The clinical and laboratory features of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies
10
0
0
Texte intégral
(2) Neuropathy with anti-disialosyl IgM antibodies. 1969. Introduction Over the last 20 years, acquired polyneuropathies have been increasingly recognized in association with benign monoclonal gammopathies, and this now forms a significant subset of predominantly late-onset neuropathy (Kahn et al., 1980; Thomas and Willison, 1994; Kyle, 1995; Quarles, 1997; Ponsford et al., 2000). The antigen specificities of the paraproteins are directed most frequently to carbohydrate determinants present on different glycoproteins and glycolipids distributed in neural tissue. The first clinical– serological association to be identified, and since studied in detail, was the demyelinating neuropathy associated with immunoglobulin M (IgM) paraproteinaemia reactive with myelin-associated glycoprotein and the cross-reactive glycolipid, sulphated glucuronyl paragloboside (Chassande et al., 1998; Latov et al., 1988; Quarles and Weiss, 1999). Chronic motor neuropathies were then identified in association with polyclonal or monoclonal IgM antibodies directed to GM1 and other Gal(β1–3)GalNAc-bearing gangliosides: these are now found in ~25–50% of cases of multifocal motor neuropathy with conduction block (Pestronk, 1991) depending upon the clinical definition and detection methodology used (Leger et al., 2001). In 1985, the first case of IgM paraproteinaemic neuropathy in which the paraprotein reacted with NeuAc(α2–8)NeuAc(α2–3)Galconfigured disialylated gangliosides including GD1b, GD3, GD2 and GT1b was reported (Ilyas et al., 1985), followed by other single case reports (Arai et al., 1992; Duane et al., 1992; Obi et al., 1992; Younes-Chennoufi et al., 1992; Yuki et al., 1992; Willison et al., 1993; Brindel et al., 1994; Herron et al., 1994; Hitoshi et al., 1994). Although there still remain fewer than 10 cases in the published literature, it appears that this syndrome presents a strikingly consistent clinical and serological picture, the main feature being chronic sensory ataxic neuropathy. In 1996, a patient with this phenotype who had the additional clinical features of ophthalmoplegia and cold agglutinins was described under the acronym, CANOMAD—chronic ataxic neuropathy with ophthalmoplegia, M-protein, agglutination and disialosyl antibodies (Willison et al., 1996). In the course of serum referrals to our routine diagnostic service for anti-glycolipid antibody estimation, we have identified and characterized further cases. Here we delineate the clinical syndrome and laboratory features associated with this antibody specificity in a series of 18 patients.. Patients and methods Study population This study is a retrospective analysis of patients whose sera contained IgM antibodies to disialylated gangliosides including GD1b, GD3, GT1b and GQ1b, identified as a result of requests for serological characterization of suspected or previously recognized antibodies with this specificity. Although cases have been collected on an ad hoc basis, no. cases of neuropathy, other neurological diseases or normal control subjects known to have anti-disialosyl IgM antibodies were excluded from this analysis. A standardized questionnaire on demographic, historical, clinical, investigative and electrophysiological data was issued to all referring clinicians for completion on 20 identified cases. Cases 16 (Willison et al., 1993) and 18 (Herron et al., 1994) have been published previously.. Serological methods Serological investigations, including high resolution agarose gel electrophoresis (HRAGE) coupled with immunofixation electrophoresis (IFE) for paraproteins, cold agglutinin estimation, serum and CSF analysis for protein levels, cell counts and local IgG synthesis in the CSF were performed by the referring clinicians. Results of these tests were supplied to the data coordinator as part of the standardized questionnaire. In addition, all sera were assessed for anti-disialosyl antibodies, cold agglutinins and paraproteins in the Neuroimmunology Labororatory, Glasgow. Antiganglioside antibodies were detected using enzyme-linked immunosorbent assay (ELISA) as described (Willison et al., 1999). Thinlayer chromatography (TLC) immuno-overlay was performed as previously described (Willison et al., 1996). In addition to the standard use of HRAGE coupled with IFE which are both required for identifying paraproteins, identification of serum IgM paraproteins and light chain typing was also conducted by isoelectric focusing (IEF) with Western blot analysis, using µ-, κ- and λ-specific secondary antibodies as described (Willison et al., 1996). The haemagglutinin assay was conducted as follows: a standard concentration (1 ⫻ 109/ml) of control red cells, free of serum and buffy coat, was established by measuring the OD541 of lysed samples (based on OD541 of 1 ⫻ 109/ml lysed red cells ⫽ 0.385). Sample sera were diluted to 1 in 20 with normal saline and red cell concentrate was added to achieve a final red cell concentration of 1 ⫻ 109/ml. The samples were placed on ice for 60 min, with occasional mixing. The presence of agglutination was assessed both by direct observation and microscopically.. Electrophysiology and other investigations Nerve conduction studies had been carried out in all cases and these data were analysed in a standardized format by S.P. In most cases (15 out of 18), data were recorded on one side only. In Cases 1, 9 and 14, both sides were examined for some nerves and, if major differences between left and right were observed, the more normal side was analysed and described here. As the results obtained were from many different clinical centres, modified criteria were used for the diagnosis of demyelination (Ad Hoc Subcommittee of the American Academy of Neurology AIDS Task Force, 1991). Definite evidence of demyelination is accepted when three of the four criteria are.
