien que de nouvelles techniques aient permis de déterminer la structure tridimensionnelle pour un nombre croissant de protéines membranaires au cours des dernières années.
Les protéines globulaires comprennent le type le plus varié de protéines. Les protéines globulaires sont solubles en solution aqueuse, elles ont généralement des résidus polaires à la surface et des résidus hydrophobes à l'intérieur. Les protéines globulaires ont souvent des structures stables qui se prêtent à la détermination de structure d‘elles-mêmes. Cela signifie qu‘il y a plus d'informations sur la structure tridimensionnelle pour les protéines globulaires que pour toutes les autres classes de protéines combinées.
1.2.3.
Les acides aminés
Un acide aminé est une petite molécule élémentaire des protéines. Il en existe 20 formes différentes synthétisées par voie ribosomale dans le monde du vivant [KER 08].
Code en une lettre Abréviation Nom
A Ala. Alanine
R Arg. Arginine
N Asn. Asparagine
D Asp. Acide aspartique
C Cys. Cystéine
Q Gln. Glutamine
E Glu. Acide glutamique
G Gly. Glycine H His. Histidine I Ile. Isoleucine L Len. Leucine K Lys. Lysine M Met. Méthionine F Phe. Phénylalanine P Pro. Proline S Ser. Sérine T Thr. Thréonine W Trp. Tryptophane Y Tyr. Tyrosine V Val. Valine
La configuration générale des acides aminés naturels est caractérisée par un groupe amine et un groupe carboxyle autour d'un atome de carbone central α (Figure 1.3). La chaîne latérale respective de chaque acide aminé détermine les propriétés chimiques, telles que l‘hydrophobie, la polarité, ou l‘acidité (Figure 1.4).
Figure 1.3. Configuration générale d‘un acide aminé [ALB 08].
Des liaisons peptidiques connectent des acides aminés individuels dans une chaîne polypeptidique. Chaque acide aminé est lié via la liaison amide de l'acide de son groupe α carboxylique au groupe amine α de l'autre [ALB 08]. Par conséquent, ils ont des extrémités N- et C- terminales. La structure primaire polypeptidique, c'est-à-dire, la séquence d'acides aminés de l'extrémité N- à l'extrémité C-terminale, peut contenir entre trois et plusieurs centaines d'acides aminés. Chaque acide aminé dans la chaîne polypeptidique est abrégé soit par un code à trois lettres ou une lettre (voir Tableau 1.1).
Les propriétés structurales et physico-chimiques de chaque acide aminé sont très variées [KER 08]. Cependant, en se basant sur la composition chimique, on peut regrouper les acides aminés en 8 familles :
1. Les acides aminés aliphatiques dont le radical est une chaîne hydrogéno carbonée apolaire.
2. Les acides aminés hydroxylés qui portent un groupe alcool. Ils sont polaires, mais non chargés et neutres.
3. Les acides aminés représentés uniquement par la proline. La chaîne latérale est repliée et établit une liaison covalente avec l‘atome d‘azote du groupement amine.
4. Les acides aminés soufrés qui comportent un atome de soufre dans la chaîne latérale. L‘un d‘eux, la cystéine est un thiol, deux molécules de cystéine peuvent établir une liaison covalente entre leurs atomes de soufre et établir une liaison supplémentaire dans la chaîne protéique.
5. Les acides dicarboxyliques portent un groupement acide organique à l‘extrémité de leur chaîne latérale. Ils sont donc polaires, chargés négativement (à pH neutre) et également acides. 𝐻 𝑹 𝐻2𝑁 𝐶 𝐶𝑂𝑂𝐻 Carbonne centrale α Groupe amine Groupe de chaîne latérale Groupe carboxyle
6. Les acides amidés, il s‘agit des versions amidées des acides aminés du groupe précédent. Le groupement OH de l‘acide carboxylique est remplacé par un groupement NH3.
7. Les acides diaminés. La chaîne latérale porte un groupement aminé. Ils sont donc polaires, chargés positivement (à pH neutre) et basiques.
8. Les acides aminés aromatiques comportent un cycle aromatique dans leur chaîne latérale. Ces molécules sont non chargées et fortement apolaires.
Figure 1.4. Diagramme de Venn des propriétés des acides aminés [SEL 08].
La machinerie cellulaire produit des protéines en assemblant des acides aminés à partir d‘un code génétique situé dans les chromosomes. Les chromosomes sont des brins d‘ADN (Acide Désoxyribonucléique) dont les bases élémentaires sont des nucléotides. L‘ADN est une longue molécule formée de deux brins complémentaires dont la structure forme une double hélice qui lui permet de se dupliquer et de se répliquer lors de divisions cellulaires. En d‘autres termes, une séquence nucléotidique d‘un gène détermine la séquence d‘acides aminés de la protéine.
