Automated structure generation for first-principles
transition-metal catalysis
by
Efthymios Ioannis Ioannidis
Diploma Chemical Engineering
National Technical University of Athens (2013)
M.S. Chemical Engineering Practice
Massachusetts Institute of Technology (2014)
Submitted to the Department of Chemical Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Chemical Engineering Practice
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2018
c
⃝ Massachusetts Institute of Technology 2018. All rights reserved.
Author . . . .
Department of Chemical Engineering
June 2018
Certified by . . . .
Heather J. Kulik
Joseph R. Mares ’24 Career Development Professor in Chemical
Engineering
Thesis Supervisor
Accepted by . . . .
Patrick S. Doyle
Robert T. Haslam (1911) Professor of Chemical Engineering
Chairman, Committee for Graduate Students
Automated structure generation for first-principles
transition-metal catalysis
by
Efthymios Ioannis Ioannidis
Submitted to the Department of Chemical Engineering in June 2018, in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy in Chemical Engineering Practice
Abstract
Efficient discovery of new catalytic materials necessitates the rapid but selective gen-eration of candidate structures from a very wide chemical space and the efficient estimation of their properties. We developed an efficient and reliable software utility for high-throughput screening of inorganic complexes that enables chemical discov-ery by automating molecular and intermolecular complex structure generation, job preparation as well as post-processing analysis to elucidate correlations of electronic or geometric descriptors with energetics. The developed software was then used to unveil different binding modes of small anions on organometallic complexes as well as functionalizations that allow for selective binding. We additionally employed our ma-terials design framework to study the binding of carbon monoxide on functionalized metalloporphyrins providing tuning strategies and uncertainty estimation. Compu-tational approaches such as density functional theory (DFT) that directly simulate the electronic properties have been increasingly used as tools for materials design mainly due to recent developments in computational speed and accuracy. DFT re-casts the many-body problem of interacting electrons into an equivalent problem of non-interacting electrons, greatly simplifying the solution procedure. This approach introduces certain approximations that are effectively modeled with an exchange and correlation functional that accounts for the many-body effects that are not included in the simplified problem. The functional choice is an important modeling decision and therefore computational predictions can be sensitive to user selection. This sensitivity is maximized for systems with highly localized electrons such as transition metals due to self-interaction error, where one electron interacts with its own mean field resulting in an unphysical delocalization of the electron density. We studied extensively how the incorporation of the widely employed Hartree-Fock and meta-GGA-type exchange functionals affects DFT predictions on transition metal complexes.
Thesis Supervisor: Heather J. Kulik
Acknowledgments
At the verge of completing my PhDCEP thesis I would like to express my gratitude to all the people that have helped me shape this amazing journey.
First and foremost, I would like to thank my academic advisor, Prof. Heather Kulik who despite the short 3-year research period of the PhDCEP program provided me with all I needed to jump start the thesis and then let me construct my own path with proper and invaluable feedback and guidance. Being one of the first students in the Kulik lab and helping start up the group was a great experience and watching it gradually grow and take its own shape and character has been very fulfilling for me. I would like to additionally thank my thesis committee members Prof. Bill Green and Prof. Yuriy Roman for their continuous support in our thesis committee meetings and the advice they offered me during the term of this research project.
I have been extremely fortunate to be part of a very special research group for the past 3 years. I would like to thank everybody who is or has been a member of the Kulik lab including Natasha, Lisi, Niladri, Qing, Helena, Yusu, Jeong Yun, JP and especially Terry for their valuable feedback and fruitful discussions on matters concerning various aspects of my thesis.
I would also like to thank all my dear friends back home that have been supporting me these years I have been away. Despite the distance I am grateful that we are still close and I do consider you my life-long friends. Also, I am grateful for the new friends I made here in Boston, the Greek community and my friends from the chemical engineering department at MIT, all of them being special people that have helped me through the transition of living and studying in the US. I cherish every moment that I have spent with you and I am looking forward to even more exciting ones.
I would not have been here if it weren’t for my parents, Liana and Ippokratis and my sisters Dioni and Eleonora. They have always been my driving force, my source of energy and motivation and have supported every decision that I have made in my life. This thesis is dedicated to you.
Contents
1 Introduction 19
1.1 Density functional theory (DFT) . . . 19
1.1.1 Introduction to density functional theory . . . 19
1.1.2 Spin density functional theory . . . 23
1.2 DFT for transition metal catalysis . . . 24
1.3 Ligand field theories . . . 26
1.3.1 Crystal field theory . . . 26
1.3.2 Ligand field theory . . . 28
1.4 High-throughput screening . . . 30
1.5 Thesis outline . . . 33
2 Effect of Hartree-Fock exchange 35 2.1 Computational details . . . 38
2.2 Dependence of spin-state ordering on functional choice . . . 40
2.3 Dependence of spin-state ordering on HF exchange . . . 43
2.3.1 Spin-state ordering dependence with Fe(III) complex test cases 44 2.3.2 Spin-state ordering: comparison with Fe(II) complexes . . . . 47
2.4 Trends in charge localization measures . . . 50
2.5 Corroborating geometric and energetic relationships . . . 56
2.6 Quantitative vs. qualitative spin-state ordering . . . 57
3 Effect of meta-GGA exchange 61
3.1 Theory . . . 62
3.2 Computational details . . . 65
3.3 Effect of meta-GGA exchange on single ions spin-state splittings . . . 68
3.4 Dependence of spin-state ordering on meta-GGA exchange . . . 69
3.5 Trends in charge localization measures . . . 74
3.6 Combined effect of HF exchange and meta-GGA exchange . . . 79
3.7 Conclusions . . . 83
4 Automatic structure generation 85 4.1 Code overview . . . 86 4.2 Code architecture . . . 87 4.3 Structure generation . . . 89 4.3.1 General approach . . . 89 4.3.2 Customized cores . . . 95 4.3.3 Modify function . . . 96 4.4 Additional features . . . 97 4.4.1 Random generation . . . 97 4.4.2 Database search . . . 98
4.4.3 Supramolecular complex building . . . 99
4.4.4 Simulation automation . . . 101
4.4.5 Structure-property correlation and analysis . . . 102
4.5 Benchmarking molSimplify . . . 105
4.6 Conclusions . . . 111
5 Selective anion binding by functionalized organometallics 113 5.1 Computational details . . . 114
5.2 Binding modes . . . 115
5.3 Selective binding . . . 118
5.3.1 Hydrogen bonding . . . 120
5.4 Conclusions . . . 126 6 CO binding on metalloporphyrins 129 6.1 Computational details . . . 131 6.2 Structures . . . 133 6.3 Binding energies . . . 135 6.4 Charge measures . . . 141 6.5 Sensitivity analysis . . . 143 6.6 Conclusions . . . 146 7 Concluding remarks 149 8 Market analysis of the Catalysis industry: Capstone paper 153 8.1 Introduction . . . 153
8.2 Types of Catalysts . . . 154
8.3 Catalyst market segments . . . 156
8.4 Environmental catalysts . . . 157
8.5 Refining catalysts . . . 160
8.6 Polymer catalysts . . . 161
8.7 Chemical catalysts . . . 163
8.8 Global Catalysis Market Trends . . . 163
8.8.1 Toward higher activity and selectivity of catalysts . . . 164
8.8.2 Changes in feedstock and more effective use of feedstock . . . 165
8.8.3 Lower Operating Temperatures . . . 166
8.8.4 Energy efficiency . . . 166
8.8.5 Creation of processes around catalyst technologies . . . 167
8.8.6 Gas-to-liquids (GTL) technologies . . . 168
List of Figures
1-1 Crystal field theory picture of orbital interaction . . . 27
1-2 Orbital splitting in an octahedral crystal field . . . 27
