HAL Id: hal-02661835
https://hal.inrae.fr/hal-02661835
Submitted on 30 May 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
phosphorylation
Cristel Archambaud, Marie-Anne Nahori, Javier Pizzaro-Cerda, Pascale
Cossart
To cite this version:
Cristel Archambaud, Marie-Anne Nahori, Javier Pizzaro-Cerda, Pascale Cossart. Control of Listeria
superoxide dismutase by phosphorylation. Journal of Biological Chemistry, American Society for
Biochemistry and Molecular Biology, 2006, 281 (42), pp.31812-31822. �10.1074/jbc.M606249200�.
�hal-02661835�
Control of Listeria Superoxide Dismutase
by Phosphorylation
*
□SReceived for publication, June 29, 2006, and in revised form, July 27, 2006Published, JBC Papers in Press, August 11, 2006, DOI 10.1074/jbc.M606249200 Cristel Archambaud1, Marie-Anne Nahori, Javier Pizarro-Cerda, Pascale Cossart2, and Olivier Dussurget3 From the Institut Pasteur, Unite´ des Interactions Bacte´ries-Cellules, Inserm, U604, INRA, USC2020, F-75015 Paris, France
Superoxide dismutases (SODs) are enzymes that protect or-ganisms against superoxides and reactive oxygen species (ROS) produced during their active metabolism. ROS are major medi-ators of phagocytes microbicidal activity. Here we show that the cytoplasmic Listeria monocytogenes MnSOD is phosphorylated on serine and threonine residues and less active when bacteria reach the stationary phase. We also provide evidence that the most active nonphosphorylated form of MnSOD can be secreted via the SecA2 pathway in culture supernatants and in infected
cells, where it becomes phosphorylated. A⌬sod deletion mutant
is impaired in survival within macrophages and is dramatically attenuated in mice. Together, our results demonstrate that the capacity to counteract ROS is an essential component of
L. monocytogenes virulence. This is the first example of a
bac-terial SOD post-translationally controlled by phosphoryla-tion, suggesting a possible new host innate mechanism to counteract a virulence factor.
Reactive oxygen species (ROS)4are continuously produced
by multiple enzymes within cells. Whereas a significant amount of ROS is generated in the cytosol of eukaryotic cells, in peroxi-somes and at the plasma membrane, oxidative phosphorylation by the mitochondrial respiratory chain is the main cellular source of ROS. Cells beneficially use ROS as antimicrobial agents (1) and regulators of stress signaling pathways, e.g. heat
shock response, NF-B, and p53 activation, phosphatidylinosi-tol 3-kinase/Akt and mitogen-activated protein kinase cascades (2, 3). However, uncontrolled production of ROS is deleterious to cells because they can damage nucleic acids, proteins, and lipids (4). Cellular response to oxidative stress is believed to be a major determinant of lifespan (5, 6). Moreover, accumulation of ROS is associated to human pathology including hyperglyce-mic damages (7), carcinogenesis and tumor progression (8), and neurodegenerative disorders, e.g. Alzheimers, Parkinsons (9), and prion diseases (10). To prevent oxidative damage, cells synthesize antioxidant systems. Superoxide dismutase (SOD) catalyzes the conversion of superoxide radical anions to hydrogen peroxide, using as cofactors manganese in the mitochondria or copper and zinc, extracellularly and in the cytosol. Mutations in the human copper-zinc SOD (CuZn-SOD) are associated with a dramatic genetic disease, i.e. familial amyotrophic lateral sclerosis (11), highlighting the importance and key role of SOD in life. Bacterial SODs are cytoplasmic, periplasmic, or secreted enzymes. They can bind nickel or iron in addition to manganese, copper, and zinc and are involved in basic processes such as growth, senescence, sporulation, and also virulence (12, 13). The expression of both eukaryotic and bacterial SODs is tightly controlled at the transcriptional and post-transcriptional levels (14 –18). To our knowledge, post-translational regula-tion of bacterial SODs has not been reported.
Listeria monocytogenesis a facultative intracellular pathogen causing a severe food-borne disease in humans and animals (19). Neutrophiles and macrophages are critical cells of host defense against L. monocytogenes (20, 21). Once phagocytosed,
L. monocytogenesfaces the phagosomal oxidative burst (22) and then escapes from the phagosome because of the secretion of listeriolysin O (23), phosphatidylinositol-specific phospho-lipase C (24), and other proteins (25). However, how L.
mono-cytogenesreacts to this bactericidal aggression has remained elusive. A single sod gene, which encodes a functional manga-nese-SOD (MnSOD) has been identified (26 –28) but it has remained poorly characterized.
Here, we report that L. monocytogenes MnSOD activity is down-regulated by serine/threonine phosphorylation during the stationary phase. We show that the most active, nonphos-phorylated form of MnSOD is secreted via the SecA2 pathway in bacterial culture and in infected cells where it is phosphoryl-ated. Inactivation of MnSOD by gene deletion resulted in increased bacterial death within macrophages and dramatic attenuation in mice, demonstrating that the antioxidant poten-tial is a critical factor for L. monocytogenes pathogenesis. *This work was supported in part from Institut Pasteur (GPH N°9); INRA,
INSERM, and the French Ministry of Research (Programme de Microbiolo-gie Fondamentale et Applique´e, Maladies Infectieuses, Environment et Bioterrorisme ACI N° MIC 0312). The costs of publication of this article were defrayed in part by the payment of page charges. This article must there-fore be hereby marked “advertisement” in accordance with 18 U.S.C. Sec-tion 1734 solely to indicate this fact.
□S The on-line version of this article (available at http://www.jbc.org) contains
two supplemental experimental procedures, one supplemental Table S1, and four supplemental Figs. S1–S4.
1Supported by fellowships from the Ministe`re de la Recherche, the
Pasteur-Weizmann Foundation, EuroPathoGenomics (NoE, Contract N° LSHB-CT-2005-512061) and the Howard Hughes Medical Institute.
2An International Research Scholar of the Howard Hughes Medical Institute.
To whom correspondence may be addressed. Tel.: 33-1-40-61-30-32; Fax: 33-1-45-68-87-06; E-mail: [email protected].
3To whom correspondence may be addressed: Unite´ des Interactions
Bacte´-ries-Cellules, Institut Pasteur, 25 rue du Dr. Roux, 75015 Paris, France. E-mail: [email protected].
4The abbreviations used are: ROS, reactive oxygen species; SOD, superoxide
dismutase; CuZnSOD, copper and zinc superoxide dismutase; MnSOD, manganese superoxide dismutase; PKA, protein kinase A; AP, alkaline phosphatase; BSA, bovine serum albumin; RNS, reactive nitrogen species; XO, xanthine oxidase; HX, hypoxanthine; NO, nitric oxide; PEM, peptone-elicited peritoneal macrophages; IFN-␥, interferon ␥; MOI, multiplicity of infection; CFU, colony-forming unit; FeSOD, iron superoxide dismutase; PBS, phosphate-buffered saline; WT, wild type.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 42, pp. 31812–31822, October 20, 2006 © 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions—All L.
monocyto-genesstrains were routinely grown in brain heart infusion (BHI) medium (Difco) at 37 °C. When required, chloramphenicol and erythromycin were added at 7g/ml and 5 g/ml, respectively.