1.3.
Généralités sur la bioinformatique
La bioinformatique dérive des connaissances par des analyses informatiques de données biologiques et moléculaires. C‘est une branche en plein essor de la biologie, hautement interdisciplinaire, elle utilise les techniques de l‘informatique, des statistiques, des mathématiques, la chimie, la biochimie et d‘autres domaines [MAR 08]. Les données biologiques peuvent être des informations stockées dans les séquences d‘ADN, des résultats
expérimentaux provenant de diverses sources, des séquences protéiques tridimensionnelles, des tableaux d‘expression des gènes, des statistiques de patients, etc. Une partie importante de la recherche en bioinformatique est dédiée au développement de méthodes de stockage, d‘extraction et d‘analyse de ces données.
1.3.1.
Historique de la bioinformatique
Le terme de bioinformatique date du début des années 80. Cependant, le concept sous-jacent de traitement de l'information biologique est bien plus ancien. Durant les années 60 (voir Tableau 1.2), la biologie moléculaire a eu besoin de modélisation formelle, ce qui a mené à la création des biomathématiques. L'apparition de la bioinformatique n'est donc pas une conséquence de la génomique (séquençage d'un génome et son interprétation), mais plutôt une de ses fondations.
Année Evènement
1965 Première compilation de protéines (―Atlas of Protein Sequences‖) : Margaret Dayhoff et al.
1967 Article : ―Construction of Phylogenetic Trees‖– Fitch & Margoliash
1970 Algorithme pour l‘alignement global des séquences : Needleman & Wunsch 1974 Programme de prédiction de structures secondaires des protéines : ―Prediction
of Protein Conformation‖ – Chou & Fasman
1978 Premières bases de données : EMBL, GenBank, PIR 1981 370.000 nucléotides et 270 séquences à GenBank
Programme d‘alignement local de séquences : Smith & Waterman
1985 ―FASTA‖: Programme d‘alignement local de séquences – Pearson & Lipman 1990 ―BLAST‖: Programme d‘alignement local de séquences – Altschul et al. 1991 ―Grail‖: Programme pour la localisation de gènes – Mural et al.
1992 - Fondation du Centre de recherche SANGER: il réalise la moitié de la ―production‖ mondiale.
- Publication de la 2e carte génétique du génome humain, établie par le Généthon à partir de 814 fragments génomiques
1993 ―SRS‖ : logiciel d'interrogation multi-banques accessible sur le web – Etzold & Argos
1998 Séquençage de 2 millions de nucléotides par jour 2001 Séquence ―premier jet‖ complète du génome humain Février 2015 Plus de 1.143.000.000.000 nucléotides !
Tableau 1.2. Historique de la bioinformatique.
La bioinformatique a gagné une importance programmatique significative au cours du programme « European Commission‘s Fifth Framework Programme (FP5 2007) » de 1998
jusqu‘en 2002, et dans la communauté scientifique, où l‘activité dans ce domaine est principalement liée au développement du stockage et l‘organisation de quantités croissantes de données produites par des technologies génétiques de plus en plus sophistiquées, en liaison avec les besoins en infrastructures qui accompagnent la recherche génétique de base [MAR 08].
1.3.2.
Les objectifs de la bioinformatique
Le rôle de la bioinformatique est d‘aider les biologistes dans la collecte et le traitement des données génomiques afin d‘étudier la fonction des gènes et des protéines. Un autre rôle important de la bioinformatique est d‘aider les chercheurs des compagnies pharmaceutiques à élaborer des études détaillées des fonctions des protéines (voir Figure 1.5) afin de faciliter la conception de médicaments [COH 04, RAO 08].
Les objectifs de la bioinformatique peuvent se résumer dans ce qui suit :
Collecter et stocker des informations dans des bases de données, accessibles en ligne. - Explosion de la quantité de données biologiques nécessitant des outils de stockage
adaptés.
Fournir des outils de comparaison de séquences protéiques et nucléotidiques. - Identifier une séquence en la comparant aux séquences d‘une base de données. - Déterminer le degré de similitude entre deux séquences.
- Repérer des motifs structuraux.
Fournir des outils de traduction de séquences. - Simplifier les tâches de traduction.
- Proposer plusieurs possibilités de protéines pour une même séquence. - Repérer les exons2/introns.
Fournir des outils de prédiction physiologique et fonctionnelle et de prédiction expérimentale.
La recherche de similarité est au centre de la bioinformatique. Quand une séquence est donnée (nucléotides ou d'acides aminés), on effectue généralement une recherche de similitude avec les bases de données qui comprennent tous les génomes disponibles et les protéines connues. Habituellement, la recherche donne de nombreuses séquences avec des degrés variés de similitudes. Il appartient à l'utilisateur de sélectionner celles qui pourraient bien se révéler être homologues.