1-3 High- and low-spin configurations for mid-row transition metals . . . 28
1-4 Ligand field theory molecular orbital diagram . . . 29
1-5 Examples of coordination complexes . . . 31
1-6 Example of SMILES string and corresponding structure . . . 32
2-1 Octahedral iron complex structures . . . 41
2-2 Relative spin state ordering for Fe(III) octahedral complexes . . . 45
2-3 Relative spin state ordering for Fe(II) octahedral complexes . . . 48
2-4 Relative spin state ordering for Fe(II/III)-N complexes . . . 49
2-5 Augmented data set with 5 additional octahedral structures . . . 50
2-6 Spin-state sensitivity against NBO charges for Fe(III) complexes . . . 52
2-7 Spin-state sensitivity against NBO charges for Fe(II) complexes . . . 54
2-8 Spin-state dependence on HFX against NBO charge dependence . . . 55
2-9 Derivative of spin-state splitting with HFX vs spin-state splitting . . 59
3-1 TPSS and mTPSS enhancement factors . . . 65
3-2 Error of spin-state energies at 0% and 100% meta-GGAX for atoms . 70 3-3 Spin sensitivity to meta-GGA exchange . . . 72
3-4 Spin sensitivity versus splitting for Cr(II)-Co(III) . . . 73
3-5 Spin sensitivity versus splitting for early- late-row TM . . . 74
3-7 Splitting versus charge sensitivity . . . 78
3-8 HFX and meta-GGAX composite spin-state splitting . . . 80
3-9 Complexes of the extended data set . . . 82
4-1 Flowchart for molSimplify process . . . 88
4-2 Example octahedral complex generation . . . 91
4-3 Alignment procedure for bidentate and monodentate ligands . . . 93
4-4 Example of custom core functionalization . . . 95
4-5 Chemical discovery workflow . . . 97
4-6 Supramolecular complex generation . . . 100
4-7 RMS gradients for benchmark data set . . . 106
4-8 Energy differences for benchmark data set with and without FF . . . 108
4-9 Energy difference between molSimplify- and UFF-generated structures 109 4-10 Representative complexes with FF optimized lowest energy structures 110 4-11 Comparison of UFF and molSimplify structures for distorted complex 110 5-1 Formate initial guesses and binding modes . . . 116
5-2 Formate binding modes . . . 117
5-3 Functional groups for ferrocenium functionalization . . . 119
5-4 Formate and perchlorate adsorption energies histogram . . . 120
5-5 Hydrogen bonds for strongly and weakly binding Fc-anion complexes 122 5-6 Relative adsorption energies versus formate adsorption energy . . . . 124
5-7 Relative adsorption energies versus charge difference measures . . . . 126
6-1 Structure of metal tetraphenyl porphyrin (MTPP). . . 131
6-2 MTPP and CO molecular orbitals . . . 135
6-3 Bond length versus MTPP-L/CO dissociation energy . . . 139
6-4 Orbital polarization by axial ligands . . . 140
6-5 NAO descriptor for binding strength . . . 141
6-6 Metal charge transfer versus binding strength . . . 142
6-8 Binding strength sensitivity versus spin-state sensitivity . . . 145
8-1 Catalyst market shares by technology . . . 155
8-2 Catalyst market shares by application . . . 157
8-3 Catalyst market shares by application . . . 158
8-4 Polymer catalyst market growth projection by subsegment . . . 162
List of Tables
2.1 Ground states of octahedral coordination complexes . . . 42
2.2 Difference in bond lengths at 20% HFX between HS and LS complexes 57 2.3 Dependence of HS-LS bond length differences on HFX . . . 57
3.1 Experimental spin-state splittings for 8 first-row transition metals . . 69
3.2 SCAN vs TPSS results for representative Fe(II)L6 complexes . . . 71
3.3 Charge vs energy sensitivity on meta-GGA exchange . . . 79
3.4 Extended data set for meta-GGA calculations . . . 82
4.1 Reported properties by the molSimplify post-processing module . . . 103
4.2 Comparison of properties for UFF and molSimplify structures . . . . 108
4.3 Comparison of UFF and molSimplify Ni(II) structures . . . 111
6.1 Five-coordinate metal distal ligand bond lengths . . . 134
6.2 Six-coordinate metal distal ligand bond lengths . . . 136
6.3 Six-coordinate metal-proximal CO ligand bond lengths . . . 136
6.4 Relaxation energies for 6-coordinate MTPP-L/CO. . . 138
6.5 Porphyrin-CO bond dissociation energies . . . 140
Abbreviations
BCP Bond critical point
B3LYP Becke, 3-parameter Lee-Yang-Parr CFT Crystal field theory
CASPT2 Complete active space perturbation theory CLI Command line interface
CN Coordination number COM Center of mass
CT Coordination template
CSD Cambridge structural database DFT Density functional theory ELF Electron localization function FG Functional group
GGA Generalized gradient approximation GUI Graphical user interface
HF Hartree-Fock
HOMO Highest occupied molecular orbital HS High-spin
IS Intermediate-spin
KS Kohn-Sham
LDA Local density approximation LFT Ligand field theory
LS Low-spin
LSDA Local spin density approximation LUMO Lowest unoccupied molecular orbital meta-GGA meta generalized gradient approximation ML Metal-ligand
modB3LYP modified B3LYP NAO Natural atomic orbital NBO Natural bonding orbital
NIST National institute of standards and technology NPA Natural population analysis
QTAIM Quantum theory of atoms in molecules RMSD Root-mean square deviation
SCAN Strongly constrained and appropriately normed SCO Spin crossover
SDF Structure data file SIE Self interaction error
SMARTS Smiles arbitrary target specification
SMILES Simplified molecular input line entry string TM Transition metal
TPP tetraphenylporphyrin
TPSS Tao, Perdew, Scuseria, Staroverov UEG Uniform electron gas
UFF Universal force field UKS Unrestricted Kohn-Sham
Chapter 1
Introduction
Efficient discovery of new materials necessitates the rapid but selective generation of candidate structures from a very wide chemical space and the efficient estimation of their properties. Experimental discovery of new materials is limited by the cost and the time required to perform the experiments. Computational approaches [1] such as density functional theory (DFT) that directly simulate the electronic properties have been increasingly used as tools for materials design mainly due to recent develop-ments in computational speed and accuracy. DFT methods are now able to rapidly characterize candidate materials before they are synthesized reducing both the cost and the time required for their development.
1.1
Density functional theory (DFT)
1.1.1
Introduction to density functional theory
The first principles method we will primarily use in this work is density functional theory. DFT is among the most popular and versatile methods available in com-putational chemistry, since it combines accuracy with comcom-putational efficiency. The theoretical foundations for its development were set by the Hohenberg-Kohn theo-rems [2] which argued that the ground state properties of a system can be described in terms of its ground state electronic density instead of the far more complicated
wavefunction. Starting from the many-electron time-independent Schrödinger equa-tion and employing the Born-Oppenheimer approximaequa-tion the equaequa-tion becomes (in atomic units): –1 2 N ∑ i=1 ∇2 i – N ∑ i M ∑ A ZA |ri– RA| + N ∑ i<j 1 |ri– rj| Ψ = EΨ, (1.1)
where the Hamiltonian operator includes contributions from the kinetic energy of the electrons, the electrostatic attraction between electrons and nuclei as well as the repulsion between electrons. M is the number of nuclei and N is the number of electrons in the system. However, the fact that the wavefunction depends on the positions of all the electrons for a given configuration of nuclei, thus on 3N variables, makes the direct solution of the equation intractable in practice. Reformulating the problem using the three dimensional electron density,ρ(r) =∑i∥ψi(r)∥2, significantly simplifies the solution procedure.
Furthermore, as demonstrated by Hohenberg and Kohn [2], all the physical quan-tities of interest are a function of an external potential, v(r), and therefore the energy of the system can be defined as a functional of the electron density:
E[ρ(r)] = F[ρ(r)] + ∫
v(r)ρ(r) dr, (1.2)
where F[ρ(r)] is a universal functional, independent of the system in question, that contains the contributions from the kinetic energy and the Coulomb interactions between the electrons, whereas v(r) represents the external potential (in our case the Coulomb interactions of electrons with the nuclei). This reformulation of the problem allows us to obtain the ground state energy by variationally minimizing the energy under the constraint that the total number of particles is preserved.