Escherichia coli strains were grown in Luria-Bertani (LB) medium (Difco) at 37 °C. When required, antibiotics were included at the following concentrations: ampicillin, 100 g/ml, chloramphenicol 7 g/ml.
Antibodies and Western Blot Techniques—Murine poly-clonal anti-MnSOD serum, produced as described (29), was used at 1:1000 in 4% Blotto. Rabbit polyclonal antiphospho-serine and antiphosphothreonine antibodies (Zymed Laborato-ries Inc.) were diluted at 1:1000 in blocking buffer (Zymed Lab-oratories Inc.). Rabbit polyclonal anti-Stp R96 (30), anti-ActA R32 (31) purified antibodies and Listeria R11 (32), anti-InlC R117 sera5were used at 1:1000 in 4% Blotto. After
separa-tion on 10% SDS-PAGE, proteins were detected by Western blotting as previously described (30).
Expression and Purification of the L. monocytogenes MnSOD— The coding region of sod (lmo1439), excluding the start codon, was amplified by PCR using genomic DNA from
L. monocytogenesEGDe (BUG 1600) and the oligonucleo-tides AC38F and AC39F (supplemental Table S1). The PCR product was digested by NdeI and XhoI and cloned into the expression vector pET-28b (Novagen), creating pET-28b(sod), that was maintained in E. coli XL-1 blue (BUG 2126). E. coli BL21 (DE3) was transformed with pET-28b(sod) and grown at 37 °C to A600 nm⫽ 0.8. Overexpression of the N-terminal
histi-dine-tagged MnSOD was induced by addition of isopropyl-1-thio--D-galactopyranoside (0.5 mM). After 4 h, cultures (200
ml) were harvested. The bacterial pellet was resuspended in 20 ml of binding buffer (20 mMTris, pH 7.4, 300 mMNaCl, 1 mM
AEBSF, 1 tablet of Complete protease inhibitor mixture, Roche Applied Science). Bacteria were lysed by 5 cycles of sonication for 30 s with 15% of amplitude. The recombinant MnSOD was then purified on Probond nickel affinity column (Invitrogen) according to the manufacturer’s instructions.
Phosphorylation Analysis—Phosphoproteome analysis was performed as described (30). Briefly, the L. monocytogenes EGDe wild-type and⌬stp cultures were grown overnight in BHI then diluted at 1:10 in 200 ml of improved minimal medium (33). Bacteria were harvested when A600 nm⫽ 0.8. Sequential extraction of bacterial proteins, first dimension separation and electrophoresis in the second dimension were performed as previously described (30). For each strain, the protein extract was loaded on three gels. One gel was colored with silver stain-ing. The two other gels were analyzed by Western blotting using either the antiphosphoserine antibodies or the antiphos-phothreonine antibodies. Spots excision from silver-stained gels and protein identification by mass spectrometry were per-formed by Proteomic Platform (Genopole, Institut Pasteur, Paris, France) (34). For in vitro phosphorylation, L.
monocyto-genespurified MnSOD (60g) was incubated with 2700 units of purified cAMP-dependent protein kinase (PKA) catalytic
sub-unit from bovine heart (Sigma) in PKA buffer (50 mMTris-HCl,
pH 7.5, 10 mMMgCl2, 1 mMEGTA, 2 mMdithiothreitol, 0.01%
Brij 35) overnight at 30 °C in the presence of 1 mMATP. Two
dialysis were performed in 2 liters of Stp buffer (50 mM Tris-HCl, pH 7.5, 1 mMof MnCl2, 0.1 mMNa2EDTA, 5 mM
dithio-threitol, 0.01% Brij 35) for 4 h at 4 °C to eliminate residual ATP. Phosphorylated MnSOD (30g) was dephosphorylated by Stp (6g) for 1 h at 37 °C in Stp buffer. Ten micrograms of phos-phorylated P-␣-casein (Sigma) were dephosphos-phorylated using 1 g of alkaline phosphatase (AP, Roche Applied Science) in phosphatase buffer (Roche Applied Science) for 1 h at 37 °C. The phosphorylation level of the resulting␣-casein was com-pared with that of the initial P-␣-casein. BSA (Pierce) was used as a nonphosphorylated protein control for Pro-Q diamond staining. MnSOD, PKA, and Stp-purified proteins, MnSOD samples after PKA dephosphorylation and Stp dephosphoryla-tion, P-␣-casein-, ␣-casein-, and BSA-purified control proteins (2g) were separated by SDS-PAGE. Gels were stained with the Pro-Q and Sypro Ruby protein gel dyes (Molecular Probes) as previously described (35).
Quantification of L. monocytogenes MnSOD Activity—SOD activity was measured using the Bioxytech SOD-525 kit (Oxis research). Briefly, 4g of purified MnSOD were phosphoryla-ted with PKA and dephosphorylaphosphoryla-ted by Stp as described above. Samples and blanks were diluted in 40l of H2O and incubated
in 900l of SOD-525 buffer at 37 °C. Thirty microliters of the R2 reagent were added and incubated at 37 °C for 1 min before addition of 30l of the R1 reagent. A525 nmwas measured every
3 s for 2 min.
Preparation of Total Bacterial Extracts and Supernatant Precipitation—Cultures (10 ml) of the L. monocytogenes EGDe wild-type and⌬stp mutant strains were harvested at different time points. Bacterial pellets were recovered after centrifuga-tion at 4000 rpm for 20 min, washed twice in PBS and resus-pended in 100l of B-PERII Bacterial Protein Extraction Rea-gent (Pierce) to extract total bacterial proteins. Bacterial supernatants were precipitated in 16% trichloroacetic acid on ice at 4 °C overnight. After centrifugation, the protein pellets were washed twice with 5 ml of cold acetone, dried, and resus-pended in 350l of 1MTris (pH 8.8). Protein concentration of
total bacterial extract and supernatant was determined by the conventional BCA assay (Pierce).
MnSOD Immunoprecipitation—Proteins (250g) from total bacterial extracts or from bacterial supernatants were immu-noprecipitated with 2.5l of the anti-MnSOD serum using the protein G immunoprecipitation kit (Sigma) according to the manufacturer’s instructions. Briefly, after overnight incubation of protein samples with anti-MnSOD serum, protein G beads were transferred in the spin column and incubated for 4 h at 4 °C. Beads were washed six times with 1⫻ IP buffer, one time with 0.1⫻ IP buffer and incubated with 60l of Laemmli buffer. Immunoprecipitates were recovered after centrifugation. Equivalent volumes (30l) of immunoprecipitate were sepa-rated by SDS-PAGE and analyzed by Western blotting using anti-MnSOD serum or antiphosphothreonine antibodies.
Immunofluorescence—One milliliter of cultures of the
L. monocytogenes strains growing in BHI at 6% O2was
har-vested at A600 nm⫽ 0.8. Pellets were washed and resuspended in
5E. Gouin, unpublished data.
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
1 ml of PBS. Fifty microliters of suspension were loaded on a coverslip and placed in a 24-well microplate. Bacteria were fixed in 10% paraformaldehyde for 10 min. Cells were washed in PBS and incubated 5 min in 50 mMNH4Cl. After blocking in
PBS, 0.5% BSA for 1 h, anti-MnSOD, or anti-Listeria R11 sera, both diluted at 1:100 in 200l of PBS, 0.5% BSA, were added for 1 h. After washes in PBS, anti-mouse IgG-Alexa-conjugated or anti-rabbit IgG-FITC-conjugated secondary antibodies (Molecular Probes) diluted at 1:200 in 200l of PBS, 0.5% BSA, were used to detect L. monocytogenes MnSOD or total bacteria, respectively. Cover slips were mounted with Mowiol and ana-lyzed with an AxioVert microscope (Zeiss) equipped with the Metamorph software (Universal Imaging Corporation).