(3) 1970. H. J. Willison et al.. met in one or more nerves, and the diagnosis is probable if any one of the four criteria is met in two or more nerves. In addition to the above analysis, individual centres were asked to grade the cases based on their overall impression of the electrophysiological findings as severe, mildly deranged or normal with respect to demyelinating, axonal, motor and sensory features. The electrophysiological techniques were considered to be consistent between the referral centres. Other ancillary investigations were carried out at the referral centres, both during the diagnostic phase and subsequently, and were neither standardized nor universal. These included CSF analysis, neuroimaging and nerve biopsy. Routine screening investigations to exclude other causes of neuropathy were negative and are not reported here.. Results Clinical presentation and course of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies Twenty sera which were positive for anti-disalosyl IgM antibodies were identified over a 7-year period (1992–9). Sufficient clinical data for inclusion in this survey were available from 18 cases, and the data are summarized in Table 1. Although the two excluded cases both had chronic peripheral neuropathies, clinical details were scanty. Of the 18 cases, 14 were male and four female. The mean age at onset of symptoms was 53 years, with a range of 40–72 years. Mean age of onset in the female cases was 48 years, compared with 55 years in the male group. The mean duration of symptoms in the group studied was 13 years (range 4– 29 years). On the background of a slowly progressive disorder, 13 out of 18 cases had a history of relapsing symptoms, the most frequent relapsing symptom being ocular (10 out of 18), followed by sensory (nine out of 18) and bulbar (eight out of 18). Four patients described motor relapses, and one had a respiratory relapse requiring temporary mechanically assisted ventilation. At the time of their last assessment, five patients were wheelchair bound and only three walked unaided. Sixteen of the 18 cases had upper and lower limb ataxia, which profoundly affected the lower limbs in eight. Fourteen of the 18 cases had no, or only mild, limb weakness, with the remaining four having significant motor disability. Only two out of 18 patients complained of significant paraesthesiae, 13 having minor symptoms. Eight cases experienced both limb and trigeminal paraesthesiae and one out of 18 had trigeminal symptoms alone. Sixteen of the 18 patients had symptoms and/or signs of extraocular muscle weakness. Case 15 had neither ataxia nor ophthalmoplegia. Bulbar features were present in 12 cases and facial weakness in four. Only four out of 18 cases had significant limb weakness (MRC grade 艋3, in two or more muscle