In practice, however, the universal functional F[ρ(r)] is not known. Kohn and Sham [3] bypassed this problem by mapping the original system onto a system of non-interacting electrons that has the same electron density and thus the same energy. This system can be described by a Slater determinant of single particle orbitals. The
universal functional F[ρ(r)] of the real system can now be expressed as: F[ρ(r)] =∑ i –1 2ψ ∗ i(r)∇2ψi(r) dr | {z } T0 +1 2 ∫ ρ(r)ρ(r′) |r – r′| dr dr′ | {z } EH +Exc[ρ(r)], (1.3)
where the first term is the kinetic energy of the fictitious system of non-interacting electrons, the second term is the classical Coulomb interaction described through their density (also called Hartree term, EH), whereas Exc[ρ(r)] or exchange-correlation
energy accounts for the many-body effects that are not included in the rest of the functional. This last contribution to the energy sums up everything that we don’t know about the universal functional in one term and in practical DFT we try to account for it using several approximations.
The simplest of these approximations is called local density approximation (LDA) [4] in which we assume that the electrons behave locally as a uniform electron gas (UEG) with constant density. Therefore, the exchange-correlation term can be ex-pressed as:
ELDAxc [ρ(r)] = ∫
εUEG
xc (r)ρ(r) dr, (1.4)
where εUEGxc (r) is the exchange-correlation energy of the homogeneous electron gas and is calculated using high accuracy quantum Monte Carlo simulations [5]. The exchange part of the functional can be calculated exactly as:
ELDAx [ρ] = –3 2 ( 3 4π )1/3 ρ(r)4/3dr. (1.5)
For quickly varying densities, as in molecules with localized subshells, LDA ex-change provides a particularly poor estimate of the exex-change energy.
Beyond the LDA, gradients of the density may be directly incorporated into semi-local descriptions of exchange [6], typically rescaled by the absolute value of the density as in the so-called generalized gradient approximation (GGA). The B88 [7]
GGA exchange energy is given by: EGGAx = ELDAx –β ∫ ρ4/3σ χ 2 σ ( 1 + 6βχσsinh–1χσ )dr, (1.6)
where this exchange energy is referenced with respect to the LDA energy and is a rescaled integral of the spin density (spin index σ, see Section 1.1.2) with a semi-empirical parameter β=0.0042a.u. Here, the variable χσ is the rescaled gradient of the density:
χσ= |∇ρσ| ρ4/3σ
. (1.7)
Higher level approximations introduce the usage of the Laplacian of the density or equivalently the kinetic energy of the electrons (meta-GGA) [8], all of them trying to provide more accurate results by incorporating more complexity in the functional ex-pression. However it is not yet clear that these functionals provide improved accuracy in all cases, since all of them are based on a mean-field formalism that is expected to work well only for systems with delocalized electrons.
On the other hand, the Hartree-Fock (HF) method accounts for the exchange in-teraction exactly while neglecting fully the electron correlation. The exchange energy in this method is explicitly given by:
EHFx [ρ(r)] = –1 2 occ ∑ i,j ∫ dr dr′ψ ∗ i(r)ψ∗j(r′)ψ∗j(r)ψ∗i(r′) |r – r′| , (1.8)
whereψi(r) is the single particle orbital i and the sum is over all occupied orbitals.
Since HF includes the exact exchange energy, hybrid functionals [9], another class of exchange-correlation functionals, incorporate a portion of exact HF in attempt to better account for this type of interaction. Hybrid functionals are very popular due to their good performance for a range of different systems, however it should be pointed out that the results obtained with these functionals depend strongly on the amount of exact exchange included and therefore they should be used with caution.
The mathematical form of hybrid functionals is given by: Exc= ELDAx +a0 ( EHFx – ELDAx ) +ax ( EGGAx – ELDAx ) +ELDAc +ac ( EGGAc – ELDAc ) . (1.9) By varying the 3 parameters, it is possible to tune the hybrid functional and adjust the HF, LDA and GGA parts. The most popular hybrid functionals in chemistry are B3LYP [9–11] with a0 = 0.2, ax = 0.72 and ac = 0.81 and PBE0 [12, 13] with
a0 = 0.25, ax = 0.75 and ac= 1.00.
1.1.2
Spin density functional theory
A useful extension of the KS approach treats separately the densitiesρα(r) andρβ(r) of electrons with spin projection up and down. Equivalently one can deal with
ρ(r) = ρα(r) +ρβ(r), (1.10)
together with the polarization function:
ζ(r) = ρρα(r) –ρβ(r)
α(r) +ρβ(r), (1.11)
that takes values between -1 (fully polarized downwards) and +1 (fully polarized upwards). The up and -down densities are generated from up and spin-down KS wavefunctions, ρα(r) = occ ∑ i |ψi,α|2 , ρβ(r) = occ ∑ i |ψi,β|2. (1.12)
The local density approximation can be extended to the local spin density approxi-mation (LSDA) based on the spin-polarized uniform gas in a manner analogous to the LDA approach. LSDA represents a considerable improvement over LDA for atomic and molecular systems with unpaired spins or open-shell systems, for which the un-polarized electron gas is clearly not a very good model. This approach is commonly
referred to as unrestricted Kohn-Sham (UKS) [14] and it can be employed in systems where a standard KS calculation would unphysically restrict the true symmetry (e.g., dissociation of the H2 molecule).
1.2
DFT for transition metal catalysis
Efficient design and discovery of catalysts is central to solving modern challenges in energy and resource utilization [15]. In the field of heterogeneous catalysis, it has been shown that a wide range of catalysts can be screened [16–18] using suitable chemical descriptors such as binding energies calculated with ab initio methods. A similar approach has been proposed for molecular catalysts [19] where binding ener-gies of specific molecules can be used as indicators for catalytic activity. Molecular catalysts enhance reaction rates and selectivities at metal-centers coordinated with specific ligands. These coordination complexes have well-defined geometric and elec-tronic structures that foster selective and targeted interactions with various classes of molecules.
The complexity of these interactions makes a molecular-level understanding of the underlying interplay between the atoms essential for the design of effective molecular catalysts. By understanding the properties that determine their catalytic activity, it is possible to tune the structure of the catalysts in order to achieve the highest possible turnover frequency.
Transition metals are present as catalytic reactive centers in a wide range of bi-ological [20] and inorganic systems [21–23]. In addition, the majority of molecular catalysts that have been studied contain transition metals. Common approaches in-cluding density functional theory techniques utilize mean field approximations to de-scribe them, with their accuracy strongly depending on the underlying assumptions of each approximation. It becomes necessary therefore to develop a better understand-ing of how the choice of different first-principles approaches can affect the calculated properties of transition metal complexes and provide estimates about the uncertainty in our predictions.
Transition metals are unique due to their open-shell character. They include partially occupied valence d or f orbitals that impart special properties such as para-magnetism [24], vivid color of their compounds [25] or electrical conductivity [26]. Transition metals exhibit a wide range of oxidation states that allow them to form many different compounds.