Mutagenesis and Complementation—Upstream and down-stream 1-kb flanking sequences of the sod gene were ampli-fied by PCR from genomic DNA from L. monocytogenes EGDe using the oligonucleotides SodKO1/SodKO2 and SodKO3/SodKO4 (supplemental Table S1). The upstream MluI-EcoRI and downstream EcoRI-NcoI fragments were cloned sequentially into the thermosensitive vector pMAD (36). The recombinant pMAD was electroporated into
L. monocytogenesEGDe to generate the sod deletion as pre-viously described (30) except that L. monocytogenes was grown at 6% O2. Deletion of sod in the mutant strain (BUG
2225) was analyzed by RT-PCR (data not shown) and West-ern blotting (supplemental Fig. S1). For complementation, a DNA fragment containing the sod gene and the 600-bp upstream sequence was amplified by PCR from L.
monocy-togenesEGDe genomic DNA with the oligonucleotides Sod-CplmUP and SodCplmDOWN (supplemental Table S1). The BamHI-XbaI PCR product was cloned into the integrative vector pPL2 digested by BamHI and SpeI (37). The resulting recombinant plasmid was introduced in E. coli S17-1, which was used for conjugation in L. monocytogenes⌬sod as previ-ously described (37), constructing the L. monocytogenes ⌬sod⫹sod strain (BUG 2226). MnSOD expression in the ⌬sod⫹sod complemented strain was analyzed by Western blotting (supplemental Fig. S1).
Sensitivity to ROS and RNS—For the disk assay, L.
monocyto-genescultures were grown at 6% O2until A600 nm⫽ 0.6 and
plated onto BHI agar plates. Filter disks (6 mm) were placed on the agar and loaded with paraquat (570g, Sigma) and hydro-gen peroxide (210g, Sigma). Plates were incubated at 37 °C at 6% O2. For the liquid assay, L. monocytogenes cultures were
grown in BHI at 37 °C and 6% O2until stationary phase. Cul-tures were then diluted in PBS at 1:100 and incubated at 37 °C with hypoxanthine (500M, Sigma) and xanthine oxidase (0.2
units/ml, Sigma) or spermine/NO (2 mM, Sigma). Number of
CFU was assessed by plating serial dilutions in duplicate on BHI agar plates, which were incubated at 37 °C and 6% O2. Student’s
ttest was used for statistical analyses.
Infection of Murine Peritoneal Macrophages—Peritoneal macrophages (PEM) were isolated from 8-week-old female BALB/c mice (Charles River) as described (38). 72 h before the bacterial survival assay, 1⫻ 106PEM per well were
acti-vated or not using interferon␥ (IFN-␥, 100 units/ml). PEM were infected with L. monocytogenes strains (MOI ⫽ 10) growing at 6% O2to an A600 nm⫽ 0.8. PEM were centrifuged
and incubated at 37 °C for 15 min to allow bacterial phago-cytosis. Non-internalized bacteria were eliminated by three washes in RPMI supplemented with 10% fetal calf serum and 10g/ml gentamicin was added for 15 min, 1 h, and 4 h. PEM were lysed with 0.2% Triton X-100 for 10 min, and the num-ber of CFU was assessed as described in the previous section. For immunofluorescence labeling, PEM adhering onto glass coverslips were loaded over 20 min with 7.5 nM of
Lyso-tracker Red DND-99 (Molecular Probes) at 37 °C. PEM were then infected as above for 4 h at 37 °C, washed once with PBS pH 7.5, and fixed with 4% paraformaldehyde for 20 min. After quenching with 50 mMNH4Cl containing 0.05%
sapo-nin and 1% BSA for 10 min, nonspecific binding sites were blocked with 0.05% saponin and 5% horse serum during 45 min. Some coverslips were incubated for 30 min with
anti-L. monocytogenesserum R11 followed by incubation during 30 min with secondary donkey anti-rabbit antibodies cou-pled to Alexa 647 and phalloidin coucou-pled to Alexa 488 (Molecular Probes). All coverslips were mounted on Mowiol and analyzed with an AxioVert microscope (Zeiss) equipped with the Metamorph software (Universal Imaging Corpora-tion). Student’s t and ANOVA tests were used for statistical analyses.
Immunoprecipitation of MnSOD from Infected Macro-phages—PEM were activated with IFN-␥ and infected with L. monocytogeneswild-type and⌬sod mutant strains (MOI ⫽ 10) as described above. 15 min after adding gentamicin, PEM were lysed with 0.2% Triton X-100 for 10 min. Bacteria were recovered from cell lysates as previously described (39), and bacterial proteins were extracted with 100l of B-PER II Bac-terial Protein Extraction Reagent. Proteins present in PEM were recovered in 500l of Tris, 1M(pH 8.8) after precipitation
of cell lysates with 16% trichloroacetic acid. Bacterial extracts (15g) and cellular extracts (250 g) were immunoprecipitated with 2.5l of anti-MnSOD serum using the protein G immu-noprecipitation kit (Sigma) as described above. Equivalent vol-umes (30l) of immunoprecipitates were separated by SDS-PAGE and analyzed by Western blotting.
Animal Studies—L. monocytogenes EGDe cultures were grown at 6% O2 atmosphere. For quantification of bacterial
multiplication, 8-week-old female BALB/c mice (Charles River) were injected intravenously with⬃8 ⫻ 103CFU. Liver and
spleen were recovered and disrupted in 3 ml PBS at 24 h, 48 h and 72 h after infection. Serial dilutions of organ homogenates were plated on BHI agar plates and CFU determined. Animal experiments were performed according to the Institut Pasteur guidelines for laboratory animal husbandry which comply with European regulations.
RESULTS
L. monocytogenes MnSOD Can Be Phosphorylated and Phos-phorylation Down-regulates Its Activity—We previously dem-onstrated that Stp is a serine-threonine phosphatase involved in
L. monocytogenesvirulence and identified EF-Tu as its first tar-get (30). Here, we identified L. monocytogenes MnSOD as the second target of Stp using a phosphoproteomic approach. Analysis of protein extracts of the L. monocytogenes ⌬stp mutant revealed the presence of a protein phosphorylated on
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
threonine and serine residues, which was not phosphorylated in protein extracts of the wild-type strain (Fig. 1A). Mass spec-trometry analysis of the corresponding spot identified the MnSOD.
To demonstrate a direct dephosphorylation of L.
monocyto-genesMnSOD by Stp, we produced and purified a recombinant MnSOD in E. coli. The recombinant MnSOD could be phos-phorylated using PKA, a cAMP-dependent serine-threonine
kinase (Fig. 1B). Phosphorylated MnSOD could be fully dephospho-rylated in vitro by Stp (Fig. 1B).
We next examined the influence of MnSOD phosphorylation state on its activity. Dephosphorylation of the PKA-phosphorylated MnSOD more than doubled its activity, revealing that MnSOD activity can be down regulated by phosphorylation (Fig. 1C). Together, these results demon-strate that L. monocytogenes MnSOD which can be present in a phospho-rylated form in bacteria is dephos-phorylated by Stp, which thus increases its activity.
