Thermodynamics of Supramolecular Associations with Macrocyclic
Water-Soluble Hosts
Ludovic Garnier, Christine Bonal,
*
and Patrice Malfreyt
CNRS, SIGMA Clermont, Institut de Chimie de Clermont-Ferrand, Université Clermont Auvergne, F63000 Clermont-Ferrand, France
*
S Supporting InformationABSTRACT: The thermodynamic study of the complexation of the β-cyclodextrins and p-sulfonatocalix[n]arenes (CnS) with the 4-aminoazobenzene was reported and was carried out by molecular dynamics simulations. We determined the whole thermodynamic properties (K,ΔrG°, ΔrH°, and TΔrS°) using the potential of mean
force (PMF) technique and more precisely the adaptive biasing force method. Depending on both the nature of the host molecule and the pH of the solution, the PMF profiles present different shapes and energy minima. Considering the complexity of these PMF profiles, we are also interested in the structural properties of these
associations. Hence, we calculated hydrogen bonds, Lennard-Jones and electrostatic energies, the number of atoms of the guest molecule inserted inside the cagelike host molecule, and the number of water molecules expelled from the cavity.
1. INTRODUCTION
Host−guest systems consist generally of two parts, the molecular receptors (usually small molecules with a hydro-phobic cavity such as cyclodextrins (CD),1 calixarenes,2 or cucurbiturils3−5) and the guests that are able to bind with both high affinity and/or selectivity. For technological applications in the field of photoswitchable materials,6−11 azobenzene derivatives have been widely investigated and cyclodextrins have been largely used as hosts for these derivatives. In the literature, some stability constants are available revealing some discrepancies.12−18In fact, supramolecular assemblies typically provide weak force associations that are quite difficult to detect and quantify with both precision and accuracy. Moreover, the experimental conditions are usually so different that the values can hardly be compared.
We previously reported experimental and computational investigations on the association of water-soluble macrocyclic hosts (β-cyclodextrin and p-sulfonatocalixarenes) with 4-aminoazobenzene (4AA)19 in water (Figure 1). UV−visible
spectroscopy was used to provide the whole thermodynamic characterizations of the association (ΔrG, ΔrH, and TΔrS). Molecular simulations were also performed to interpret experimental results and to obtain structural data for these
complexes. The pH effect was studied. Whereas the
thermodynamic parameters found for β-cyclodextrin were almost identical at the two pH values studied (pH 1 and 7), these parameters differed significantly for p-sulfonatocalixar-enes. Stable inclusion complexes with calixarenes were found at acidic pH, but our investigations were unable to confirm the inclusion of the guest at neutral pH. Knowing that the binding abilities of such calixarenes are dramatically pH-controlled, a weaker association was indeed expected.20 However, a
clarification is required to provide a definitive answer regarding the possible binding of the 4AA to calixarenes at neutral pH. Molecular description combined with thermodynamic characterization using numerical experiments is an efficient approach for the study of the binding process.21−26 In the present paper, we have re-examined the association process using the free energy approach to obtain a thermodynamic characterization using numerical experiments. This approach corresponds to the calculation of the free energy along a reaction coordinate. The calculation of the potential of mean force (PMF) along complex chemical reactions was originally Received: July 11, 2019
Accepted: September 11, 2019
Published: October 1, 2019
Figure 1. Representation of (a) β-Cd, (b) 4AA, and (c) p-sulfonatocalix[4]arene.
Article http://pubs.acs.org/journal/acsodf Cite This:ACS Omega 2019, 4, 16899−16905
copying and redistribution of the article or any adaptations for non-commercial purposes.
Downloaded via 118.70.52.165 on June 24, 2021 at 14:15:38 (UTC).
introduced by Kirkwood in 1935.27The comparison between the binding properties previously obtained from UV−visible spectroscopy for β-cyclodextrin and p-sulfonatocalixarenes with 4-aminoazobenzene in water at acidic pH allowed the validation of the computational procedures. As a result, we have given a quantitative answer to the possible association between p-sulfonatocalixarenes and 4-aminoazobenzene in water at neutral pH.
Section 2discusses the main results, and our conclusions are given inSection 3. Section 4describes the calculation of the potential of mean force using the adaptive biasing force (ABF) and of the corresponding thermodynamic properties of association. The computational details of the molecular dynamics are also provided.