Transition metal compounds are currently one of the biggest challenges for theo-retical chemistry [21,27]. The high localization of the d electrons can not be described adequately by most exchange-correlation functionals that tend to delocalize the elec-tron density [28]. The main source of this problem is self-interaction error (SIE). As we can see from Eq. 1.3, the Hartree term, EH, includes the repulsion of each refer-ence electron in the mean field of the rest. However, the calculated mean field density includes the charge of the reference electron as well, thus causing the electron to in-teract with its own mean field. For a simple, one-electron system with wavefunction ϕ(r), the Hartree term from eq. 1.3 becomes:
EH = 1 2
∫ |ϕ(r)|2|ϕ(r′)|2
|r – r′| dr dr′ ̸= 0, (1.13)
which indicates that the Hartree term even in an one-electron system predicts the unphysical repulsion of an electron by its own mean field. Including the exact exchange energy of Eq. 1.8 The sum becomes:
EH+ Ex = 1 2 ∫ |ϕ(r)|2|ϕ(r′)|2 |r – r′| dr dr′– 1 2 ∫ |ϕ(r)|2|ϕ(r′)|2 |r – r′| dr dr′= 0. (1.14)
Thus, in Hartree-Fock theory self-interaction cancels exactly, however the lack of dynamic correlation makes the method unable to accurately describe transition-metal containing systems. Most exchange and correlation functionals, including hybrids, still fail to systematically localize d electrons [29–31] correcting this way for self-interaction and thus alternative approaches are still the topic of ongoing interest [32].
1.3
Ligand field theories
Fundamental for the description of open-shell transition metal coordination complexes is an understanding of the interactions between the central metal atom and the co-ordinating ligands. Various theories have been proposed to describe the bonding in coordination complexes with the most popular amongst them being crystal and ligand field theory.
1.3.1
Crystal field theory
Within crystal field theory (CFT) we assume that the 6 ligands in an octahedral complex behave as negative point charges that are brought near the metal in an oc-tahedral array along the three principle axes of the d orbitals. These point charges represent lone pairs on the ligands that are considered to be fully localized and there-fore are not involved in any type of covalent bonding with the metal. As a result, CFT assumes a purely ionic type of interaction between the metal and the ligands.
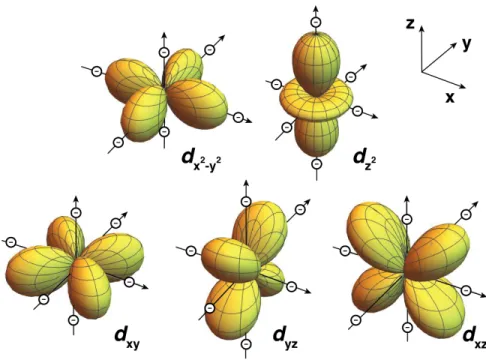
The dx2–y2 and dz2 orbitals of the metal are affected the most by the negative charges that point directly at their corresponding electron clouds (Figure 1-1). Any electrons in these orbitals will be strongly repelled by the corresponding point charges, thus raising the energy levels of the two orbitals. In contrast, the dxy, dxz and dyz
orbitals have their lobes directed between the ligands (Figure 1-1) increasing the stability of these orbitals and thus lowering their energy.
The net result of this purely electrostatic interaction is that the 5 degenerate metal
d orbitals are split into two groups, a set of 2 orbitals, eg, with high energy and a set
of 3 orbitals, t2g, with low energy, separated by the so-called crystal-field splitting energy,Δo (Figure 1-2).
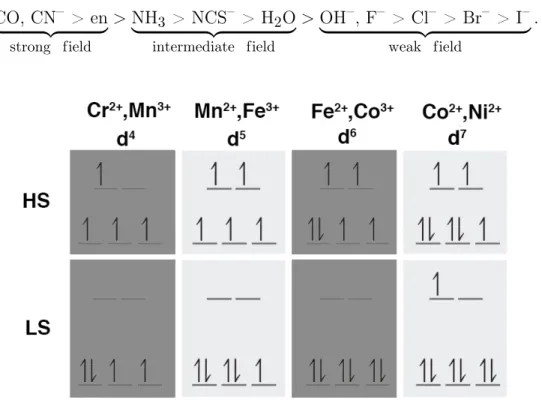
The splitting energy is crucial in accounting for magnetic properties in coordina-tion complexes. Pairing electrons on the same orbital requires energy input and if Δo is smaller than this pairing energy then a configuration where unpaired electrons
occupy eg orbitals will be preferred in a so-called high-spin configuration. If Δo is
Figure 1-1: Crystal field theory representation of the repulsion between the metal d orbitals and the 6 point charges that represent the ligands.
Figure 1-2: Splitting of the degenerate metal d orbitals in the presence of an octahe-dral crystal field.
orbitals t2g (Figure 1-3) obtaining a low-spin configuration.
The magnitude of the splitting energy will depend on the electrostatic or crystal field created by the ligands. A strong crystal field will result in stronger repulsion and a larger Δo that favors low-spin configurations. Weaker crystal fields will result in
weaker repulsion, lowering theΔo and thus favoring high-spin electron configurations.
strength as indicated by the spectrochemical series: CO, CN– > en | {z } strong field > NH| 3 > NCS{z– > H2O} intermediate field > OH| –, F– > Cl{z– > Br– > I–} weak field .
Figure 1-3: High- (HS) and low-spin (LS) configurations for common mid-row tran-sition metals.
1.3.2
Ligand field theory
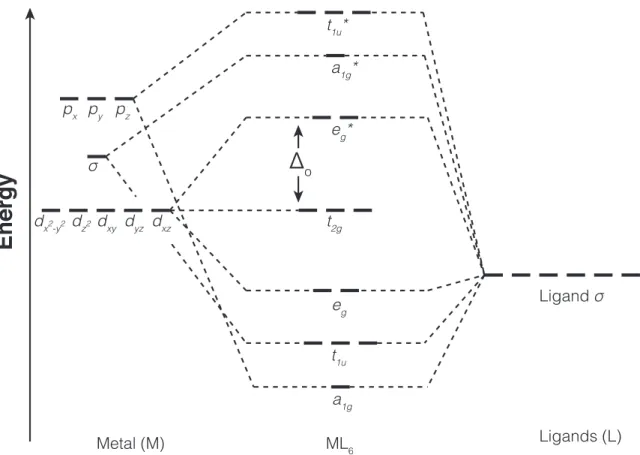
Extending on the concepts that were developed in crystal field theory, ligand field theory (LFT) takes explicitly into account the orbitals of the ligands. Within LFT, six ligand orbitals are initially assumed to haveσ symmetry around the metal-ligand bond lines and are allowed to interact with six of the nine valence metal orbitals, namely s, px, py, pz, dx2–y2 and dz2. The dxy, dyz, dxz orbitals have the wrong
symmetry for combining with σ-type ligand orbitals and are therefore nonbonding in character. The resulting energy-level diagram includes 6 bonding, 6 antibonding and 3 nonbonding orbitals (Figure 1-4). The nonbonding level and the lowest antibonding level correspond to the two levels, t2g and eg predicted by CFT.
Including the effect of ligand π-type orbitals, the molecular orbital diagram is not changing significantly since they mainly interact with the nonbonding t2g molecular orbitals. If the π-type ligand orbitals are populated, they will repel the t2g orbitals
Δ
oEn
e
rg
y
dxy dyz dxz dz2 dx2-y2 Ligand σ σ px py pz eg* Metal (M) ML6 Ligands (L) eg a1g* t1u* t1u a1g t2gFigure 1-4: Simplified molecular orbital diagram for an octahedral complex as pre-dicted by ligand field theory.
that have strong metal dxy, dxz and dyz character, raising their energy level and
thus decreasing the Δo. Any ligand with filled orbitals having such π symmetry
around the ligand-metal axis (e.g., Cl–, OH–) is classified as weak-field under LFT. On the other hand, ligands that have an unfilled antibonding orbital withπ symmetry behave differently. The antibondingπ∗orbitals in this molecular group accept electron density from the nonbonding t2g orbitals that become partially delocalized. This delocalization stabilizes them and lowers their energy resulting in increased Δo and
favored low-spin configurations. The interaction between the metal t2g orbitals and empty ligand π∗ orbitals is called π back-bonding and strengthens the metal-ligand bond. Ligands that increase the splitting of the levels in this way (e.g., CO, NO–2) are classified as strong-field under LFT. Both theories can be extended to other geometries using the same methodology that was initially developed to explain properties of octahedral coordination complexes.