L. monocytogenes Cytoplasmic MnSOD Is Phosphorylated upon Entry in Stationary Phase and Is Secreted in a Nonphosphorylated State by the SecA2-dependent Machinery—We investigated MnSOD phosphorylation during L.
mono-cytogenes growth (Fig. 2A) and performed immunoprecipitation experiments on bacterial extracts and culture supernatants. MnSOD could be immunoprecipitated from both bacterial extracts and culture supernatants, in both exponential and stationary phases (Fig. 2B, panel 1). Whereas the secreted MnSOD was constantly found in its nonphosphorylated state, MnSOD from bacterial extracts was nonphosphorylated in exponential phase and became phosphorylated upon entry in sta-tionary phase (Fig. 2B, panel 1). Increased MnSOD phosphoryla-tion was concomitant with de-creased Stp production in bacteria (Fig. 2B, panel 2). We analyzed the global production of MnSOD dur-ing growth. The MnSOD level, which was high in bacterial ex-tracts in exponential phase, de-creased in stationary phase while the amount of MnSOD detected in culture supernatants increased (Fig. 2B, panel 2). Thus, upon entry in stationary phase, non-phosphorylated MnSOD is increasingly secreted while the remaining cytoplasmic MnSOD is progressively phosphorylated.
Using the⌬stp mutant, we next addressed the role of Stp on MnSOD dephosphorylation and production (Fig. 2B,
panel 3and panel 4). As expected, in the bacterial extracts of the⌬stp mutant, the phosphorylated form of MnSOD was
FIGURE 1. Regulation of L. monocytogenes MnSOD activity by phosphorylation. A, phosphoproteome analysis of protein extracts from wild-type EGDe strain (WT) and⌬stp mutant (30). Phosphorylation of the L.
monocytogenes MnSOD (dotted circle) on threonine residues (central panel ) and on serine residues (right panel )
was only detectable in the⌬stp mutant by Western blotting. The spots, corresponding to phosphorylated MnSOD present in silver-stained two-dimensional gels (left panel ), were analyzed by MALDI-TOF. Molecular masses are indicated on the left. B, phosphorylation and dephosphorylation of purified MnSOD in vitro. Phos-phoproteins were detected on SDS-PAGE gel stained with Pro-Q Diamond fluorescent dye. The gel was further processed for total protein staining using Sypro Ruby fluorescent stain. Recombinant MnSOD was phospho-rylated using PKA and dephosphophospho-rylated by Stp. Phosphophospho-rylated␣-casein (P-␣-casein), alkaline phosphatase (AP)-dephosphorylated␣-casein and nonphosphorylated BSA were used as phosphoprotein controls. Molec-ular masses are indicated on the left. C, MnSOD activity in vitro. SOD activity was quantified by spectrophoto-metric measurement at 525 nm every 3 s for 2 min, using the Bioxytech SOD-525 kit. Recombinant MnSOD (4 g) was phosphorylated by PKA and dephosphorylated by Stp. Mean values are expressed as the measure of
A525 nm⫾ S.D. (n ⫽ 3).
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
detected earlier, i.e. at mid-log phase, and at a higher level com-pared with the wild-type strain (Fig. 2B, panel 3). Strikingly, Mn-SOD immunoprecipitated from culture supernatants was still con-stantly detected in its nonphospho-rylated form in the⌬stp mutant. Because phosphorylated listerioly-sin O and phosphorylated EF-Tu control proteins could be detected in supernatants from the wild-type and ⌬stp mutant respectively (supplemental Fig. S2), these re-sults strongly suggest that secretion of the phosphorylated MnSOD can-not occur. We also observed that the level of MnSOD in both bacterial ex-tracts and culture supernatants was higher in the⌬stp mutant than in the wild-type strain while two controls proteins, ActA, the actin-based motil-ity protein, and the secreted interna-lin InlC, were detected in similar amounts in both strains (Fig. 2B,
panel 4). Thus, the presence of Stp controls not only MnSOD phospho-rylation but also its production.
We then investigated the secre-tion mechanism of the MnSOD, whose amino acid sequence does not contain any signal peptide. Since the accessory secretion pro-tein SecA2 had been shown to medi-ate secretion of proteins, which lack a signal peptide, in Gram-positive bacterial pathogens we analyzed supernatants from L. monocyto-genes wild-type 10403S, from a
⌬secA2 mutant, and from a complemented strain (40) and found that the MnSOD was secreted by a SecA2-dependent machinery (Fig. 2C). Thus, MnSOD belongs, along with
L. monocytogenesFbpA (41), to the increasing list of proteins lacking a signal peptide and secreted by the SecA2 pathway in pathogenic bacteria (40).
MnSOD Protects L. monocytogenes against Reactive Oxygen and Nitrogen Species-mediated Toxicity in Vitro—Because SOD is a major antioxidant enzyme, we investigated the sensi-tivity to ROS and reactive nitrogen species (RNS) of an isogenic ⌬sod mutant compared with the wild-type strain. We observed a large increase in the doubling time of the⌬sod mutant grown
FIGURE 2. Analysis of MnSOD phosphorylation, secretion, and production during L. monocytogenes growth. A, growth curve of wild-type EGDe strain (WT) and⌬stp mutant in BHI at 37 °C. Bacteria were harvested at various time points (arrows). Total bacterial proteins were extracted, and proteins from culture supernatants were precipitated. B, analysis of MnSOD phosphorylation and production in total extracts and culture supernatants from WT and⌬stp mutant. At the chosen time points, MnSOD was immunoprecipitated from WT (panel 1) and ⌬stp mutant (panel 3) bacteria, using the anti-MnSOD serum. Immunoprecipitates from total bacterial extracts and culture supernatants were separated by SDS-PAGE. MnSOD was detected by Western blotting with the anti-MnSOD serum. The IgG small chain is indicated (*). Phosphorylated MnSOD was detected by Western blotting using the antiphosphothreonine antibodies. Three additional phosphorylated proteins that could correspond to MnSOD partners, are indicated (**). Positions of the molecular mass markers are indicated on the right. At the time points indicated above, WT (panel 2) and⌬stp mutant (panel 4) bacteria were harvested. Proteins from total extracts (20g) and culture supernatants (70 g) were separated by SDS-PAGE. Anti-MnSOD serum and anti-Stp antibodies were used to detect MnSOD and Stp by Western blotting, respectively. Immunodetection of ActA and InlC control proteins was performed using the anti-ActA antibodies and the anti-InlC serum on the same samples. C, SecA2-dependent secretion of MnSOD. Proteins were precipitated from culture supernatants (80g) of the wild-type L. monocytogenes 10403S (WT), the ⌬secA2 mutant and complemented strains (⌬secA2⫹secA2) (40). MnSOD was detected by immunoblotting using the anti-MnSOD serum. Immunodetection of the InlC control protein was performed using the anti-InlC antiserum.