2. RESULTS AND DISCUSSION
2.1. β-Cyclodextrin/4AA. The potential of mean force characterizing the association process of the 4AA with the β-cyclodextrin is shown inFigure 2 at pH = 7. No significant
changes are observed by changing the pH in concordance with our experimental results.19 The Gibbs free energy profile in
Figure 2shows some local minima characterized by separation distances that clearly implies the insertion of the 4AA into the CD cavity in line with our previous results. Because the azobenzene moiety is longer than the cyclodextrin cavity, the 4AA is included longitudinally in theβ-cyclodextrin cavity in perfect agreement with some reported NMR results.15 Interestingly, three quite different local minima were found; the first at the distance of −1.3 Å is the deeper free energy minimum. It corresponds to the configuration in which the first phenyl ring of the azobenzene moiety is inside the cavity with the hydrophilic head, the amino group, exposed to water. The second minimum at 0.75 Å corresponds to the configuration in which the two phenyls rings of azobenzene are exposed to the bulk water. Finally, in the third minimum at 3.95 Å, it is the second phenyl ring that is included in the cavity of β-cyclodextrin.
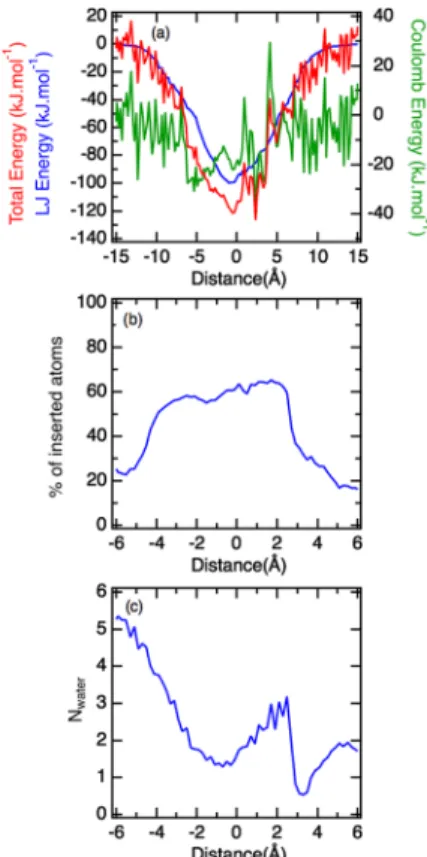
To explain the driving forces ofβ-cyclodextrin complexation, we report inFigure 3a the energy contributions and inFigure 3b the percentage of inserted atoms into the CD cavity at different separation distances. The number of water molecules inside the cavity of CD is also detailed along the coordinate reaction in Figure 3c. As expected for an insertion complex with theβ-CD, the van der Waals interaction is predominant
over the electrostatic energy contribution. A strong correlation was also observed between the LJ contributions and the number of inserted atoms. This contribution is more favorable between the distance of−4 and 4 Å of the two centers of mass when the percentage of inserted atoms is maximum. Note that the Lennard-Jones (LJ) energy minimum corresponds to the first deeper Gibbs free energy minimum of the PMF at −1.3 Å (Figure 2). Regarding the number of water molecules inserted into the cavity, two minimums are shown at−0.7 and 3.3 Å, respectively (Figure 3b). These separation distances are in line with the two local minima of the potential of mean force corresponding to the locations of the two phenyl rings of azobenzene within the cavity. Clearly, these conformations involve larger dehydration of the host. We also observe that the cavity recovers some water molecules when the two phenyl groups of 4AAB are exposed to water; this situation corresponds to the second minimum at 0.75 Å in the free energy profile (Figure 2).
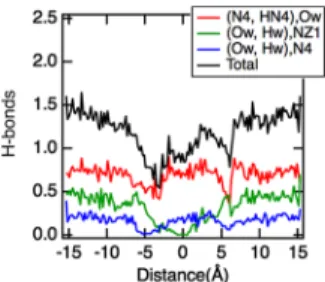
To conclude on these aspects, these results indicate that the more negative free energy minimum at −1.3 Å in the free energy profile given inFigure 2is a result of the combination of both the larger dehydration of the cavity and the more negative LJ energy contributions. Finally, for the same system, we have also reported the number of hydrogen bonds formed by the 4AA with water molecules as a function of the separation distance of the two centers of mass as illustrated in
Figure 4. Obviously, as 4AA penetrates into the cavity of the host, the total number of hydrogen bonds formed by the 4AA
Figure 2.Gibbs free energy profile obtained for the inclusion complex (β-Cd, 4AA) as a function of the reaction coordinate. In the inset, the PMF is shown in the whole range of sampled separation distances.
Figure 3. (a) Energy contributions as a function of the distance between centers of mass for (β-Cd, 4AA). The blue curve corresponds to the Lennard-Jones energy, the green to the electrostatic part, and the red is the sum of both contributions. (b) Percentage of 4AA atoms inserted inside theβ-Cd cavity as a function of the distance between centers of mass. (c) Number of water molecules inside the host cavity as a function of the distance between the host and the guest.
decreases. As shown inFigure 4, this decrease is mainly due to the azo group that is deeply inserted into the cavity, preventing any formation of hydrogen bonds.