Transition metals are present as catalytic reactive centers in a wide range of bi-ological [20] and inorganic systems [22, 23]. In addition, the majority of molecular catalysts that have been stu
1.4
High-throughput screening
Catalysts, be they molecular [33], heterogeneous [34], or biological [20], selectively and efficiently convert abundant feedstocks to more useful, functionalized products. Therefore, efficient design and discovery of catalysts is central to solving modern challenges in energy and resource utilization. Nevertheless, experimental discovery of new catalysts is slow, and computational study is often limited to rationalizing experimental observations. While experimental catalyst design efforts have focused on developing new synthesis techniques [35], characterizing alloys [36], using chemical intuition [37], or taking inspiration from biology [38], there are essentially infinite candidate catalysts that have not yet been synthesized or characterized. Catalyst discovery often relies on a make-first, explain-later approach, where reactivity is ra-tionalized after compounds are synthesized.
Exponential growth in computational power has enabled major first-principles screening efforts in heterogeneous catalysis [16, 17, 39, 40] and materials [41–43]. Part of the success of these solid-state screening efforts lies in the fact that for a given composition, a relatively limited number of possible crystal structures exists that may be efficiently enumerated, for instance by evolutionary algorithms [42, 43]. Molecular catalysts are another attractive target for computational screening efforts due to their high selectivity, activity, and wide range of tuning made possible through variation of metal and ligand identities. While some experimental [44, 45] or computational [33, 46–51] screens have been carried out for the discovery of molecular catalysts, robust and broadly applicable tools for the rapid generation and assessment of inorganic complexes (Figure 1-5) are not yet available. Recent work [19] has shown that the binding energy relations used in the solid state can be generalized into an energetic span model that relates turnover frequencies to binding energies across all catalytic
systems, thus enabling the fast assessment of the catalytic properties for molecular systems with an approach similar to the volcano plots used in heterogeneous catalysis.
Figure 1-5: Three coordination complexes: Zn(NH3)4,Co(CO)5,Fe(phen)2(NCS)2, with Zn, Co and Fe as metal centers and tetrahedral, trigonal-bipyramidal and octa-hedral coordinations respectively.
Unlike transition-metal complexes, organic molecules are much more straightfor-ward targets for computational screening. Often, first-principles simulation is not even needed to evaluate chemical properties of simple organic molecules. Instead, cheminformatics tools have been developed to store [52–55] and analyze [56–60] large quantities of chemical data for organic molecules including chemical formulas, struc-tures, and connectivity information. The development of these methodologies has also enabled encoding of all structural information in a molecule into a two-dimensional (2D) representation of a molecule such as a connection table, a simplified molecular input line entry string (SMILES) [61] string (Fig. 1-6) or a Smiles ARbitrary Target Specification (SMARTS) string [62]. Most tools evaluate chemical properties solely on the basis of connectivity definitions [63–65], while some also work with three-dimensional (3D) structures [66–68]. The interface between cheminformatics and simulation is made possible by generators that can turn these 2D strings into three-dimensional (3D) coordinates in a process called structure diagram generation [69]. These coordinates can then be used as input for the calculation of various properties of interest with first-principles simulation methods such as DFT. However, for these structure generation tools to succeed, atom valency and connectivity should be well defined, which is seldom the case for transition metal complexes.
O = Cc1 ccc|{z}
c:c
(O)c(OC)c1 –→
Figure 1-6: Example of SMILES string and the SMARTS pattern c:c that indicates aromatic carbons joined by an aromatic bond. The resulting 3D structure of vanillin is also illustrated.
While the generation of 3D coordinates from a 2D representation is straight-forward for small organic molecules, this process becomes challenging for inorganic complexes. Here, structure prediction is complicated by the fact that the number of bonds and valency are variable for inorganic complexes and not well defined as in organic molecules. However, the open-shell character, large number of electrons, and diversity of chemical bonds observed in transition metal complexes [70–73] that make it difficult to predict 3D structures are also the properties that impart interesting cat-alytic activity and chemical properties to these systems. Therefore, there is a clear need to develop automatic structure generation tools that can robustly obtain 3D properties of inorganic complexes. Additionally, strategies [74] for generating multi-ple conformers of a given 2D formula are immulti-plemented in several codes [67, 68, 75–77] and work reasonably well for organic molecules. However, generating conformers of inorganic complexes becomes more challenging, and few tools exist for weakly bound complexes. Molecular catalysis screening efforts necessitate evaluation of binding en-ergetics with adsorbates and non-covalent interactions. Initial guesses for such studies typically require painstaking hands-on generation or customized, user-built scripts. Crucially, first-principles geometry optimizations will only find the closest local mini-mum to a user-guessed geometry, and there may be many such local minima for most chemical systems of interest.
re-lies on databases of experimentally characterized chemical compounds [78–85]. While mining such databases for existing complexes that can be repurposed for other objec-tives in catalysis [86] often proves fruitful, discovery of new materials also necessitates generating molecules that are not already experimentally known. Automatic struc-ture generation tools that combine both new strucstruc-tures as well as fragments from experimental structures will instead allow the extension of the surveyed chemical space in a combinatorial search to previously uncharacterized molecules. Some more flexible 3D structure generation approaches [87–89] for inorganic complexes have in-stead decomposed the molecule into elementary fragments that are matched against 3D structures in experimental libraries. However, the reliance of most of these tools on commercial databases [78] has not been without controversy [90, 91], leading to their limited distribution.
For a screening effort to succeed, both the calculations and structure generation should be automated. However, there are only a few examples (e.g., the Atomistic Simulation Environment python toolkit in the solid state [92] or the Avogadro code [93] for quantum chemistry) are available that both generate structures and aid the preparation of first-principles calculations through the generation of input files and analysis of results. In order to accelerate discovery in transition metal chemistry, there is a clear need to both enable the flexible generation of 3D structures and the evaluation of their properties with first-principles simulations in a reliable and automatic framework.
1.5
Thesis outline
The remainder of this thesis is outlined as follows:
• Chapter 2 discusses the effect and role of Hartree-Fock exchange in describing
the electronic structure of transition-metal complexes. Detailed results on the spin-state energetics of iron octahedral complexes are reported including charge analysis and structural properties. The relative performance of hybrid density functionals is also compared against alternative exchange-correlation functionals
and literature results.
• Chapter 3 considers the effect of meta-GGA exchange on the spin-state
energet-ics of a wide range of single metal ions and octahedral coordination complexes. Correlations between electronic structure and functional performance are drawn with the results compared against reference values. Furthermore, the combined effect of Hartree-Fock exchange and meta-GGA exchange is discussed for a set of iron coordination complexes, suggesting differing effects that depend on the ligand-field strength.
• Chapter 4 discusses the development of molSimplify, a structure generation
soft-ware that utilizes freely available modules and custom routines for the accurate generation of 3D coordinates in a wide range of transition metal complexes. The program employs an array of geometric manipulation routines that offer significant advantages against alternative methods for generating coordination complexes. Furthermore, additional tools such as post-processing modules or database searching routines that are embedded in the program are presented and their functionality discussed.
• Chapters 5 and 6 present two applications of this framework. Initially, the
se-lective binding of anions on functionalized ferrocenium complexes is discussed, with the results suggesting that hydrogen-bonding plays an important role in the interactions between organometallics and anions. Finally, results regarding the binding strength of carbon monoxide on model catalytic metalloporphyrins functionalized with an additional axial ligand are presented. Correlations be-tween electronic and structural properties are obtained and a binding strength descriptor is suggested. Additionally, uncertainty estimates on the results are introduced based on sensitivity analysis.
Chapter 2
Effect of Hartree-Fock exchange
Reprinted (adapted) with permission from [94]. Copyright 2015 American Chemical Society.