groups), and in three of these cases there was also profound ataxia.. Serological studies All 18 cases had high titre IgM anti-ganglioside antibodies (the upper limit of normal being ⬍1/500) as summarized in Table 2. Samples were collected at a single time point and no serial data are available. In some cases, the volume of serum available was insufficient to conduct all investigations. In 16 out of 18 cases, the sera reacted with three or more of the four disialosylcontaining gangliosides against which these were tested: GD3, GD1b, GT1b and GQ1b. Case 10 reacted with GD3 and GQ1b; Case 17 with GD3 only. Eight of the 18 sera also reacted to some extent with GD1a and other gangliosides which lack disialosyl epitopes. Examples of ganglioside immunoreactivity as demonstrated by TLC overlay are shown in Fig. 1. Immunoglobulin light chain (kappa, κ; lambda, λ) analysis of the anti-ganglioside antibodies was performed by ELISA in 16 cases (Cases 3 and 9 were omitted due to insufficient sample). In 12 cases, this demonstrated that the antibodies were of a single light chain type (eight κ, four λ). In four cases (Cases 6, 7, 16, and 18), both κ and λ light chains were identified, indicating that at least two distinct antibody clones were responsible for the anti-ganglioside antibody activity. Testing for the presence of serum paraproteins, firstly by HRAGE coupled with IFE (all 18 cases, in the referring institutions) and also by IEF with Western blotting (17 out of 18 cases in Glasgow; insufficient sample in Case 3), demonstrated an IgM monoclonal protein (M-protein) in 17 out of 18 samples (Table 2 and Fig. 2). In Case 15, an IgM paraprotein was not detectable by either HRAGE, IFE or IEF, indicating that the anti-disialosyl antibody activity could not be accounted for by a paraprotein. Light chain typing of the M-protein(s) conducted on 16 sera showed a single type (κ or λ) in 12 out of 16 cases, and both κ and λ in four cases (Cases 4, 6, 7 and 16) as shown in Table 2 and Fig. 2. Light chain comparison of the anti-ganglioside antibody activity and the M-protein showed concordance of a single light chain subtype in 10 cases (seven κ/κ, three λ/λ). In three cases (6, 7 and 16), there were both κ and λ light chains in the anti-ganglioside antibody fraction and the M-protein bands. In Case 18, κ and λ chains were present in the anti-ganglioside fraction, with only a λ chain demonstrable in the M-protein band. Conversely, in Case 4, all of the anti-ganglioside antibody was λ light chain, with both λ and κ M-proteins being identified, indicating that the IgMκ paraprotein did not have antidisialosyl antibody activity. Cold agglutination was demonstrated in eight out of 17 cases. The sub-specificity of the cold agglutinins was not investigated further, but in a previous analysis of two cases these had been confirmed as anti-Pr2 (Case 16) and anti-Pr1d (Case 18).. Clinical electrophysiology Electrophysiological data from studies conducted in individual centres are summarized in Table 3. The interval from the onset of the symptoms to the time of the electrodiagnostic examination reported here was known for seven cases (Cases 2,.