2.2. Calixarenes/4AA. Now, we focus on the potential of mean force characterizing the association process of the 4AA into p-sulfonatocalixarenes in both neutral and acidic solutions (Figure 5). It should be noted that compared to the previous
system, the 4AA does not pass through the cavity of the calixarenes. The results obtained for C4S and C6S are rather similar in terms of the shape and well depth of the PMF curve. However, the PMFs are strongly dependent on the pH. Indeed, the PMF obtained in acidic solution shows deeper minimum than in neutral medium and the location of 4AA into the cavity is always deeper at pH = 1 (at 4.1 and 2.5 Å for C4S and C6S, respectively) than that at pH = 7 (at 5.35 and 3.35 Å for C4S and C6S, respectively). Interestingly, for both C4S and C6S at pH = 7, one can observe a small energy barrier (about 5 kJ mol−1) that precedes the local free energy minimum.
We propose here to focus our attention on the C4S. To further understand these PMFs, the partitioning of the free energy profiles into the Lennard-Jones and electrostatic parts of the energy contributions is reported as a function of the separation distance inFigure 6for (C4S, 4AA) in water. The total energy contributions are also reported in the samefigure. At both pH values, the variations of the LJ contributions as a
function of the distance are similar. When the 4AA approaches the C4S, these contributions become largely favorable, with the insertion of the 4AA into the cavity starting at a distance of about 8 Å.Figure 7a shows the percentage of inserted atoms
into the C4S cavity, andFigure 7b shows the number of water molecules inside the host cavity. Interestingly, the more negative LJ contribution at the Gibbs free energy minima in acidic medium corresponds to 40% of the guest insertion, whereas it is only 20% in neutral solution. In both cases, at the Gibbs free energy minima, the dehydration of the cavity represents at least 50% of the total number of the water molecules inside the cavity.
According to the PMFs discussed above, it is clear that changing the pH impacts significantly the electrostatic
Figure 4.Number of hydrogen bonds (H-bonds) formed by the 4AA with water molecules. The colored curves correspond to a given type of H-bond, whereas the black curve corresponds to the sum of all contributions.
Figure 5.Free energy profiles obtained for (a) (C4S, 4AA) and (b) (C6S, 4AA). The plain line corresponds to acidic pH, whereas the dotted line refers to the neutral medium.
Figure 6.Energy contributions as a function of the distance between centers of mass for (C4S, 4AA) at (a) acidic pH and (b) neutral pH. The blue curve corresponds to the Lennard-Jones energy, the green to the electrostatic part, and the red is the sum of all contributions. The dotted lines represent the location of the deep minimum in the PMF profile for (C4S, 4AA).
Figure 7.(a) Percentage of 4AA atoms inserted inside the C4S cavity as a function of the distance between centers of mass. (b) Number of water molecules inside the host cavity as a function of the distance between the host and the guest. The dotted lines represent the location of the deep minimum in the PMF profile for (C4S, 4AA).
interactions. In acidic medium, these contributions are negative and gradually decrease when the guest approaches the cavity. On the contrary, the electrostatic part is always positive along the host−guest separation distance in the neutral medium. Interestingly, the penetration of 4AA into the cavity (from a distance of 8 Å) is associated with an electrostatic energy cost in both cases. Note that this cost is in the range of 400 kJ mol−1at pH = 1, whereas it is rather on the order of 40 kJ mol−1at pH = 7. However, the electrostatic energy remains largely negative and it is partially compensated by the negative Lennard-Jones energy contribution in acidic solution. This is no longer true in the neutral medium as shown in the total energy contributions (Figure 6), as the energy balance is clearly unfavorable. This leads to the small energy barrier mainly observed in the PMF curves obtained in the neutral medium.
We propose here to explain these unfavorable electrostatic terms observed at 8 Å at both pH values and discussed above. To do so, we assume that the guest changes its orientation to form the energetically favored inclusion structure for complexes. In acidic solutions, the approach of the guest to the cavity allows strong electrostatic interactions of protonated 4AA with the sulfonate groups, with the amino group of the guest being oriented toward the sulfonate groups of the C4S. This orientation may also promote hydrogen bond formation between the amino group of 4AA and oxygen atoms of sulfonate groups. Upon complexation, the penetration into the host cavity of the phenyl part of the guest rather than the amino group will enhance its hydrophobic interactions with C4S. As a consequence, the orientation of the guest must change. To validate this hypothesis, we will deeply investigate the orientation coefficient α and the hydrogen bonds in the following part.