Density functional theory has seen widespread use and exponential growth [95] ow-ing to its relatively computationally efficient description of short-range, dynamic cor-relation. Ease of entry for new users has made practical DFT one of the most popular "black box" computational chemistry techniques, despite well-known shortcomings. Namely, predictions are sensitive to user selection of the exchange-correlation func-tional amongst a "zoo" of choices. Decisions about funcfunc-tional choice are in turn often influenced by word of mouth or popular opinion [96] and availability in a localized basis or plane wave electronic structure code. Extensive optimization of exchange-correlation functional parameters in DFT against test sets with a large number of parameters [97] has improved accuracy, though reduction in parameters [98] or lim-ited use of parameters in some functionals [12,13] can provide improved transparency. Nevertheless, mathematical expressions for exchange and correlation still prevent a clear understanding of how accuracy may be systematically and globally improved. Established test and training sets for functional development primarily focus on ther-mochemistry of main group molecules [99], and accuracy is not necessarily transferable to other properties or elements.
Importantly, exchange-correlation functionals that work well for main group ele-ments may not work as well for transition metals [100], which are central to homo-geneous [21–23], heterohomo-geneous [101], or enzymatic [20] catalysis. Transition metals are increasingly prominent in computational design screens [101, 102] for which high-accuracy and high-efficiency black box DFT predictions are needed. Nevertheless, transition metals remain a challenge due to the close spacing of electron configura-tions (e.g. 3d74s1 vs. 3d64s2 in neutral Fe) that leads to several accessible spin states and oxidation states [103]. Spin-crossover (SCO) complexes [104, 105], which typically contain Fe(II) or Fe(III) centers [106], represent a particularly challenging class of molecules because the spin state can change with small changes in tempera-ture. SCO molecules have shown promise in nanoscale storage devices [107], spintron-ics [108, 109], and catalysis [110–112]. Nevertheless, common exchange-correlation functionals struggle to reproduce critical features for the spin-dependent potential energy surfaces [113–116].
In transition metal complexes, low-spin states are known to be favored by gen-eralized gradient approximation exchange-correlation functionals, while hybrid func-tionals that include a fraction of Hartree-Fock exchange often prefer high-spin states [116–121], and different energy gaps are obtained with different exchange-correlation functionals [122,123]. One suggested reason for the failure of practical DFT in describ-ing transition metal complexes is that relatively localized 3d valence electrons suffer strongly from self-interaction error (SIE) present in pure DFT functionals and only approximately corrected in hybrid functionals. Strides have been made in systematic removal of SIE [124, 125] and identification of paths to improve balance in spin-state ordering [116, 117, 126–130] but hybrid functionals remain a popular and straightfor-ward approach to approximately correct for energetic errors driven by imbalances in SIE between spin states.
It is worthwhile to note that mixing in of Hartree-Fock exchange may poten-tially trade reduction in self-interaction errors for an increase in static correlation errors, which tend to plague Hartree-Fock more than density functional theory ap-proaches. For a balanced treatment of both static correlation and in the absence of
self-interaction error, multireference wavefunction techniques have been used [131– 138] to study spin crossover complexes up to around 45 atoms in size [136]. The predominant method employed in the study of spin-crossover complexes is CASPT2, and, while it scales more expensively than density functional theory approaches, re-cent improvements in scaling [132] have made larger systems tractable. Other studies have applied the even more expensively-scaling CCSD(T) to smaller spin crossover complexes [137]. Wavefunction approaches are not without challenges and in some cases still produce sizeable, 5 kcal/mol energetic errors in spin-state ordering [136]. However, they are typically in very good agreement with experimental spin crossover properties and are a suitable reference for benchmarking of exchange-correlation func-tionals for higher throughput studies. Despite advances in wavefunction theory, ap-proximate density functionals are still preferred by most computational researchers due to ease of use and lower scaling that makes geometry optimization and high-throughput calculations feasible.
Extending study [119–121] of how inorganic complex spin states vary with func-tional choice can broaden an understanding of the ways in which hybrid funcfunc-tionals improve predictions of spin-state orderings, especially since both low [120, 139, 140] and high [119, 141, 142] percentages of Hartree-Fock exchange have been proposed for the description of transition metal complexes. Rather than focusing on finding one prescription for exchange, we aim to understand the way in which relative energetic, electronic, and structural properties of spin states are sensitive to these descriptions. Understanding this variability unifies many functionals and can provide a useful guide for interpreting the prediction bias introduced through functional choice in DFT liter-ature. Finally, we aim to enlarge a quantitative understanding of how self-interaction error manifests and is balanced through the use of Hartree-Fock exchange.
While B3LYP is commonly employed to successfully describe organic systems, its direct application to organometallics leads to mixed results. One approach is to ad-just the extent of Hartree-Fock exchange in a functional in order to reproduce key energetics and spin-state orderings in organometallic systems where multiple spin mul-tiplicities lie close in energy [115, 119, 139–142]. However, such exchange-correlation
functional tuning is then constrained by the availability of experimental data or well-converged correlated quantum chemistry results. Importantly, the outcome from these fitting studies are often contradictory: alternative mixings of 0% [120], 15% [139,140], 25% [141, 142], and 30-50% [119] HF exchange have all been proposed for Fe(II) oc-tahedral complexes alone. Such broad outcomes suggest that the mixing of exact exchange in a functional is highly dependent on the underlying chemistry of the system, and a one-size-fits-all approach is not likely to be successful. Preliminary success has been made in identifying chemically-motivated ways to tune functional parameters [143–145] outside of organometallic chemistry. The appropriate tuning for organometallic complexes or correlated materials is largely still approximated on local measures of the chemical potential of the subshell [130] of interest, and efforts to improve functionals on transition metal test sets have indicated no clear path to optimization [146].
2.1
Computational details
Calculations were carried out using the TeraChem [147] package for all LDA, GGA, and GGA hybrid calculations. The default B3LYP definition in TeraChem uses the VWN1-RPA form for the LDA VWN [4] component of LYP correlation [10]. Initial calculations on GGA hybrids also considered the effect of using instead the 3-parameter or 5-parameter forms of the VWN correlation (B3LYP3, B3LYP5 key-words) as well as using other forms of the correlation with the B3P86 [9,148], B3PW91 [6,9], PBE0 [12,13] (25% HF exchange vs. 20% in B3LYP), or B97 [149] (19% HF ex-change) GGA hybrids. Overall qualitative GGA hybrid predictions were unchanged and therefore B3LYP1 is chosen as the representative functional.
Altered Hartree-Fock exchange percentages in a modified form of B3LYP were implemented in TeraChem for this work. Meta-GGA calculations were carried out with Q-Chem 4.2. All calculations were performed using the LANL2DZ effective core potential basis for the iron atom and the 6-31G* basis for the other atoms. Geometry optimizations were carried out using the L-BFGS algorithm in Cartesian coordinates,
as implemented in DL-FIND [150], to default thresholds of 4.5x10–4 hartree/bohr for the maximum gradient and 1x10–6 hartree for the change in SCF energy between steps.