(4) Neuropathy with anti-disialosyl IgM antibodies. 1971. Table 1 Clinical data Case Referral Sex source*. Age at onset (years). Duration Mode (years) of onset. 1. 2. F. 57. 8. A. 2 3. 9 3. M M. 72 67. 15 4. C S. 4. 3. M. 41. 9. S. 5 6. 3 6. M M. 72 28. 7 27. C C. 7. 6. F. 40. 7. C. 8 9. 7 12. M M. 46 42. 13 20. C A. 10 11. 4 8. M M. 58 64. 12 6. C C. 12. 10. F. 41. 25. A. 13. 8. M. 58. 7. C. 14. 11. M. 64. 19. C. 15 16. 6 1. M F. 42 56. 6 13. C C. 17. 1. M. 59. 6. C. 18. 5. M. 51. 29. A. Relapses Cranial nerve involvement III, IV, VI. V. ⫹ (smb). ⫹. ⫹ (so) ⫹ (sbo) ⫹ (o) ⫹ (bo) ⫹ (smbor) ⫹ (so) ⫹ (mbor) ⫹ (s) ⫹ (smb) ⫹ (so) ⫹ (sbo) ⫹ (smbo). VII. Limb involvement Bulbar. Paresis Paraesth. UL ataxia. ⫹. ⫹. ⫹. ⫹. ⫹⫹. ⫹. ⫹. ⫹. ⫹ ⫹. ⫹. ⫹. ⫹. ⫹. ⫹. ⫹ ⫹. LL ataxia. Gait ataxia. ⫹⫹ ⫹. ⫹⫹ ⫹. ⫹⫹ ⫹. ⫹. ⫹⫹. ⫹⫹. ⫹⫹. ⫹⫹. ⫹ ⫹. ⫹ ⫹⫹. ⫹ ⫹. ⫹. ⫹. ⫹. ⫹. ⫹⫹. ⫹. ⫹⫹. ⫹⫹. ⫹. ⫹ ⫹. ⫹. ⫹ ⫹. ⫹⫹ ⫹. ⫹ ⫹. ⫹ ⫹. ⫹ ⫹. ⫹ ⫹. ⫹⫹ ⫹. ⫹⫹. ⫹⫹ ⫹. ⫹⫹ ⫹. ⫹⫹ ⫹. ⫹. ⫹⫹. ⫹⫹. ⫹⫹. ⫹. ⫹. ⫹. ⫹⫹. ⫹. ⫹. ⫹. ⫹⫹. ⫹. ⫹⫹. ⫹⫹. ⫹. ⫹. ⫹. ⫹⫹. ⫹⫹. ⫹. ⫹⫹ ⫹. ⫹. ⫹⫹. ⫹. ⫹⫹. ⫹⫹. ⫹ ⫹ ⫹. ⫹ ⫹. ⫹⫹ ⫹. ⫹ ⫹. ⫹. ⫹. ⫹ ⫹. ⫹. ⫹. ⫹. ⫹. ⫹. ⫹. ⫹. *Referral source numbered as per the list of authors. A ⫽ acute; S ⫽ subacute; C ⫽ chronic; b ⫽ bulbar; m ⫽ motor; o ⫽ ophthalmoplegia; r ⫽ respiratory; s ⫽ sensory; ⫹ ⫽ present; ⫹⫹ ⫽ substantive feature.. Table 2 Serological data Case. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18. IgM anti-ganglioside antibodies. Immunoglobulin analysis. GD1b. GD3. GT1b. GQ1b. Type. Others. M-protein. Type. IgM g/l. ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹ ⫹⫹ ⫹⫹ ⫹ ⫹⫹ ⫺ ⫹ ⫹⫹ ⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫺ ⫹⫹. ⫺ ⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫺ ⫹⫹ ⫹⫹ ⫺ ⫹⫹ ⫹ ⫺ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹. ⫹ ⫺ ⫹⫹ ⫹⫹ – ⫹⫹ ⫹⫹ ⫹⫹ ⫹ ⫺ ⫹ ⫹⫹ ⫹ ⫹ ⫹⫹ ⫹⫹ ⫺ ⫹⫹. ⫹ ⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫹ ⫹ ⫹⫹ ⫹ ⫹⫹ ⫹ ⫹⫹ ⫹⫹ ⫹⫹ ⫺ ⫹⫹. κ κ ND λ λ κ, λ κ, λ κ ND λ κ κ λ λ κ κ, λ κ κ, λ. GD1a – GD1a GA1, GM3, GD1a – GA1, GM1, GM2, GM3, GD1a GM3, GD1a – GA1, GM1, GM2, GM3, GD1a – – – – – – GM3, GD1a – GD1a. ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ – ⫹ ⫹ ⫹. κ κ ND κ, λ κ κ, λ κ, λ κ ND λ κ κ λ λ – κ, κ λ. 4.5 WNR 5.8 3.9 WNR 9.0 4.5 2.8 17.2 6.1 1.3 9.0 4.4 13.0 WNR 6.3 3.46 5.7. Agglutinins. ⫺ ⫺ ⫹ ⫺ ⫹ ⫹ ⫺ ⫺ ND ⫺ ⫺ ⫺ ⫹ ⫺ ⫹ ⫹ ⫹ ⫹. ND ⫽ not determined; WNR ⫽ within normal range. Anti-ganglioside antibodies: ⫹ ⫽ titre ⬎1/500 ⬍1/5000; ⫹⫹ ⫽ titre ⬎1/5000; – ⫽ ⬍1/500..