Further insight into the orientation of the guest as a function of the separation distance may be obtained from the calculation of α value. This parameter depends on the orientation of the guest within the calixarene cavity as explained in Figure S5 (Supporting Information). Three zones are defined, and at larger separation distances of the two centers of mass (zone C), a significantly larger α value (α > 1) is calculated. This result confirms a privileged position of the amino group toward the sulfonate groups, especially true in acidic solution (with the protonated 4AA). In zone B, theα value fluctuates in agreement with the change in the guest orientation upon complexation. Finally, the insertion of the guest inside the cavity leads to a smaller value ofα (Zone A) that is thoroughly consistent with the penetration of the phenyl part of 4AA rather than the amino group.
Finally, we also report in Figure 8 the total number of hydrogen bonds as a function of the separation distance for both pH values. We observe that the total number of hydrogen bonds is fewer at pH = 7 than that at pH = 1. From 10 Å, the amino group of 4AA forms hydrogen bonds with the sulfonate group of C4S. Upon complexation, these H-bonds are replaced by H-bonds with the oxygens of the water due to the change in the 4AA orientation in agreement with thefluctuation of the α value. Once the insertion has been made, the amino group is able to recover hydrogen bonds with the sulfonate groups as shown in Figure 8. This leads to a favorable negative electrostatic energy obtained from 5 Å as shown inFigure 6. As a conclusion, the formation of H-bonds and the evolution ofα validate our hypothesis previously made.
Unambiguously, the small energy barriers obtained at 7.75 and 7.5 Å for C4S and C6S, respectively, at pH = 7 are related to the fact that the orientation of the guest is changed upon complexation. As already mentioned in theIntroductionpart, our previous results19 tend to show that the 4AA is not significantly complexed by both C4S and C6S in neutral medium. The calculation of the free energy profiles obtained here and the presence of these barriers give a quantitative answer regarding this complexation and confirm that there is no association between the calixarene and the 4AA in water, in perfect agreement with our experimental and simulation results.19 The same conclusions can be drawn for the (C6S, 4AA) association from the analysis ofFigures S2−S4(see the Supporting Information).
2.3. Thermodynamics Properties. The thermodynamic properties of the association calculated from the PMFs are reported in Table 1. As underlined in the methodological
section, the calculation of the thermodynamic properties requires the knowledge of the d parameter. The dependence of the calculation on the value of d is shown for each complex in
Figures S7−S9(see the Supporting Information). In the case of the complex studied here, the use ofeqs 7−9represents an approximation because of the nonsphericity of the
conforma-Figure 8. H-bonds formed as a function of the distance between centers of mass for (C4S, 4AA) association at (a) acidic pH and (b) neutral pH. The colored curves correspond to a given type of H-bond, whereas the black curve corresponds to the sum of all H-bonds. The dotted lines represent the location of the deep minimum in the PMF profile for (C4S, 4AA).
Table 1. Thermodynamic Properties of (Host, 4AA) Complexes Obtained from Free Energy Profilesa
host ΔrG° (kJ mol−1) ΔrH° (kJ mol−1) TΔrS° (kJ mol−1)
β-Cdb(pH = 7) −14 −21 −7 β-Cd (exp) −19 −8 11 C4Sc(pH = 1) −14 −25 −11 C4S (exp) −11 −25 −13 C6Sd(pH = 1) −10 −20 −10 C6S (exp) −14 −18 −4
aValue of d is given as a function of r
mindefined as the position of the
Gibbs free energy minimum.bd = rmin.cd = rmin+ 2.5 Å.dd = rmin+
tions of the guest and the host. However, we can estimate the thermodynamic properties that are listed inTable 1. For each calculation, we report the value of d, which remains reasonable for an association in terms of separation distances.
The calculation of the thermodynamic properties of the association from the PMFs shows a weak association in line with experimental interpretations. The complexes are charac-terized by the insertion of the guest into the host in line with negative values ofΔrH°. The values of the Gibbs free energy of
association are on the same order of magnitude as those obtained from spectroscopy. In the case of the calixarenes, the deviation between simulations and experiments does not exceed 10% on the values of the enthalpy. The largest deviation is obtained with theβ-Cd and can be explained by the fact that the reaction path is much more complicated for this association process. Indeed, the host molecule is then accessible from both sides and the guest is longitudinally inserted. These two features make the original assumptions of the spherical shapes of the guest and the host no longer valid. The deviation of the value of the enthalpy explains the change in the sign of the entropic term.