High-spin states (quintet multiplicity for Fe(II) and sextet for Fe(III)) are com-pared against low-spin states (singlet for Fe(II) and doublet for Fe(III)). Intermediate spin states were not considered. Oxidation states are qualitative and obtained by constraining total charge to correspond to the net charge on the respective ligands along with a positive (+2 or +3) charge for the iron center. Quantitative determina-tion of the charges and occupadetermina-tion of subshells (i.e. 3d and 4s) was obtained from the TeraChem interface with the Natural Bond Orbital (NBO) v6.0 package [151]. NBO calculates the natural atomic orbitals (NAOs) for each atom by computing the orthogonal eigenorbitals of the atomic blocks in the density matrix. After the set of NAOs is defined, NAO occupancy is obtained using natural population analysis (NPA) [152], which permits estimation of 3d and 4s subshell occupation. The NBO partial charge (q) on an atom is calculated by taking the difference between the atomic number (Z) and the total population (N) for the NAOs for each atom (i):
qi = Zi– Ni. (2.1)
Several octahedral complex structures (ligands: CO, CN–, CNH, NCH, NH3,
H2O) were generated from simplified molecular input line entry system (SMILES) [61] strings. Using OpenBabel [68], the SMILES strings were converted to structures that were starting points for TeraChem geometry optimizations. The larger octahedral complex structures (ligands: (phen)2(NCS)2, PEPXEP, HICPEQ, bpy, terpy), were
obtained from the Cambridge Structural Database (CSD) [78]. PEPXEP denotes the CSD accession code for a compound with N6C26H38 stoichiometry, while HICPEQ
corresponds to a N8C18H26 compound. The (phen)2(NCS)2 structure was previously identified as a good test case [139]. The other ligands were selected by using the CCDC ConQuest web-screening tool with a query limiting elements to Fe, C, N, H in an octahedral complex with symmetric Fe-N bonds, as was previously used for
catalyst screening [153].
2.2
Dependence of spin-state ordering on functional
choice
We have considered a test set of representative Fe(II) and Fe(III) octahedral com-plexes (Fig. 2-1) for various exchange-correlation functionals. In all cases, the ground state spin is known experimentally or may be suggested from ligand field theory. Fe(II) and Fe(III) have nominally 3d6 and 3d5 electron configurations, giving rise to low-spin (LS) singlet or doublet spin multiplicity or high-spin (HS) quintet or sextet electronic states. The adiabatic electronic energy gap between HS and LS states is:
ΔEHS–LS
= EHS(RHS) – ELS(RLS), (2.2)
where EHS(RHS) is the electronic energy of the HS state at its geometry optimized coordinates and ELS(RLS) is the equivalent for the LS state. The initial set of struc-tures includes two carbon ligand sets (CO and CNH), three nitrogen ligand sets (NH3, NCH, and (phen)2(SCN)2), and one oxygen ligand set (H2O) (see structures in Fig. 2-1). One representative functional is chosen for each class: LDA (PZ81 [125]), GGA (PBE [153]), GGA hybrid (B3LYP) and meta-GGA (M06-L [154]) to compare qualitative relative high-spin/low-spin energetics. Reliance on a single representative functional for each class is motivated by preliminary findings in comparing a wider array of functionals. Pure density functionals (LDA or GGA) consistently predict low-spin ground states in nine of the ten cases (six are Fe(II) and four are Fe(III)) considered (Table 2.1), although only half of the ten cases are expected to be low spin.
Pure GGA preference for low-spin Fe(II)/Fe(III) complexes is consistent with earlier observations [29, 119, 157]. Including higher order dependence on the density as in a meta-GGA improves identification of some high-spin states: Fe(II)(NH3)6 and Fe(III)(NCH)6 are predicted to be high spin with a meta-GGA, while they were
Figure 2-1: Structures of octahedral iron complexes classified by direct ligand identity: carbon (top), nitrogen (middle), or oxygen (bottom).
predicted to be low-spin with a GGA. However, meta-GGA results are inconsistent: Fe(III)(NH3)6and Fe(II)(NCH)6have high-spin ground states [119,156] but the
meta-GGA predicts both to be low-spin. Identification of how the higher-order terms of the density may be systematically incorporated to improve predictions of magnetic ordering or spin states is of ongoing interest since meta-GGAs have the potential to improve predictions in extended systems where explicit incorporation of
Hartree-Structure Reference LDA GGA hybrid meta-GGA FeII(CO)6 LS LS LS LS LS FeII(H2O)6 HS HS HS HS HS FeII(CNH)6 LS LS LS LS LS FeII(NCH)6 HS LS LS HS LS FeII(NH3)6 HS LS LS HS HS FeII(phen)2(NCS)2 LS LS LS HS LS FeIII(NH3)6 HS LS LS LS LS FeIII(NCH)6 HS LS LS HS HS FeIII(CNH)6 LS LS LS LS LS FeIII(CO)6 LS LS LS LS LS
Table 2.1: Ground states of octahedral Fe(II) and Fe(III) complexes with speci-fied ligand sets for LDA, GGA, GGA hybrid, and meta-GGA classes of exchange-correlation functionals. Reference data are from experiment (indicated in bold), oth-erwise approximations from ligand-field theory are provided. Incorrect predictions of the ground state spin for a functional are indicated by red color. The experimen-tal data are from those collected in Ref. [119], except for Fe(II)(H2O)6 (Ref. [155]), Fe(II/III)(NH3)6 (Ref. [156]), and Fe(II)(phen)2(NCS)2 (Ref. [139]).
Fock exchange may be prohibitive (see Chapter 3). For the GGA hybrid class of functionals, correct qualitative identification of spin states is achieved in eight out of ten cases. However, in the case of (phen)2(NCS)2, a high-spin ground state is predicted despite experimental observation [139] of a low-spin ground state. While this test set is relatively small, it reinforces general observations that GGA hybrids tend to over-predict high-spin ground states, while pure density functionals predict low-spin ground states. This trend will be investigated on an expanded molecule test set in Sec. 2.3.
Qualitative spin-state assignment is difficult in weak ligand cases where the quan-titative gap falls below 5 kcal/mol due to basis set dependence or zero-point energy and vibrational entropy effects [119, 121] not considered here but covered in detail in the recent work by Mortensen and Kepp [121]. For the GGA, Fe(II)(NH3)6 is close to
crossover to high-spin, which would improve agreement with experiment. Three of the meta-GGA predictions: Fe(II)(NCH)6, Fe(III)(NH3)6, and Fe(II)(phen)2(NCS)2, are
close to the spin crossover point to HS states, which would improve agreement with experiment in the first two cases but worsen agreement for the last case. We also note
that these meta-GGA results may be more substantially sensitive to the functional form since M06-L, for instance, is highly parameterized. We thus compare against TPSS [8], a meta-GGA with fewer adjustable parameters. Comparing the TPSS and M06-L meta-GGAs, we find the two are qualitatively consistent, but TPSS has a stronger bias for high spin systems. This bias leads to improved qualitative agree-ment for two compounds (Fe(II)(NCH)6 and Fe(III)(NH3)6) as high-spin but also reduced qualitative agreement for two low-spin compounds that TPSS predicts to be high-spin (Fe(II)(phen)2(NCS)2 and Fe(III)(CO)6).
2.3
Dependence of spin-state ordering on HF
ex-change
In order to broadly investigate the effect of HF exchange on spin-state ordering, we vary the amount of HF exchange included in a modified B3LYP (modB3LYP) functional. The DFT exchange for the modB3LYP functional is calculated using the following expression:
EmodB3LYPx =αHFEHFx + (1 –αHF)ELDAx + 0.9(1 –αHF)(EGGAx – ELDAx ), (2.3)
where αHF is the amount of HF exchange. For αHF → 0, the exchange is pure DFT-GGA (as in BLYP), while for αHF → 1, the exchange becomes pure HF. The factor 0.9 was introduced so that the ratio
EGGAx
ELDAx = 9 (2.4)
is equal to that of the original B3LYP functional (0.72 for EGGAx and 0.08 for ELDAx ) and constant for all αHF. We apply the modB3LYP functional (with HF exchange = 0-50%) to select octahedral complexes from the initial test set (Sec. 2.2) as well as an expanded test set. Throughout, we also compare to a narrow range of 12.7-28.3%, which corresponds to 3σ confidence interval on the normal distribution fit to
the votes for standard hybrid exchange-correlation functionals in a popular density functional theory poll [96]. While the narrower range indicates the most common hybrid exchange ratios, the wider range permits connection to both pure GGA and high HF exchange functionals.