(5) 1972. H. J. Willison et al.. Fig. 1 TLC of gangliosides (1 µg/lane each of GM1, GM2, GM3, GD3, GD1a, GD1b and GT1b), followed by immuno-overlay with sera from four chronic sensory ataxic neuropathy cases (diluted 1 : 100 to 1 : 500) showing the spectrum of ganglioside reactivity between cases as follows: lane 1, Case 6; lane 2, Case 16; lane 3, Case 11; lane 4, Case 2.. 7, 12, 14, 15, 16 and 17), ranging from 1 to 25 years, with a mean of 8 years. According to the criteria defined above (see Patients and methods), definite evidence of demyelination was present in five cases (27%; Cases 1, 6, 7, 8 and 9), and probable evidence in four (Cases 2, 3, 4 and 16). The limited results available in Cases 12 and 18 were consistent with demyelination. Initial nerve conduction studies in Case 7 (3 years after the onset of symptoms) were consistent with axonal degeneration. Two years later, demyelination was evident, and still present in a further study at 4 years. Three patients showed changes clearly consistent with axonal degeneration (Cases 13, 15 and 17), and three cases (Cases 5, 10 and 11) had sensory abnormalities only. Clinical electrophysiological reporting from individual centres concluded the presence of sensory abnormalities in all (18 out of 18) cases (16 severe; two mild), motor abnormalities in 13 out of 18 cases (four severe; 10 mild), and an equal proportion of cases exhibiting demyelinating (five severe; eight mild) and/or axonal (six severe; seven mild) features.. Fig. 2 IEF followed by western blot analysis of serum IgM illustrated for five chronic sensory ataxic neuropathy cases as follows: lane 1, Case 13; lane 2, Case 12; lane 3, Case 6; lane 4, Case 4; lane 5, Case 15. Paraproteins migrate to appear as ‘ladders’ in IEF gels. One or more IgM paraproteins were identified by µ chain-specific antibodies in all sera, except Case 15 (lane 5, A). λ (B) and κ (C) light chain typing either revealed a single type (shown here for κ, Case 12, lane 3; λ, Case 13, lane 1) or revealed both κ and λ light chain types (shown here for Case 6, lane 2; Case 4, lane 4) indicating at least biclonal paraproteins to be present in these latter sera.. CSF analysis. Neuroimaging. Sixteen patients (Cases 7 and 13 excepted) had CSF analysis at their referral centre. CSF protein was ⬍0.5 g/l in five cases (Cases 1, 9, 10, 14 and 16), ⬎0.5 ⬍1.0 g/l in nine cases (Cases 3–6, 8, 11, 12, 15 and 17) and ⬎1.0 g/l in two cases (Case 2, 1.45 g/l; Case 18, 1.2 g/l). In three cases, CSF lymphocyte counts were elevated (Cases 3 and 4, 7 cells/mm3; Case 18, 16 cells/mm3). CSF glucose was normal in all 16 cases. Local synthesis of oligoclonal bands within the CNS compartment was not detected in any of the cases tested, including those with CNS white matter abnormalities on imaging.. Eight cases had no neuroimaging (Cases 4–8, 11, 13 and 18). Case 10 had a normal CT brain scan. Case 2 had a normal MRI of the spine. Eight cases (Cases 1, 3, 9, 12 and 14–17) had MRI of the brain. Two were reported as normal (Cases 9 and 15). Three scans (Cases 3, 14 and 16) were reported as showing ischaemic lesions, and one (Case 17) as showing generalized atrophy (attributed to alcohol abuse). Two scans (Cases 1 and 12) were reported as showing multifocal lesions suggestive of central white matter demyelination, and neither case had CSF oligoclonal bands..
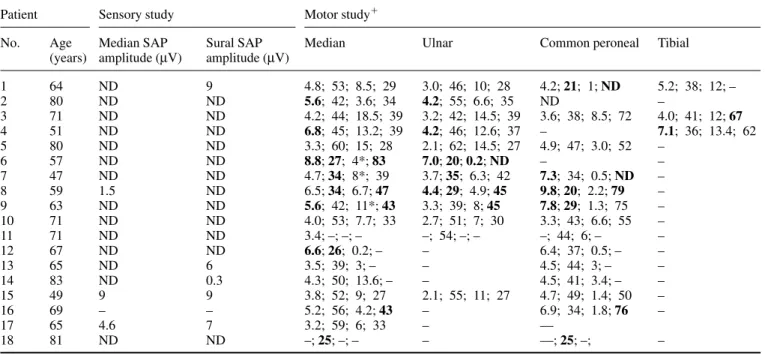
(6) Neuropathy with anti-disialosyl IgM antibodies. 1973. Table 3 Clinical electrophysiological data on all 18 cases Patient. Motor study⫹. Sensory study. No.. Age Median SAP (years) amplitude (µV). Sural SAP amplitude (µV). Median. Ulnar. Common peroneal. Tibial. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18. 64 80 71 51 80 57 47 59 63 71 71 67 65 83 49 69 65 81. 9 ND ND ND ND ND ND ND ND ND ND ND 6 0.3 9 – 7 ND. 4.8; 53; 8.5; 29 5.6; 42; 3.6; 34 4.2; 44; 18.5; 39 6.8; 45; 13.2; 39 3.3; 60; 15; 28 8.8; 27; 4*; 83 4.7; 34; 8*; 39 6.5; 34; 6.7; 47 5.6; 42; 11*; 43 4.0; 53; 7.7; 33 3.4; –; –; – 6.6; 26; 0.2; – 3.5; 39; 3; – 4.3; 50; 13.6; – 3.8; 52; 9; 27 5.2; 56; 4.2; 43 3.2; 59; 6; 33 –; 25; –; –. 