There are two predominant effects that influence the entropy effects: the loss of degrees of freedom and the desolvation of both guest and host molecules. From our simulation results, we saw that the entropic term is negative for all complexes. The loss of degrees of freedom is rather predominant. Nevertheless, for (BCD, 4AA) association, the value of the entropy is higher than the one for (CnS, 4AA) associations. This implies that the loss of degrees of freedom is higher for CnS than for BCD. These results are in perfect agreement with the ones we previously obtained.19
3. CONCLUSIONS
Molecular simulations have been performed to calculate the potential of mean force in the association between the 4AA and C4S, C6S, and β-CD in water. The calculation of PMF represents a valuable property to definitely conclude on the association between two molecules. The PMFs have been calculated using the method of the adaptive biasing force. Indeed, these PMFs can help in the interpretation of experiments when the experimental data do not allow a definitive conclusion of the association in neutral medium. The shape of the simulated PMF was rather complex, and additional simulations were performed to investigate the changes in the structure and energy along the reaction coordinate.
In a neutral medium, we have demonstrated that an energetic barrier of about 5 kJ mol−1prevents any association, as suggested by experiments. This is the main conclusion of this work. We have also explained the reasons for the formation of the energetic barrier from the analysis of the van der Waals and electrostatic energy contributions. In acidic medium, the simulations confirm a weak association and an insertion complex in terms of thermodynamic properties. These results agree with experiments showing that the process is enthalpically favored. We conclude that only a combined approach of molecular simulations (structure and energetics) and experiments is able to elucidate the formation of these weak complexes.
4. EXPERIMENTAL SECTION
4.1. Methodology. 4.1.1. Adaptive Biasing Force. The adaptive biasing force (ABF) method is an adaptative method that computes the potential of mean force w(ξ) along a reaction coordinateξ. It was designed by Darve et al.28,29 in 2001. Since then, many improvements have been proposed. For example, we could cite the extended-system adaptive biasing force (eABF) designed by Lesage et al.30 or more recently the meta-eABF designed by Fu et al.31 In ABF method, the total reaction coordinate ξ is divided in an appropriate number of bins. For each bin, the mean force, ⟨Fξ|ξ*⟩, is evaluated from a running average
ξ ξ ξ ⟨ | *⟩ = −Fξ * w d ( ) d (1)
By applying an opposite force−⟨Fξ|ξ*⟩, the total average force acting on this system becomes close to zero after a brief equilibration. This allows avoiding significant energy barriers. The reaction coordinate is then well sampled. Now, let us look at the way we can compute the potential of mean force (PMF) with this method. By considering that nk(Nstep) is the number
of samples collected in bin k after Nstep, the running average force Fξkacting on ξ in the bin k is expressed as
∑
ξ = ξ ξ = i k jjj y{zzz F n N t m t x 1 ( ) d d d d ( ) k k l n N l k step 1 ( ) k step (2) where ξ ξ(
m)
( )x dt dt l k d dcorresponds to the lth force sample when ξ is in the bin k. In the LAMMPS collective variable module, the average force for the ABF method is calculated from
α ξ
= ∇ ̃
ξ ξ
Fk ( )N G( ) (3)
with α(Nξ) being a scaling factor (going from 0 to 1)
depending on the number of samples Nξ and ∇G̃(ξ) the estimate of the gradient of free energy for the pointξ in the reaction coordinate. Finally, the Gibbs free energy along the reaction coordinate from a state a to b,ΔGa→b, can be obtained
by integrating the total biased force according to
∫
ξ ξ ξ∑
Δ = − ≈ − − ξ ξ ξ ξ → = G F k F N d ( ) k k k a b b a max 1 step, a a b m x (4)Nevertheless, if the sampling is poor for a given bin k (typically at the beginning of the simulation), it is possible to have a nonequilibrium effect due to poor estimation of ⟨Fξ|ξ*⟩ that leads to an incorrect bias. To avoid these effects, we multiply Fξk by a function R(nk(N
step)) = min(1, nk(Nstep)/n0). In
practice, we took n0= 200. The running average force becomes
∑
ξ = ξ ξ = i k jjj y{zzz F R n N n N t m t x ( ( )) ( ) d d d d ( ) k k k l n N lk step step 1 ( ) k step (5)4.1.2. Thermodynamic Properties. The thermodynamic properties of the association are calculated from the profile of the potential of mean force (PMF). The association constant32 Kacan be calculated using the following expression
∫
π = i− k jjjjj y{zzzzz K N r w h k T h exp ( ) d d a 0 A cyl B (6)where NA corresponds to the Avogadro constant, kB to the Boltzmann constant, and w(h) to the PMF profile. rcyl
corresponds to the mean radius of the cylinder where the guest can freely evolve for its in-plane movement when it is associated inside the cavity. This mean radius is calculated at each step h of the reaction coordinate. When the guest molecule is out of the cavity, we consider rcyl as a constant equal to the highest value (i.e., when the guest is placed above the bigger rim). The parameter d, which is introduced in the different integrations, defines the upper limit of the association in terms of separation distances. This is not an easy parameter to define especially when the shapes of the guest and hosts are no longer spherical and quite different between different association processes. We took the route of representing different integrals as a function of the d parameter in the Supporting Information (Figures S7−S9).