La bioinformatique a ainsi rendu possible la cartographie complète du génome humain3 et des génomes de nombreux autres organismes en un peu plus d‘une décennie. Ces découvertes,
2
Les exons sont les fragments d‘un ARN primaire qui se retrouvent dans l‘ARN cytoplasmique après épissage (processus par lequel les ARN transcrits à partir de l'ADN génomique peuvent subir des étapes de coupure et ligature qui conduisent à l'élimination de certaines régions dans l‘ARN final), par opposition aux introns qui sont des fragments d‘ARN primaire éliminés au cours de l‘épissage.
ainsi que les efforts actuels pour déterminer les fonctions des gènes et des protéines ont amélioré les capacités de comprendre les maladies animales, végétales et humaines et de trouver de nombreux traitements [RAO 08].
Figure 1.5. Analyse des données biologiques.
1.3.3.
Les concepts et techniques de la bioinformatique
La tâche majeure de la bioinformatique est de permettre d‘identifier les fonctions d‘un gène ou d‘une protéine à partir de données existantes. Puisque les données sont variées, incomplètes, bruyantes et couvrent une variété d‘organismes, il y a un recours constant aux principes biologiques afin de filtrer les informations utiles [COH 04]. Il y a différentes techniques qui conduisent à une meilleure compréhension de la fonction des gènes et des protéines, telles que :
La construction évolutive d’arbre phylogénétique : ces arbres sont souvent construits
après comparaison de séquences appartenant à différents organismes d‘une même espèce. Les arbres regroupent les séquences selon leur degré de similitude. Ils servent de guide pour le raisonnement sur la façon dont les séquences ont été transformées au courant de l‘évolution. Par exemple : ils déduisent l‘homologie de la similitude et peuvent écarter des hypothèses erronées qui sont en contradiction avec le processus connu de l‘évolution.
3
La cartographie du génome est la détermination de la position d‘un locus (gène ou marqueur génétique) sur un chromosome en fonction du taux de recombinaison génétique.
Analyse de séquences
Séquence nucléotidique
Gène Protéine Fonction biochimique
Activité biologique
Prédiction/simulation expérimentale
Détection de motifs dans les séquences : il y a certaines parties de séquences de nucléotides et des séquences d‘acides aminés qui doivent être détectées. Il y a deux exemples principaux qui sont la recherche de gènes dans l‘ADN et la détermination des sous-composants de séquences d‘acides aminés (structure secondaire). Il existe plusieurs moyens pour effectuer ces tâches, un grand nombre d‘entre eux sont basés sur l‘apprentissage automatique et incluent les grammaires probabilistes ou les réseaux neuronaux.
Déterminer des structures 3D à partir de séquences : les problèmes, en bioinformatique, qui se rapportent aux structures tridimensionnelles impliquent des calculs difficiles à réaliser. La détermination de la forme d'ARN à partir de séquences nécessite des algorithmes de complexité cubique. L'inférence des formes de protéines à partir de séquences d'acides aminés reste, à ce jour, un problème non résolu.
Déduction de la régulation cellulaire : la fonction d‘un gène ou d‘une protéine est mieux décrite par son rôle. Les gènes interagissent les uns avec les autres, les protéines peuvent également prévenir ou aider à la production d‘autres protéines. Les modèles disponibles de la régulation cellulaire peuvent être discrets ou continus. Il y a habituellement une distinction entre la simulation et la modélisation cellulaire.
Déterminer la fonction de protéine et les voies métaboliques : c‘est l‘un des domaines les plus difficiles de la bioinformatique et pour lequel il n'y a pas beaucoup de données disponibles. L'objectif ici est d'interpréter les annotations humaines pour la fonction des protéines et également de développer des bases de données représentant des graphiques qui peuvent être interrogés pour l'existence de nœuds (les réactions à préciser) et les chemins (en précisant les séquences de réactions).
Assembler les fragments d’ADN : les fragments fournis par séquençage sont
assemblées à l'aide d'ordinateurs. La partie la plus délicate de cet assemblage est que l'ADN a de nombreuses régions répétitives et les mêmes fragments peuvent appartenir à différentes régions. Les algorithmes d'assemblage de l'ADN sont surtout utilisés par les grandes entreprises.
En plus de ses différentes techniques, la bioinformatique comprend deux concepts clés, les bases de données biologiques et la comparaison de séquences. En effet, les bases de données représentent le point de départ des recherches et à partir de ces bases de données, une comparaison de séquences peut être réalisée en comparant la séquence requête avec l‘ensemble des séquences de la base de données correspondante. Les résultats obtenus peuvent orienter le chercheur ou lui donner une idée de la fonction de la séquence requête. Nous allons donc aborder, dans ce qui suit, ces deux concepts.