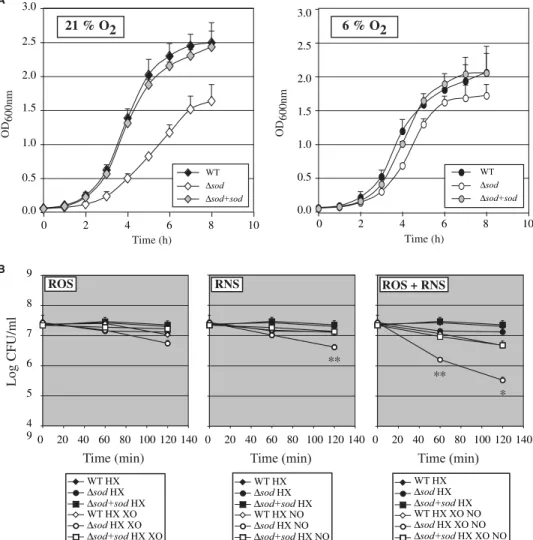
FIGURE 3. Role of L. monocytogenes MnSOD in resistance to oxidative stress in vitro. A, effect of sod deletion on aerobic and microaerophilic growth. Growth of the wild-type EGDe strain (WT), the⌬sod mutant and the complemented strain (⌬sod⫹sod) was measured in BHI at 37 °C and 21% O2(left panel ) or
6% O2(right panel ) atmosphere. Mean values are expressed as the measure of A600 nm⫾ S.D. (n ⫽ 3). B,
effect of the sod deletion on sensitivity to oxidants. Growth of the WT strain, the⌬sod mutant and the ⌬sod⫹sod strain was measured in the presence of the superoxide-generating HX-XO system and/or nitric oxide donor spermine/NO (NO). Mean values are expressed as the number of CFU⫾ S.D. (n ⫽ 3), with p values of p⬍ 0.05 (*) and p ⬍ 0.01 (**) corresponding to the comparison of the CFUs of the WT and ⌬sod mutant strains.
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
in normal atmosphere (pO2⫽ 21%) compared with the
wild-type strain and the⌬sod strain complemented with the sod gene that had a similar doubling time (Fig. 3A). In microaerophilic atmosphere (pO2⫽ 6%), the doubling time of the ⌬sod mutant
was similar to that of the wild-type and of the complemented strains (Fig. 3A).
We then tested the sensitivity of the⌬sod mutant to ROS in these microaerophilic growth conditions using a disk diffusion
assay. The⌬sod mutant was hyper-sensitive to paraquat, a reagent that increases the intracellular level of superoxides. Indeed, the diameter of growth inhibitory area of the ⌬sod mutant (36.3 mm ⫾ 0.8) was strongly increased compared with that of the wild-type and the com-plemented strains whose growth was not affected (0.0 mm and 0.1 mm ⫾ 0.1, respectively). Alterna-tively, addition of hydrogen perox-ide on filter disks resulted in a signif-icant albeit less severe difference between growth inhibition of the ⌬sod mutant (5.3 mm ⫾ 1.3) and that of the wild-type and the com-plemented strains (2.5 mm⫾ 0.5; 1.8 mm ⫾ 1.0, respectively). We next analyzed growth of the ⌬sod mutant in liquid culture media in the presence of exogenous ROS and/or RNS. ROS were generated by xanthine oxidase (XO) from hypox-anthine (HX). We first verified that addition of HX did not inhibit
L. monocytogenesgrowth (data not shown). Nitric oxide (NO) was pro-duced from spermine/NO. The ⌬sod mutant had a significantly higher susceptibility to RNS (Fig. 3B, central panel) compared with that of the wild-type and comple-mented strains. No drastic effect of the production of exogenous ROS was observed in these conditions on the growth of the⌬sod mutant (Fig. 3B, left panel). However, a strong synergistic effect of ROS and RNS on growth inhibition of the⌬sod mutant was observed (Fig. 3B, right panel). Collec-tively, these results show that MnSOD protects L.
monocy-togenesfrom exogenous ROS and RNS.
MnSOD Is Found in a Phosphorylated Form in Macro-phages—Because ROS and RNS are mediators of antibacterial activity in phagocytes, we next assessed the contribution of MnSOD to L. monocytogenes intracellular survival in macro-phages. The wild-type,⌬sod mutant and complemented strains had similar multiplication rates in the non listericidal macro-phage-like RAW264.7 cell line and in peritoneal macrophages (data not shown). However, the⌬sod mutant was killed more efficiently and significantly faster than the wild-type and com-plemented strains by listericidal macrophages, i.e. peritoneal macrophages activated with IFN-␥ (Fig. 4A).
To further compare the intracellular behavior of the wild-type,⌬sod mutant and complemented strains in peritoneal macrophages activated with IFN-␥, we indirectly analyzed bacterial escape from phagosomes, by counting after 4 h of
FIGURE 4. Role of L. monocytogenes MnSOD during infection. A, effect of the sod deletion on intracellular survival in macrophages. Peptone-elicited peritoneal macrophages (PEM, 106cells/well) from BALB/c mice
were activated with IFN-␥ and infected at a MOI of 10 with the wild-type strain (WT), the ⌬sod mutant and the complemented strain (⌬sod ⫹ sod). The number of CFU was determined at 15 min, 1 h, and 4 h after addition of gentamicin. Mean values are expressed as CFU⫾ S.D., with p values of p ⬍ 0.05 (*) and p ⬍ 0.01 (**) corresponding to the comparison of WT and⌬sod mutant CFUs. B, detection of MnSOD during PEM infection. PEM were activated with IFN-␥ and infected for 15 min with the WT strain and the ⌬sod mutant. Infected PEM were lyzed and centrifuged to separate L. monocytogenes from PEM extracts (PEM). Proteins were then extracted from bacteria recovered from infected PEM (BACTERIA) as described under “Experimental Proce-dures.” MnSOD immunoprecipitation was carried out on PEM extracts and bacterial extracts of infected PEM and on PEM extracts of non infected macrophages (NI PEM). Immunoprecipitates were analyzed by immuno-blotting using the anti-MnSOD serum raised against L. monocytogenes MnSOD and with antiphosphothreo-nine antibodies. Molecular masses are indicated on the right. C, effect of the sod deletion on L. monocytogenes survival in BALB/c mice. Multiplication of the WT strain, the⌬sod mutant and the ⌬sod ⫹ sod strain was determined in the spleen and liver of BALB/c mice infected intravenously with 8⫻ 103CFU. For each strain, CFU
were counted in organs of 4 BALB/c mice at 24, 48, and 72 h after infection. TABLE 1
Role of L. monocytogenes MnSOD on phagosomal escape in macrophages
WT ⌬sod ⌬sod ⴙ sod
% Infected PEM 99.2 99.2 100
% Infected PEM with intracytosolic bacteria labeled with phalloidin
22.3 14.3a 21.7
% Infected PEM with intracytosolic bacteria labeled with lysotracker
56.9 82.5b 63.8 ap⬍ 0.05.
bp⬍ 0.0001.
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
infection, the number of macrophages containing intracel-lular bacteria surrounded with polymerized actin in the cytosol. As shown in Table 1, all macrophages were infected with these bacterial strains. Significantly fewer⌬sod mutant bacteria were able to polymerize cytosolic actin compared with wild-type and complemented strains, suggesting that a lower number of⌬sod bacteria had reached the cytosol. To detect if actin-negative bacteria were present in a phagoly-sosomal compartment, we loaded macrophages with the lysosome marker LysoTracker Red DND-99. After 4 h of infection, the macrophages infected with the⌬sod mutant contained a much higher number of LysoTracker Red-posi-tive bacteria than the macrophages infected with the wild-type strain (Table 1). These results strongly suggested that MnSOD contributes to L. monocytogenes vacuole escape, possibly by preventing phagosomal bactericidal mechanisms.