The derived properties of the association such as the Gibbs free energy, enthalpy, and entropy can be calculated using the following expressions
∫
π Δ ° = − = − i − k jjjjj ikjjjjj y{zzzzz y{zzzzz G k T K k T N r w h k T h ln ln exp ( ) d d r B a B 0 A cyl B (7)∫
∫
Δ ° = = − −(
)
(
)
H k T K T w h r h r h d ln d ( ) exp d exp d d w h k T d w h k T r B 2 a 0 cyl ( ) 0 cyl ( ) B B (8)∫
∫
∫
π Δ ° = + = + − − − i k jjjjj ikjjjjj y{zzzzz y{zzzzz(
)
(
)
T S k T K T k T K w h r h r h k T N r w h k T h d ln d ln ( ) exp d exp d ln exp ( ) d d w h k T d w h k T d r B 2 a B a 0 cyl ( ) 0 cyl ( ) B 0 A cyl B B B (9)4.2. Computational Details. As shown by experimental results,19 the stoichiometry of these complexes is 1:1. As a consequence, the simulation boxes were composed of one host, e.g., a calixarene (see Figure 1c for C4S) or a β-Cd (Figure 1a); one guest, e.g., a 4AAB (Figure 1b); and 2000 water molecules. To maintain charge neutrality in the box, we may add, if needed, some Cl−or Na+. Our simulation boxes were cubic with a box length of 42 Å. The periodic boundary conditions were applied in the three directions. A scheme of each molecule is given along with the nomenclature in the Supporting Information (Figures S1−S3).
Concerning the forcefields, we chose the TIP4P2005 model for water molecules33 and the General AMBER Force Field (GAFF) for hosts and guests molecules.34 However, for the guest molecule, the dihedral angle C−NN−C was taken from the work of Heinz et al.35 MD simulations were performed with the LAMMPS package.36 The simulation parameters were chosen as follows:
• The SHAKE algorithm37
was used to constrain H-based bonds and the HOH angle for water.
• Lennard-Jones crossing parameters were calculated using Lorentz−Berthelot rules (i.e., ϵ =ij ϵ ϵii jj and
σ =ij σ+σ
2
ii jj
, where i and j refer to the force centers andϵ and σ are the energy parameter and diameter of atoms of types i and j, respectively).
• Our simulations were performed in the NPT ensemble with a velocity-Verlet integrator and a timestep of 2 fs. • We used a Langevin38,39
thermostat with a relaxation time of 2 ps and kept the temperaturefixed at 300 K. • A Berendsen barostat40
was used with a relaxation time of 2 ps and a bulk modulus of 1000 atm. The pressure was keptfixed at 1 atm.
• We considered both long-range dispersion−repulsion tail correction41 and long-range electrostatic interac-tions. Long-range Coulomb interactions were calculated using the PPPM style (particle-particle particle-mesh) with a relative error force of 10−4 which maps atom charge to a three-dimensional mesh.42,43
Our simulations were composed of an equilibration phase of 1 ns and an acquisition phase of 140 ns for a PMF going from 0 to 15 Å (280 ns for PMF going from−15 to 15 Å).
■
ASSOCIATED CONTENT*
S Supporting InformationThe Supporting Information is available free of charge on the
ACS Publications website at DOI: 10.1021/acsome-ga.9b02136.
Nomenclature of studied molecules; (C6S, 4AA) association: results; inversion of the guest molecules; Thermodynamic properties (PDF)
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: Christine.Bonal@uca.fr. Tel: +33 (0)4.73.40.71.65. Fax: +33 (0)4.73.40.53.28.
ORCID
Christine Bonal:0000-0002-5612-3152
Patrice Malfreyt:0000-0002-3710-5418
Notes
The authors declare no competingfinancial interest.
■
REFERENCES(1) Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743−1754.
(2) Vicens, J.; Bohmer, V. Calixarenes: a Versatile Class of Macrocyclic Compounds; Topics in inclusion science v. 3; Kluwer Academic Publishers: Dordrecht, Boston, 1991.
(3) Ko, Y. H.; Kim, E.; Hwang, I.; Kim, K. Supramolecular Assemblies Built With Host-Stabilized Charge-Transfer Interactions. Chem. Commun. 2007, 1305−1315.
