Application of synchrotron radiation for measurement of iron red-ox speciation in atmospherically processed aerosols
Texte intégral
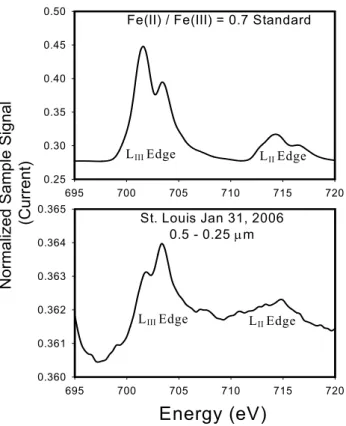
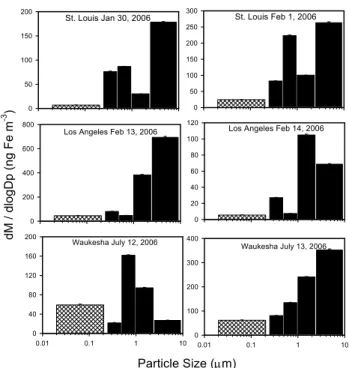
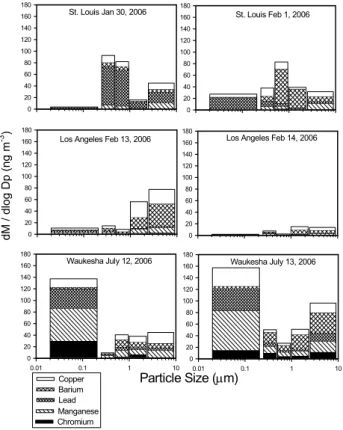
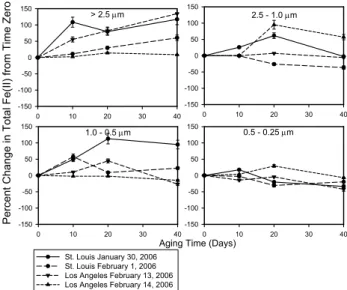
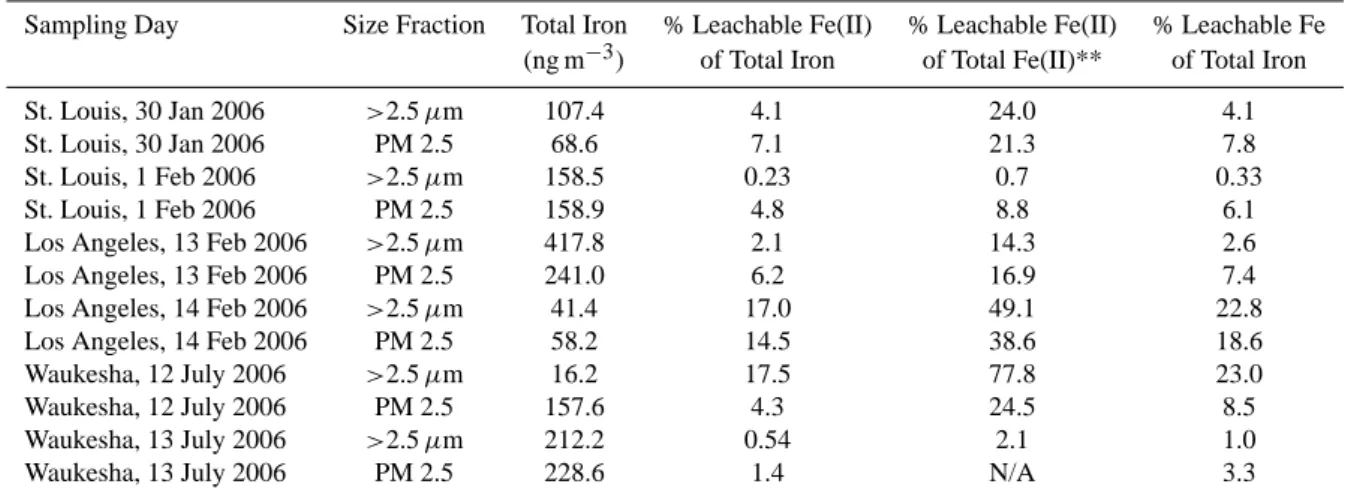
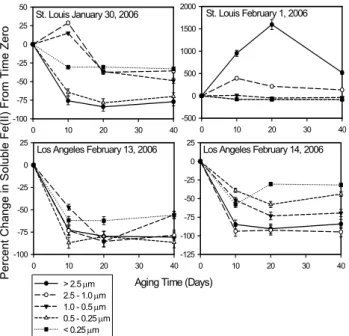
Figure


Documents relatifs
Therefore, an educational resource manual related to Lewy body dementia is a beneficial resource that can be utilized by nursing staff in long-term care to promote
On figure 1C, we see the following behavior: after a trigger, the value is correctly updated in short-term memory and remains stable until C is updated (after 100 time steps) and
In this study, LSTM (Long-Short Term Memory) neural network models are constructed to predict travel time for both long term and short term using real world data of New York
o The results imply that the microencapsulation technique could reduce potential interaction of iron with other food components, reduce its exposure to
AMM (année de commercialisation en France) Posologie (instauration du traitement) Nombre de prises par jour Principaux effets secondaires Interactions médica-
Under ambient conditions, the thiol groups from DMSA are not stable and do not allow a direct functionalization without storage in stringent conditions or a chemical regeneration
In addition to this study, in order to describe the composition of small eukaryotes and potentially to observe changes in their structure, we used a similar microcosm experiment
Soil corrosion can also be driven locally by galvanic coupling between ferrite and cementite (Fe 3 C, enriched in carbon). 3) At a certain stage of corrosion, cracks are formed in