We then determined the phosphorylation state of L.
mono-cytogenesMnSOD in peritoneal macrophages activated with IFN-␥. We immunoprecipitated MnSOD from protein extracts of bacteria recovered from infected macrophages and from cel-lular extracts of infected macrophages (Fig. 4B). As shown in the control experiment with non infected macrophages, our anti-MnSOD antibodies cross-reacted with a cellular protein, probably a SOD. However, as expected, MnSOD was detected in the bacterial extracts from cells infected with the wild-type bacteria and it was not the case when⌬sod bacteria had been used to infect cells. Strikingly, we detected a signal correspond-ing to a phosphorylated SOD in infected macrophages. Because this phosphorylated SOD was only present in macrophages infected with the wild-type strain and not with the ⌬sod mutant, it most likely corresponds to L. monocytogenes phos-phorylated MnSOD (Fig. 4B), suggesting that L. monocytogenes MnSOD can be phosphorylated in macrophages. It is unlikely that phosphorylated MnSOD was secreted from the bacteria, as this has never been observed in vitro.
MnSOD Is Required for L. monocytogenes Pathogenesis—All our results were converging to indicate that MnSOD was criti-cal for virulence of L. monocytogenes. We thus addressed the role of MnSOD in vivo. BALB/c mice were challenged intrave-nously with the wild-type, the⌬sod mutant and the comple-mented strains. In spleen and liver, which are early sites of
L. monocytogenesreplication, growth of the⌬sod mutant was strongly impaired compared with that of the wild-type strain (Fig. 4C). This difference could be detected as soon as 1 day after infection in the spleen and 2 days post-infection in the liver (Fig. 4C). At 3 days post-infection, there were 10 times fewer mutant bacteria in the spleen compared with the wild-type strain and growth of the mutant was controlled by the animals in the liver. The wild-type and complemented strains behaved similarly in animals. Thus, MnSOD is a novel virulence factor of L. monocytogenes significantly contributing to listeri-osis in mice.
DISCUSSION
In this article, we have characterized a new virulence factor in
Listeriaand the first example of a SOD down-regulated by phos-phorylation. In addition, we provide evidence that MnSOD, which is secreted as an active, nonphosphorylated form by the
SecA2-dependent machinery, plays a critical role in intracellu-lar survival in macrophages and is required for full virulence of
L. monocytogenes. The first unexpected result and the starting point of this study was that Listeria MnSOD can be phospho-rylated on serine and threonine residues. The second important result was that Listeria MnSOD can be secreted and that only the nonphosphorylated and most active form is secreted. Strik-ingly, MnSOD can become phosphorylated inside infected cells, highlighting a possible cross-talk between the host cell and the incoming microbe.
That L. monocytogenes MnSOD was present in culture supernatants as well as in the bacterial cytoplasm was at first unexpected since its amino acid sequence does not reveal any standard signal peptide. However, several pathogenic bacteria, including L. monocytogenes and Mycobacterium tuberculosis, have now been shown to secrete proteins lacking a signal pep-tide, through the SecA2 auxilliary secretion system (40 – 43). Thus MnSOD secretion occurs by the well established mecha-nism, the SecA2-dependent mechanism.
ROS are produced during growth in all living cells and thus the cytoplasmic form of MnSOD protects Listeria from endogenously produced ROS. During the oxidative burst that occurs after phagocytosis, ROS are among the main mediators of the antibacterial activity of phagocytes. Super-oxides produced in the acidic phagolysosome can directly damage targets on the bacterial surface (44). They can also penetrate inside bacteria and react with cytosolic targets such as [4Fe-4S] cluster proteins or DNA (45). In addition, superoxides can indirectly be deleterious by reacting with nitric oxide to form peroxynitrite, a toxic oxidant that can freely enter bacteria. Thus targets of ROS and RNS are pres-ent both inside bacteria and on its surface. Interestingly, our preliminary results show that L. monocytogenes MnSOD can be detected on the bacterial surface (supplemental Fig. S3), as reported for SODs from Mycobacterium leprae (46),
Mycobacterium avium(47), and Mycobacterium bovis BCG (48). Therefore Listeria and mycobacterial MnSODs may protect bacterial surface components from superoxide dam-age as documented for well-characterized periplasmic CuZnSOD from pathogenic Gram-negative bacteria (13). As shown by the increase in the bacterial doubling time in vitro, ROS and RNS are indeed toxic for Listeria and the ⌬sod mutant was much more sensitive than the wild type to ROS and RNS, definitively establishing the role of the MnSOD in protection against these radicals.
The generation of superoxides into the phagocytic vacuole has been reported to induce a potassium influx which increases the pH to the optimal value for activation of bactericidal pro-teases (49), which also participate to the phagocytic process. We observed that the⌬sod mutant was associated with acidic compartments of infected macrophages, failing to escape from phagosomes, strongly suggesting that MnSOD allows L.
mono-cytogenes to resist phagolysosomal degradation in perfect agreement with the report that localized reactive oxygen and nitrogen intermediates inhibit escape from vacuoles in acti-vated macrophages (22). As expected from its location in the infected cell, survival of the⌬sod mutant was impaired in list-ericidal macrophages. Together these results demonstrate that
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
MnSOD counteracts a host defense mechanism by destroying superoxide radicals generated by the activated macrophages, as reported for SODs produced by other pathogenic bacteria (50 – 53). In line with the role of MnSOD in intracellular survival in macrophages, this enzyme was required for full virulence in mice.
We also provide here evidence that Listeria MnSOD can be phosphorylated and that phosphorylation controls its activity. Post-translational modifications of SODs affecting their enzy-matic activity have been reported in eukaryotes. For instance, tyrosine nitration of human MnSOD decreases its activity (54) and glycation of human CuZnSOD, which is associated to dia-betis and aging, inactivates the enzyme (55–57). More recently, Csar et al. (58) reported for the first time, through a proteomic analysis, transient phosphorylation of a cytoplasmic CuZnSOD after treatment of myeloı¨d cells by the granulocyte colony stim-ulating factor. However, whether phosphorylation could regu-late SOD activity remained elusive. We demonstrate here that complete dephosphorylation of L. monocytogenes MnSOD by the serine-threonine phosphatase Stp dramatically increases its activity, possibly through conformational changes
affect-ing the active site. Listeria growaffect-ing exponentially produced the most active, nonphosphorylated MnSOD, thereby preventing dam-age from ROS produced during aerobic respiratory metabolism (Fig. 5A) (59). In contrast, when bacterial division and metabolism slow down upon entry in station-ary phase, MnSOD is found in its less active phosphorylated state. The increased phosphorylation of MnSOD observed in stationary phase is concomitant with a decreased production of the serine-threonine phosphatase Stp, con-firming the regulatory link between the two proteins (Fig. 5A). Along these lines, MnSOD phosphoryla-tion, which occurs earlier and at a higher level during growth of a⌬stp mutant, correlated with an in-creased, possibly compensatory, production of MnSOD. Interest-ingly, in mammalian cells, a link between phosphorylation of anti-oxidant proteins and growth seems to exist as inactivation of the antioxidant human peroxire-doxin I by Cdc2-dependent phos-phorylation, has been hypothe-sized to be important for cell cycle progression (60). Strikingly, a phosphorylated form of L.
mono-cytogenesMnSOD was detected in the cytoplasm of infected macro-phages. Since secreted MnSOD is constantly found in its nonphosphorylated form in L.
mono-cytogenes, we propose that a cellular kinase down-regulates MnSOD activity by phosphorylation (Fig. 5B). Secretion of proteins interfering with phagosomal maturation, e.g. M.
bo-vis BCG serine-threonine kinase G (61) and Salmonella pathogenicity island 2 proteins (62), is a strategy used by pathogenic bacteria to promote intracellular survival. Inac-tivation of secreted proteins important for bacterial survival could be a strategy used by the host phagocytes to control intracellular infection. Up to now, and to our knowledge, this is the first reported case of bacterial protein down-regula-tion through phosphoryladown-regula-tion in the host. A previous report has nevertheless documented the differential ubiquitina-tion/degradation of Salmonella type III effectors (63).