(4) Yuan, L.; Wang, R.; Macartney, D. H. Binding Modes of Cucurbit[6]uril and Cucurbit[7]uril with a Tetracationic Bis-(viologen) Guest. J. Org. Chem. 2007, 72, 4539−4542.
(5) Márquez, C.; Hudgins, R. R.; Nau, W. M. Mechanism of Host-Guest Complexation by Cucurbituril. J. Am. Chem. Soc. 2004, 126, 5806−5816.
(6) Wang, Y.; Ma, N.; Wang, Z.; Zhang, X. Photocontrolled Reversible Supramolecular Assemblies of an Azobenzene-Containing Surfactant withα-Cyclodextrin. Angew. Chem. 2007, 119, 2881−2884. (7) Griffiths, J. Photochemistry of Azobenzene and its Derivatives. Chem. Soc. Rev. 1972, 1, 481−493.
(8) Yamaguchi, H.; Kobayashi, Y.; Kobayashi, R.; Takashima, Y.; Hashidzume, A.; Harada, A. Photoswitchable Gel Assembly Based on Molecular Recognition. Nat. Commun. 2012, 3, No. 603.
(9) Nachtigall, O.; Kördel, C.; Urner, L. H.; Haag, R. Photo-responsive Switches at Surfaces Based on Supramolecular Function-alization with Azobenzene-Oligoglycerol Conjugates. Angew. Chem., Int. Ed. 2014, 53, 9669−9673.
(10) Deng, J.; Liu, X.; Shi, W.; Cheng, C.; He, C.; Zhao, C. Light-Triggered Switching of Reversible and Alterable Biofunctionality via β-Cyclodextrin/Azobenzene-Based Host-Guest Interaction. ACS Macro Lett. 2014, 3, 1130−1133.
(11) Patra, D.; Zhang, H.; Sengupta, S.; Sen, A. Dual Stimuli-Responsive, Rechargeable Micropumps via“Host-Guest” Interactions. ACS Nano 2013, 7, 7674−7679.
(12) Venkatesh, G.; Prabhu, A. A. M.; Rajendiran, N. Azonium-Ammonium Tautomerism and Inclusion Complexation of 1-(2,4-diamino phenylazo) Naphthalene and 4-aminoazobenzene. J. Fluoresc. 2011, 21, 1485−1497.
(13) Sanchez, A. M.; de Rossi, R. H. Effect ofβ-Cyclodextrin on the Thermal Cis-Trans Isomerization of Azobenzenes. J. Org. Chem. 1996, 61, 3446−3451.
(14) Sueishi, Y.; Kasahara, M.; Inoue, M.; Matsueda, K. Effects of Substituent and Solvent on Inclusion Complexation of β-Cyclo-dextrins with Azobenzene Derivatives. J. Inclusion Phenom. Macrocyclic Chem. 2003, 46, 71−75.
(15) Liu, Y.; Zhao, Y.-L.; Chen, Y.; Guo, D.-S. Assembly Behavior of Inclusion Complexes of β-Cyclodextrin with 4-Hydroxyazobenzene and 4-Aminoazobenzene. Org. Biomol. Chem. 2005, 3, 584−591.
(16) Zhang, L.; Zhang, H.; Gao, F.; Peng, H.; Ruan, Y.; Xu, Y.; Weng, W. Host-Guest Interaction Between Fluoro-Substituted Azobenzene Derivative and Cyclodextrins. RSC Adv. 2015, 5, 12007−12014.
(17) Bortolus, P.; Monti, S. Cis-Trans Photoisomerization of Azobenzene-Cyclodextrin Inclusion complexes. J. Phys. Chem. A 1987, 91, 5046−5050.
(18) Wu, J.; Isaacs, L. Cucurbit[7]uril Complexation Drives Thermal trans-cis-Azobenzene Isomerization and Enables Colorimetric Amine Detection. Chem. - Eur. J. 2009, 15, 11675−11680.
(19) Garnier, L.; Sarraute, S.; Israëli, Y.; Bonal, C.; Malfreyt, P. Associations of Water-Soluble Macrocyclic Hosts with 4-Amino-azobenzene: Impact of pH. J. Phys. Chem. B 2018, 122, 11953−11961. (20) Liu, Y.; Guo, D.-S.; Zhang, H.-Y.; Ma, Y.-H.; Yang, E.-C. The Structure and Thermodynamics of Calix[n]arene Complexes with Dipyridines and Phenanthroline in Aqueous Solution Studied by Microcalorimetry and NMR Spectroscopy. J. Phys. Chem. B 2006, 110, 3428−3434.
