Analysis of the dystrophin interactome
Texte intégral
Figure
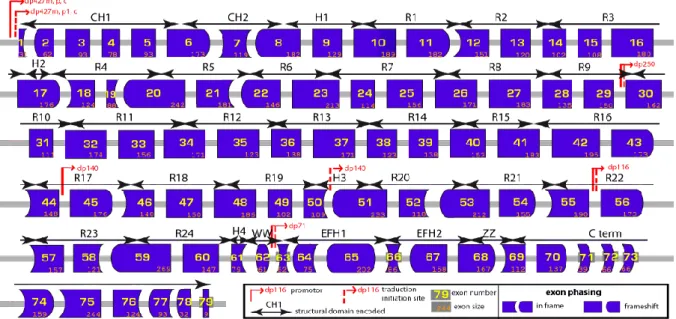


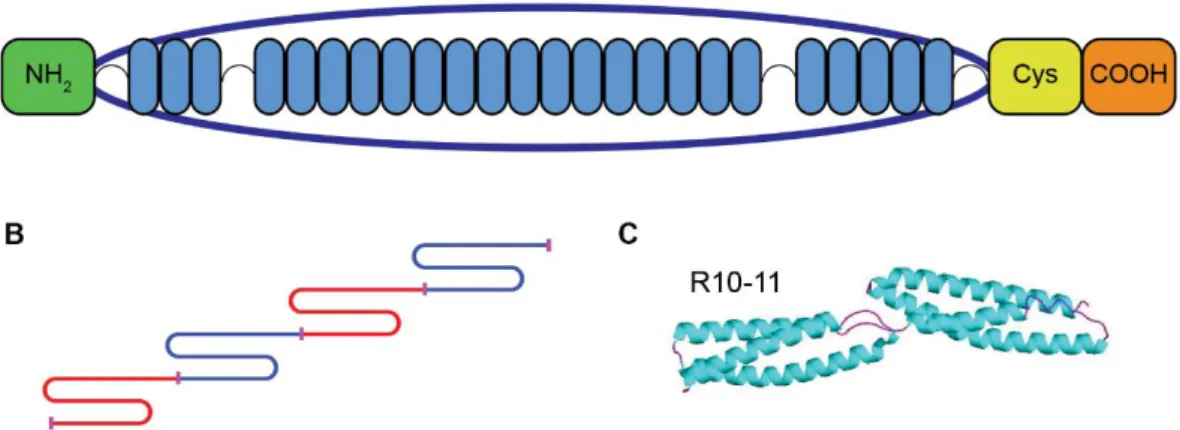

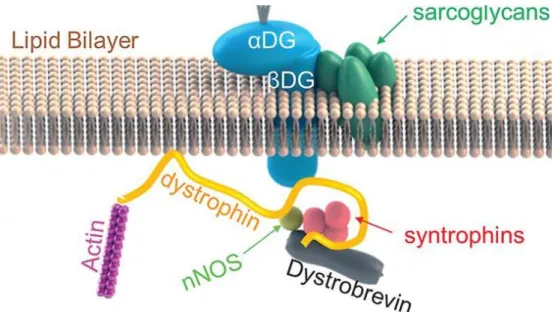


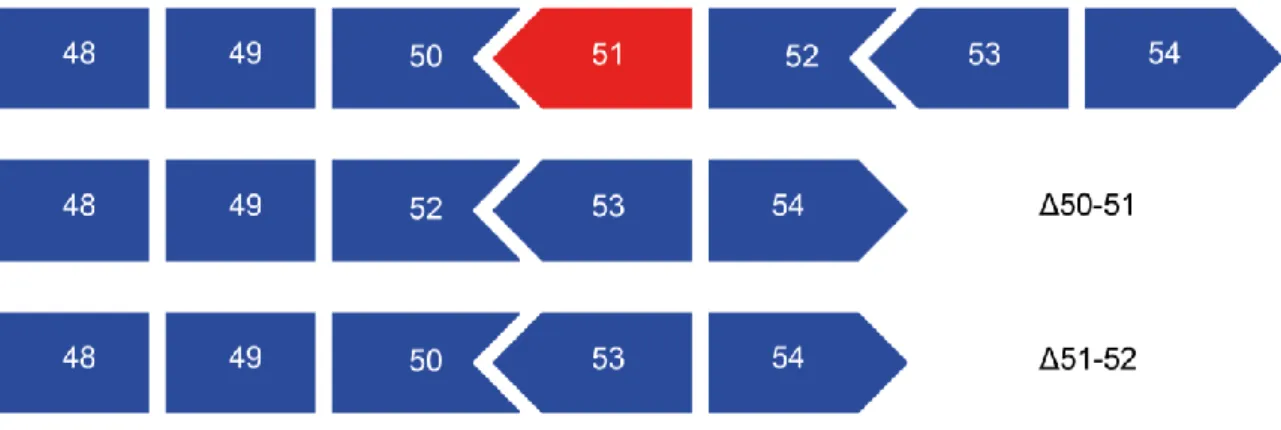
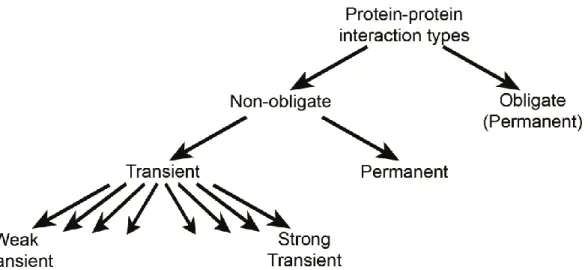
Documents relatifs
Nonlinear principal component analysis (NLPCA) is applied on detrended monthly Sea Surface Temperature Anomaly (SSTA) data from the tropical Atlantic Ocean (30°W-20°E, 26°S-22°..
The present work is a contribution to statistical analysis of PEMFC data by analysing the effects of groups of test parameters on the performance of a fuel cell and by correlating
To allow profiling the functional state of miRNA primary transcription units across commonly used human cell lines, we have generated GRO-seq libraries for 19 cell types, and
Dans ce choix, on peut lire deux volontés simultanées : celle d’un nouveau bâtiment iconique, dans la lignée de ceux qui ont fait la spécificité de la skyline de Hong
Information extracted from patients medical records in the form of descriptive data, images captured over the treatment time span and different forms of numerical
Compared to baseline, the NIV mode corrected premature cycling in four ventilators (Extend, Newport e500, Raphael, Vela), and worsened it in four others (Elysée, Raphael, Servo i,
Principal component analysis, Compressed principal component analysis, Non- Gaussian vector, Random eigenvectors, Symmetric polynomials, Random fields, Stochastic processes,
At T = 0, T = 2, T = 4 and T = 6 days after induction of differentiation, cells were fixed and stained with Oil-red-O to reveal lipid accumulation (panel on the right) or incubated