2.3.1
Spin-state ordering dependence with Fe(III) complex test
cases
We first focus on the relative electronic energy between high-spin (HS) and low-spin (LS) electronic states (ΔEHS–LS) for four Fe(III) octahedral complexes (N ligands: NCH, NH3, C ligands: CNH, CO) across the 0-50% HF exchange range (Fig. 2-2). Fe(III) complexes have a d5 configuration that will lead to complete filling of all
d levels in the high-spin case or a paired, closed-shell doublet in the low-spin case.
Linear behavior in spin-state energetics is observed over the complete range of HF exchange covered with modB3LYP exchange for Fe(III) complexes with both carbon and nitrogen ligands, extending and confirming earlier observations by Droghetti on octahedral Fe(II) complexes [119] over a range of about 15-40% HF exchange.
This linear energetic dependence is surprising because it suggests that any re-sponse that the density has to the modified HF potential is of the same magnitude in both the high-spin and low-spin state. That is, it is evident that simply mixing increasing fractions of HF exchange energy on a high-spin state will lower its energy linearly with respect to a low-spin state. However, if the density responds differently in the case of the low-spin state, e.g. through increased localization with respect to the high-spin state due to imbalances in self-interaction error, then the energetic dependence should contain higher order terms than a simple linear averaging. This linear result suggests that HF-derived localization of covalent, delocalized orbitals may not be suitable for understanding the effect of higher fractions of HF exchange. A comparison to self-interaction correction schemes [124, 125] and the delocalization-penalty +U approach [130] will likely be instructive in the future.
0 10 20 30 40 50 % HF exchange -20 0 20 40 60 80 ΔE HS-LS (kcal/mol) Fe(III)(CNH)6 Fe(III)(CO)6 Fe(III)(NH3)6 Fe(III)(NCH)6
Figure 2-2: Relative high spin (HS)-low spin (LS) energy (ΔEHS–LS) in kcal/mol of four Fe(III) octahedral complexes (two nitrogen ligands: NCH and NH3 and two carbon ligands: CNH and CO) with 3σ confidence interval from normal distribution poll data on hybrid exchange functionals, as indicated with black dashed lines and black arrow.
good approximations to the partial derivative of the energy with respect to HF ex-change (αHF), slope = ∆∆E HS–LS ∆αHF ≈ ∂∆EHS–LS ∂αHF , (2.5)
where the correlation coefficients (i.e., R2 values) for these fits are all 0.999. We introduce here the unit notation "HFX", where on unit of HFX corresponds to the range from 0% to 100% HF exchange. The identify of the directly bonded element dominates the value of ∂∆E∂αHS–LS
HF , and nitrogen-containing ligands have nearly iden-tical values: -75 molkcal·HFX for Fe(III)(NH3)6 and -77 molkcal·HFX for Fe(III)(NCH)6. For carbon-containing ligands, the correspondence is also quite close: -110 molkcal·HFX for Fe(III)(CNH)6 and -106 molkcal·HFX for Fe(III)(CO)6. Over the 3σ confidence interval, the carbon ligand sets always prefer a low-spin ground state, but ΔEHS–LS is re-duced by 17 kcal/mol over this range, which is a significant change in predictions of quantitative energetics. For comparison, the shift in ΔEHS–LS from BLYP (0%) to B3LYP (20%) is larger at around -21 kcal/mol, and the difference between 20% and 25% HF exchange shifts the prediction by -5 kcal/mol. The ratio of Hartree-Fock
exchange and the direct ligand dominates these trends, rather than the form of the DFT exchange or the associated correlation functional. When calculations are carried out with a modified PBE0 functional instead of modB3LYP, slopes are qualitatively unchanged, with an average difference of 6% in computed slopes and a maximum deviation of -9 molkcal·HFX for Fe(III)(CO)6.
While the spin-state splitting derivatives are smaller for the nitrogen-containing ligands, the proximity of the curves to the HS-LS crossover makes the qualitative spin-state assignment more challenging. Furthermore, both Fe(III)(NH3)6and Fe(III)(NCH)6
are low-spin at the lower bound of the 3σ confidence interval (αHF=0.127), while they are both high-spin at the upper bound of that same interval (αHF=0.283). If the objec-tive of a computational study is qualitaobjec-tive spin-state assignment, such an assignment would be highly sensitive to functional choice. Experimentally [8], Fe(III)(NH3)6 is
known to be high-spin, but HS-LS spin crossover occurs at 27.2% HF exchange, which is a higher fraction than is incorporated in B3LYP or PBE0. Despite challenges in qualitative assignment, quantitative changes in spin-state orderings are slightly lower: the shift inΔEHS–LS in the confidence interval is -12 kcal/mol, from BLYP to B3LYP it is -15 kcal/mol, and the difference between a 20% and 25% HF exchange is -4 kcal/mol.
Previous work in this area [121,158–160] suggests that a ligand field theory picture that focuses on ligand strength following the spectrochemical series [161] may provide some, albeit tenuous, guidance regarding observations in HS-LS splitting. The CO ligand is the strongest in the spectrochemical series and should maximize octahedral field splitting (Δo) between the three low-energy t2g and two high-energy eg states,
while the NH3 ligand is considerably weaker and should have a smaller Δo value.
Across the range of all HF exchange percentages, the LS state is relatively preferred for the strong CO with respect to the NH3 ligand. However, for high HF exchange
(40-50%), the HS state is the ground state for Fe(III)(CO)6and the relative penalty of ΔEHS–LS(CO) vs. ΔEHS–LS(NH
3) narrows. In a simplified LFT picture, increasing
HF exchange is modulating the octahedral field splitting more dramatically for strong ligands (e.g. CO) than for weak ligands (e.g. NH3). Thus, these trends suggest that
too-high ratios of exact exchange in functionals will override established ligand field concepts.
2.3.2
Spin-state ordering: comparison with Fe(II) complexes
Qualitative trends in spin-state ordering with αHF(Fig. 2-3) previously observed for Fe(III) are preserved in Fe(II), but with slightly lower correlation coefficients (R2 = 0.995-0.997). For Fe(II), carbon ligand systems have higher ∂∆E∂αHS–LS
HF values: -151
kcal
mol·HFX for Fe(II)(CNH)6 and -155
kcal
mol·HFX for Fe(II)(CO)6, and this higher slope
appears to correlate with higherΔEHS–LS versus Fe(III) obtained at a GGA reference by 20 kcal/mol. Such a difference between Fe(II) and Fe(III)ΔEHS–LS diverges from ligand field theory, since in LFT, Fe(II) does not populate any additional high-energy levels in the HS or LS state. The Fe(II)(NH3)6 and Fe(II)(NCH)6 ∂∆E
HS–LS
∂αHF values diverge slightly from their Fe(III) counterparts, reducing the slope to -63 molkcal·HFX in the former case and increasing to -86 molkcal·HFX in the latter. Nevertheless, trends are preserved: here, spin-crossover from LS to HS occurs near the lower bound of the 3σ confidence interval, while both ligands prefer high spin at the upper bound.
Despite high correlation coefficients, the fit to linear trend lines appears poorer in the case of Fe(II) compared to Fe(III). It is likely that extra degrees of freedom associ-ated with the unpaired minority-spin 3d electron in Fe(II) make energetic predictions more sensitive to HF exchange ratios. Quadratic fits of the data were thus also ob-tained, leading naturally to an improved fit. First derivatives of the second order polynomials give access to a range of instantaneous ∂∆E∂αHS–LS
HF values. By definition, the derivatives obtained at 25% HF exchange from either approach match exactly, but this single value is an underestimate for low and an overestimate for high αHF. The carbon ligand slope ranges are from -106 (αHF=0.5) to -206 (αHF=0.0) for CO and -114 (αHF=0.5) to -183 (αHF=0.0) for CNH. Nitrogen ligand ranges are slightly smaller: -48 to -81 for NH3 and -50 to -125 for NCH. Such ranges are subject to the
number of data points and nature of the higher order fit, but they provide a reference frame for evaluating the magnitude of variation of derivatives. Thus, although there