1.3.3.1. Les bases de données biologiques
Le concept le plus important pour la bioinformatique appliquée est la collecte de données de séquence et son information biologique associée. Par exemple, les projets de séquençage du génome génèrent quotidiennement une très grande quantité de données dans le monde entier. Pour utiliser ces données de façon appropriée, un système de dépôt structuré de ces données est nécessaire, mais les données devraient également être accessibles aux personnes
intéressées [SEL 08]. De ce fait, des bases de données en ligne ont vu le jour, elles sont accessibles gratuitement à tous, ce qui représente un avantage considérable. Différents chercheurs à travers le monde peuvent travailler sur le même ensemble de données et ainsi pouvoir comparer leurs résultats. Ce concept a tellement pris de l‘ampleur, que le journal
Nucleic Acids Research consacre chaque année un numéro entier à toutes les bases de
données biologiques disponibles, qui sont enregistrées sous forme de tableau avec les liens respectifs. En outre, pour un certain nombre de bases de données, des articles originaux décrivent leurs fonctions.
Selon le type de données qu‘elles contiennent, deux catégories de bases de données biologiques peuvent être distinguées. Les bases de données primaires qui contiennent des informations de séquences primaires (de nucléotides ou de protéines), des informations d'annotation relatives à la fonction, la bibliographie ainsi que des renvois vers d'autres bases de données, etc. Contrairement aux bases de données secondaires qui résument les résultats de l'analyse des bases de données de séquences de protéines primaire. L'objectif de ces analyses est de tirer des caractéristiques communes pour les classes de séquences, qui peuvent à leur tour être utilisés pour la classification des séquences inconnues (annotation).
Les bases de données primaires contiennent différentes bases de données protéiques et nucléiques. Il y a trois grandes bases de données nucléiques très connues à travers le monde, la base de données GenBank du centre national d‘information technologique américain (National Center of Biotechnology Information – NCBI), qui contient plus de 76 million de séquences nucléiques [SEL 08]. La base de données EMBL (European Molecular Biology Laboratory) de l‘institut bioinformatique européen (European Bioinfomatics Institute – EBI), et la base de données DDBJ (DNA Data Bank of Japan) du centre pour l‘information biologique du Japon (Center for Information Biology – CIB). Ces trois organismes : NCBI, EBI et CIB comprennent des bases de données nucléiques internationales et synchronisent leurs bases de données toutes les 24h.
L‘une des plus importantes bases de données protéiques est la base de données Swiss-Prot de l‘institut Suisse de bioinformatique (Swiss Institute of Bioinformatics – SIB), elle a la spécificité d‘être organisé à la main, c'est-à-dire, chaque enregistrement dans la base est vérifié par un spécialiste et, si nécessaire, par rapport aux données de la littérature, elle contient plus de 500.000 séquences annotées. Cette base est aujourd‘hui incorporée à la base de données UniProt qui comprend une autre base de données, TrEMBL dont les séquences ont été obtenues suite au processus de traduction de l‘ADN (voir Figure 1.1) et qui contient plus de 48 millions de séquences annotées automatiquement qui attendent d‘être vérifiées par des spécialistes. Une autre base de données protéique très connue et très utilisée, la base de données PDB (Protein Data Base) qui contient principalement les structures tridimensionnelles de protéines et même d‘acides nucléiques.
Les bases de données secondaires contiennent une multitude de bases de données spécifiques aux résultats recherchés telles que la base de données PROSITE, qui représente une base de données de sites fonctionnels (motifs) et de domaines, elle permet de scanner une séquence protéique dans le but d‘identifier sa région conservée (motif) ou son domaine. Dans la même

![Figure 1.4. Diagramme de Venn des propriétés des acides aminés [SEL 08].](https://thumb-eu.123doks.com/thumbv2/123doknet/2030906.4128/28.892.219.710.308.697/figure-diagramme-venn-proprietes-acides-amines-sel.webp)
![Figure 1.7. Quelques applications de la fouille de données en Protéomique [INZ 10]](https://thumb-eu.123doks.com/thumbv2/123doknet/2030906.4128/38.892.137.742.660.1125/figure-applications-fouille-donnees-proteomique-inz.webp)
![Figure 2.3. Processus de détection du NSA [DAS 09]. Début](https://thumb-eu.123doks.com/thumbv2/123doknet/2030906.4128/57.892.319.621.740.1070/figure-processus-detection-nsa-das-debut.webp)
![Figure 2.5. Modèle de la théorie du danger [AIC 14].](https://thumb-eu.123doks.com/thumbv2/123doknet/2030906.4128/62.892.289.771.144.523/figure-modele-theorie-danger-aic.webp)