Are other bacterial SODs phosphorylated and would phos-phorylation also occur during infection with other bacteria? Our preliminary results show unambiguously that the myco-bacterial FeSOD from M. bovis BCG is phosphorylated on both serine and threonine residues and can be dephosphorylated in
vitro(supplemental Fig. S4). Interestingly, it has been reported that M. tuberculosis FeSOD, which is essentially identical to
FIGURE 5. Regulation of MnSOD activity by Stp in L. monocytogenes. A, during exponential growth phase, phosphorylation of MnSOD by an unidentified bacterial kinase is counteracted by Stp, generating a pool of highly active MnSOD. In stationary phase, decrease in the level of Stp leads to an increase in phosphorylated MnSOD, while nonphosphorylated MnSOD is actively secreted by the SecA2 system. B, within host macro-phages, phagocytosed L. monocytogenes neutralizes the oxidative burst-generated ROS by secretion of MnSOD, which in turn is possibly down-regulated by phosphorylation mediated by an unknown cellular kinase.
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
M. bovisBCG FeSOD, can be secreted by the SecA2 pathway and is critical for survival in the host (43, 64). Whether this FeSOD activity is tightly regulated by phosphorylation in myco-bacteria has not been determined. Given the present high inter-est in mycobacterial kinases and phosphatases as potential drug targets, this issue clearly deserves investigation.
In conclusion, we have shown that post-translational regula-tion of Listeria MnSOD represents a key level of control of this important enzyme. It would be interesting to investigate if a similar post-translational level of regulation also exists in humans. Activation and inactivation of SODs or of proteins regulating SODs may provide tools to fight against diseases associated to superoxide-mediated cell injury such as cancer, neurodegenerative, inflammatory, and cardiovascular diseases.
Acknowledgments—We thank Daniel Portnoy for providing L. mono-cytogenes 10403S strains, Fabienne Nicolle and Nathalie Winter for M. bovis BCG culture supernatants and Douglas Young for anti-M. tuberculosis FeSOD antibodies. We are grateful to Claire Poyart, Tony Pugsley, and Jacques D’Alayer for productive discussions. We thank members of the laboratory for helpful discussion and comments.
REFERENCES
1. Heyworth, P. G., Cross, A. R., and Curnutte, J. T. (2003) Curr. Opin.
Im-munol. 15,578 –584
2. Finkel, T., and Holbrook, N. J. (2000) Nature 408, 239 –247
3. Kamata, H., Honda, S., Maeda, S., Chang, L., Hirata, H., and Karin, M. (2005) Cell 120, 649 – 661
4. Imlay, J. A. (2003) Annu. Rev. Microbiol. 57, 395– 418
5. Balaban, R. S., Nemoto, S., and Finkel, T. (2005) Cell 120, 483– 495 6. Schriner, S. E., Linford, N. J., Martin, G. M., Treuting, P., Ogburn, C. E.,
Emond, M., Coskun, P. E., Ladiges, W., Wolf, N., Van Remmen, H., Wallace, D. C., and Rabinovitch, P. S. (2005) Science 308, 1909 –1911 7. Nishikawa, T., Edelstein, D., Du, X. L., Yamagishi, S., Matsumura, T., Kaneda, Y., Yorek, M. A., Beebe, D., Oates, P. J., Hammes, H. P., Giardino, I., and Brownlee, M. (2000) Nature 404, 787–790
8. Valko, M., Rhodes, C. J., Moncol, J., Izakovic, M., and Mazur, M. (2006)
Chem. Biol. Interact. 160,1– 40
9. Barnham, K. J., Masters, C. L., and Bush, A. I. (2004) Nat. Rev. Drug Discov.
3,205–214
10. Brown, D. R. (2005) Folia Neuropathol. 43, 229 –243
11. Selverstone Valentine, J., Doucette, P. A., and Zittin Potter, S. (2005)
Annu. Rev. Biochem. 74,563–593
12. Inaoka, T., Matsumura, Y., and Tsuchido, T. (1999) J. Bacteriol. 181, 1939 –1943
13. Battistoni, A. (2003) Biochem. Soc. Trans. 31, 1326 –1329
14. Lynch, M., and Kuramitsu, H. (2000) Microbes. Infect. 2, 1245–1255 15. Knirsch, L., and Clerch, L. B. (2001) Biochemistry 40, 7890 –7895 16. Masse, E., and Gottesman, S. (2002) Proc. Natl. Acad. Sci. U. S. A. 99,
4620 – 4625
17. Zelko, I. N., Mariani, T. J., and Folz, R. J. (2002) Free Radic. Biol. Med. 33, 337–349
18. Ahn, B. E., Cha, J., Lee, E. J., Han, A. R., Thompson, C. J., and Roe, J. H. (2006) Mol. Microbiol. 59, 1848 –1858
19. Hamon, M., Bierne, H., and Cossart, P. (2006) Nat. Rev. Microbiol. 4, 423– 434
20. Cousens, L. P., and Wing, E. J. (2000) Immunol. Rev. 174, 150 –159 21. Pamer, E. G. (2004) Nat. Rev. Immunol. 4, 812– 823
22. Myers, J. T., Tsang, A. W., and Swanson, J. A. (2003) J. Immunol. 171, 5447–5453
23. Henry, R., Shaughnessy, L., Loessner, M. J., Alberti-Segui, C., Higgins,
D. E., and Swanson, J. A. (2006) Cell. Microbiol. 8, 107–119
24. Poussin, M. A., and Goldfine, H. (2005) Infect. Immun. 73, 4410 – 4413 25. Chatterjee, S. S., Hossain, H., Otten, S., Kuenne, C., Kuchmina, K.,
Machata, S., Domann, E., Chakraborty, T., and Hain, T. (2006) Infect.