(21) Mendes, A.; Bonal, C.; Morel-Desrosiers, N.; Morel, J. P.; Malfreyt, P. Molecular Dynamics Simulations of p-Sulfonatocalix[4]-arene Complexes with Inorganic and Organic Cations in Water: A Structural and Thermodynamic Study. J. Phys. Chem. B 2002, 106, 4516−4524.
(22) Ghoufi, A.; Bonal, C.; Morel, J. P.; Morel-Desrosiers, N.; Malfreyt, P. Structures and Energetics of Complexes of the p-Sulfonatocalix[4]arene with Ammonium, Alkylammonium, and Tetraalkylammonium Cations in Water Using Molecular Dynamics Simulations. J. Phys. Chem. B 2004, 108, 5095−5104.
(23) Ghoufi, A.; Malfreyt, P. Entropy and enthalpy calculations from perturbation and integration thermodynamics methods using molecular dynamics simulations: applications to the calculation of hydration and association thermodynamic properties. Mol. Phys. 2006, 104, 2929−2943.
(24) Ghoufi, A.; Pison, L.; Morel, J. P.; Morel-Desrosiers, N.; Bonal, C.; Malfreyt, P. Computational and Experimental Investigations of Supramolecular Assemblies of p-Sulfonatocalix[4]arene Organized by Weak Forces. J. Phys. Chem. B 2007, 111, 11478−11485.
(25) Filippini, G.; Bonal, C.; Malfreyt, P. Why is the Association of Supramolecular Assemblies Different Under Homogeneous and Heterogeneous Conditions? Phys. Chem. Chem. Phys. 2012, 14, 10122−10124.
(26) Filippini, G.; Goujon, F.; Bonal, C.; Malfreyt, P. Energetic Competition Effects on Thermodynamic Properties of Association
betweenβ-CD and Fc Group: A Potential of Mean Force Approach. J. Phys. Chem. C 2012, 116, 22350−22358.
(27) Kirkwood, J. G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300−313.
(28) Darve, E.; Pohorille, A. Calculating Free Energies Using Average Force. J. Chem. Phys. 2001, 115, 9169−9183.
(29) Darve, E.; Rodríguez-Gómez, D.; Pohorille, A. Adaptive Biasing Force Method for Scalar and Vector Free Energy Calculations. J. Chem. Phys. 2008, 128, No. 144120.
(30) Lesage, A.; Lelièvre, T.; Stoltz, G.; Hénin, J. Smoothed Biasing Forces Yield Unbiased Free Energies with the Extended-System Adaptive Biasing Force Method. J. Phys. Chem. B 2017, 121, 3676− 3685.
(31) Fu, H.; Zhang, H.; Chen, H.; Shao, X.; Chipot, C.; Cai, W. Zooming across the Free-Energy Landscape: Shaving Barriers, and Flooding Valleys. J. Phys. Chem. Lett. 2018, 9, 4738−4745.
(32) Prue, J. E. Ion Pairs and Complexes: Free Energies, Enthalpies, and Entropies. J. Chem. Educ. 1969, 46, 12.
(33) Abascal, J. L. F.; Vega, C. A General Purpose Model for the Condensed Phases of Water: TIP4P/2005. J. Chem. Phys. 2005, 123, No. 234505.
(34) Wang, J.; Wolf, R. M.; Caldwell, J. W.; Kollman, P. A.; Case, D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157−1174.
(35) Heinz, H.; Vaia, R. A.; Koerner, H.; Farmer, B. L. Photoisomerization of Azobenzene Grafted to Layered Silicates: Simulation and Experimental Challenges. Chem. Mater. 2008, 20, 6444−6456.
(36) Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1−19.
(37) Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H. J. C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327−341.
(38) Schneider, T.; Stoll, E. Molecular-Dynamics Study of a Three-Dimensional One-Component Model for Distortive Phase Tran-sitions. Phys. Rev. B 1978, 17, 1302−1322.
(39) Dünweg, B.; Paul, W. Brownian Dynamics Simulations Without Gaussian Random Numbers. Int. J. Mod. Phys. C 1991, 02, 817−827. (40) Berendsen, H. J. C.; Postma, J. P. M.; van Gunsteren, W. F.; DiNola, A.; Haak, J. R. Molecular Dynamics With Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684−3690.
(41) Sun, H. COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase ApplicationsOverview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1998, 102, 7338−7364.
(42) Hockney, R. W.; Eastwood, J. W. Computer Simulation Using Particles; CRC Press, 1988.
(43) Pollock, E. L.; Glosli, J. Comments on P3M, FMM, and the Ewald Method for Large Periodic Coulombic Systems. Comput. Phys. Commun. 1996, 95, 93−110.