Immun. 74,1323–1338
26. Brehm, K., Haas, A., Goebel, W., and Kreft, J. (1992) Gene (Amst.) 118, 121–125
27. Vasconcelos, J. A., and Deneer, H. G. (1994) Appl. Environ. Microbiol. 60, 2360 –2366
28. Glaser, P., Frangeul, L., Buchrieser, C., Rusniok, C., Amend, A., Baquero, F., Berche, P., Bloecker, H., Brandt, P., Chakraborty, T., Charbit, A., Chet-ouani, F., Couve, E., de Daruvar, A., Dehoux, P., Domann, E., Dominguez-Bernal, G., Duchaud, E., Durant, L., Dussurget, O., Entian, K. D., Fsihi, H., Garcia-del Portillo, F., Garrido, P., Gautier, L., Goebel, W., Gomez-Lopez, N., Hain, T., Hauf, J., Jackson, D., Jones, L. M., Kaerst, U., Kreft, J., Kuhn, M., Kunst, F., Kurapkat, G., Madueno, E., Maitournam, A., Vicente, J. M., Ng, E., Nedjari, H., Nordsiek, G., Novella, S., de Pablos, B., Perez-Diaz, J. C., Purcell, R., Remmel, B., Rose, M., Schlueter, T., Simoes, N., Tierrez, A., Vazquez-Boland, J. A., Voss, H., Wehland, J., and Cossart, P. (2001) Science
294,849 – 852
29. Cooper, H. M., and Paterson, Y. (1995) in Curr. Protocol. Immunol. 2.4, 1–9
30. Archambaud, C., Gouin, E., Pizarro-Cerda, J., Cossart, P., and Dussurget, O. (2005) Mol. Microbiol. 56, 383–396
31. Steffen, P., Schafer, D. A., David, V., Gouin, E., Cooper, J. A., and Cossart, P. (2000) Cell. Motil. Cytoskeleton 45, 58 – 66
32. Gouin, E., Dehoux, P., Mengaud, J., Kocks, C., and Cossart, P. (1995) Infect.
Immun. 63,2729 –2737
33. Phan-Thanh, L., and Gormon, T. (1997) Int. J. Food Microbiol. 35, 91–95 34. Saveanu, C., Miron, S., Borza, T., Craescu, C. T., Labesse, G., Gagyi, C., Popescu, A., Schaeffer, F., Namane, A., Laurent-Winter, C., Barzu, O., and Gilles, A. M. (2002) Protein Sci. 11, 2551–2560
35. Steinberg, T. H., Agnew, B. J., Gee, K. R., Leung, W. Y., Goodman, T., Schulenberg, B., Hendrickson, J., Beechem, J. M., Haugland, R. P., and Patton, W. F. (2003) Proteomics 3, 1128 –1144
36. Arnaud, M., Chastanet, A., and Debarbouille, M. (2004) Appl. Environ.
Microbiol. 70,6887– 6891
37. Lauer, P., Chow, M. Y., Loessner, M. J., Portnoy, D. A., and Calendar, R. (2002) J. Bacteriol. 184, 4177– 4186
38. Alford, C. E., King, T. E., Jr., and Campbell, P. A. (1991) J. Exp. Med. 174, 459 – 466
39. Saito, S., Shinomiya, H., and Nakano, M. (1994) Infect. Immun. 62, 1551–1556
40. Lenz, L. L., Mohammadi, S., Geissler, A., and Portnoy, D. A. (2003) Proc.
Natl. Acad. Sci. U. S. A. 100,12432–12437
41. Dramsi, S., Bourdichon, F., Cabanes, D., Lecuit, M., Fsihi, H., and Cossart, P. (2004) Mol. Microbiol. 53, 639 – 649
42. Lenz, L. L., and Portnoy, D. A. (2002) Mol. Microbiol. 45, 1043–1056 43. Braunstein, M., Espinosa, B. J., Chan, J., Belisle, J. T., and Jacobs, W. R., Jr.
(2003) Mol. Microbiol. 48, 453– 464
44. Miller, R. A., and Britigan, B. E. (1997) Clin. Microbiol. Rev. 10, 1–18 45. Korshunov, S. S., and Imlay, J. A. (2002) Mol. Microbiol. 43, 95–106 46. Marques, M. A., Chitale, S., Brennan, P. J., and Pessolani, M. C. (1998)
Infect. Immun. 66,2625–2631
47. Escuyer, V., Haddad, N., Frehel, C., and Berche, P. (1996) Microb. Pathog.
20,41–55
48. Kang, S. K., Chung, T. W., Lee, J. H., and Kim, C. H. (2006) Protein. Expr.
Purif. 47,52–59
49. Reeves, E. P., Lu, H., Jacobs, H. L., Messina, C. G., Bolsover, S., Gabella, G., Potma, E. O., Warley, A., Roes, J., and Segal, A. W. (2002) Nature 416, 291–297
50. Franzon, V. L., Arondel, J., and Sansonetti, P. J. (1990) Infect. Immun. 58, 529 –535
51. De Groote, M. A., Ochsner, U. A., Shiloh, M. U., Nathan, C., McCord, J. M., Dinauer, M. C., Libby, S. J., Vazquez-Torres, A., Xu, Y., and Fang, F. C. (1997) Proc. Natl. Acad. Sci. U. S. A. 94, 13997–14001
52. Piddington, D. L., Fang, F. C., Laessig, T., Cooper, A. M., Orme, I. M., and Buchmeier, N. A. (2001) Infect. Immun. 69, 4980 – 4987
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org
53. Gee, J. M., Valderas, M. W., Kovach, M. E., Grippe, V. K., Robertson, G. T., Ng, W. L., Richardson, J. M., Winkler, M. E., and Roop, R. M., 2nd. (2005)
Infect. Immun. 73,2873–2880
54. MacMillan-Crow, L. A., Crow, J. P., Kerby, J. D., Beckman, J. S., and Thompson, J. A. (1996) Proc. Natl. Acad. Sci. U. S. A. 93, 11853–11858 55. Arai, K., Maguchi, S., Fujii, S., Ishibashi, H., Oikawa, K., and Taniguchi, N.
(1987) J. Biol. Chem. 262, 16969 –16972
56. Adachi, T., Ohta, H., Hirano, K., Hayashi, K., and Marklund, S. L. (1991)
Biochem. J. 279,263–267
57. Yan, H., and Harding, J. J. (1997) Biochem. J. 328, 599 – 605
58. Csar, X. F., Wilson, N. J., Strike, P., Sparrow, L., McMahon, K. A., Ward, A. C., and Hamilton, J. A. (2001) Proteomics 1, 435– 443
59. Gonzalez-Flecha, B., and Demple, B. (1995) J. Biol. Chem. 270,
13681–13687
60. Chang, T. S., Jeong, W., Choi, S. Y., Yu, S., Kang, S. W., and Rhee, S. G. (2002) J. Biol. Chem. 277, 25370 –25376
61. Walburger, A., Koul, A., Ferrari, G., Nguyen, L., Prescianotto-Baschong, C., Huygen, K., Klebl, B., Thompson, C., Bacher, G., and Pieters, J. (2004)
Science 304,1800 –1804
62. Vazquez-Torres, A., Xu, Y., Jones-Carson, J., Holden, D. W., Lucia, S. M., Dinauer, M. C., Mastroeni, P., and Fang, F. C. (2000) Science 287, 1655–1658
63. Kubori, T., and Galan, J. E. (2003) Cell 115, 333–342
64. Edwards, K. M., Cynamon, M. H., Voladri, R. K., Hager, C. C., DeStefano, M. S., Tham, K. T., Lakey, D. L., Bochan, M. R., and Kernodle, D. S. (2001)
Am. J. Respir. Crit. Care Med. 164,2213–2219
at INRA Institut National de la Recherche Agronomique, on November 8, 2010
www.jbc.org