3.0; 46; 10; 28 4.2; 55; 6.6; 35 3.2; 42; 14.5; 39 4.2; 46; 12.6; 37 2.1; 62; 14.5; 27 7.0; 20; 0.2; ND 3.7; 35; 6.3; 42 4.4; 29; 4.9; 45 3.3; 39; 8; 45 2.7; 51; 7; 30 –; 54; –; – – – – 2.1; 55; 11; 27 – – –. 4.2; 21; 1; ND ND 3.6; 38; 8.5; 72 – 4.9; 47; 3.0; 52 – 7.3; 34; 0.5; ND 9.8; 20; 2.2; 79 7.8; 29; 1.3; 75 3.3; 43; 6.6; 55 –; 44; 6; – 6.4; 37; 0.5; – 4.5; 44; 3; – 4.5; 41; 3.4; – 4.7; 49; 1.4; 50 6.9; 34; 1.8; 76 –– ––; 25; –;. 5.2; 38; 12; – – 4.0; 41; 12; 67 7.1; 36; 13.4; 62 – – – – – – – – – – – –. ND ND ND ND ND ND ND 1.5 ND ND ND ND ND ND 9 – 4.6 ND. –. ⫹Sequential. values as follows: distal motor latency (ms); motor conduction velocity (m/s); distal compound muscle action potential amplitude (mV); minimal F wave latency (ms). SAP ⫽ sensory action potential; ND ⫽ not detected; – ⫽ value not available; * ⫽ conduction block on proximal stimulation; values considered abnormal in the demyelinating range are in bold.. Histopathology Three cases underwent nerve biopsy—three sural nerve (Cases 9, 10 and 16) and one dorsal root ganglion (Case 10). In Case 9, light and electron microscopic studies of a sural nerve biopsy showed thin myelin sheaths, small onion bulb formations and a few inflammatory cells scattered in the endoneurium. In Case 10, a sural nerve biopsy demonstrated chronic axonal sensorimotor degeneration without specific features. In the same case, a dorsal root ganglion biopsy showed no abnormality, and gastrocnemius muscle biopsy demonstrated denervation atrophy. In Case 16, a sural nerve biopsy demonstrated a severe reduction in large myelinated fibres with preservation of the unmyelinated and small myelinated fibre population. No active fibre degeneration or demyelination was observed although there were regenerative clusters with fibres surrounded by thin myelin sheaths. There were no hypertrophic changes and no inflammatory cells or immunoglobulin deposits.. Discussion This series of patients is characterized by a large number of shared clinical and immunological features. The most prominent feature of this clinical phenotype is a loss of kinaesthesia with relative preservation of muscle strength and small fibre sensation. The patients therefore typically present with gait and upper limb ataxia. Acral and perioral paraesthesiae are also frequently present. A severe sensory ataxia with pseudo-athetosis and loss of vibration and joint position sense, accompanied by near-normal limb power and. tendon areflexia, is found on clinical examination. Thus the clinical descriptor common to these cases is of a chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies. The pattern of abnormalities identified from many of our own cases and previously published case reports, which led us previously to propose the acronym CANOMAD, is also strongly reinforced by these cases. Despite the absence of significant limb weakness in most cases, the majority of cases (16 out of 18) have motor and sensory cranial nerve involvement, either fixed or as relapsing–remitting symptoms at any chronological stage of the syndrome; these comprise external ophthalmoplegia, dysphagia, dysarthria and, rarely, respiratory muscle weakness. We thus recommend screening sera for this antibody specificity in neuropathy cases with some or all of these clinical features, including those without obviously detectable IgM paraproteins, until the full extent of the phenotypic variation is delineated. The overall clinical pattern is reminiscent of the regional pattern of clinical involvement seen in Miller Fisher syndrome, in which anti-disialosyl antibodies are also found in the form of an acute-phase IgG response, principally directed against GQ1b and GT1a (Chiba et al., 1992; Willison and Veitch, 1994). In contrast to the acute, self-limiting course of Miller Fisher syndrome, the clinical course of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies often extends over decades, which is typical of other IgM paraproteinaemic neuropathies. However, the chronic course of this neuropathy is interspersed with episodes of motor and sensory cranial nerve involvement that are unusual and have not been been documented previously in chronic IgM paraproteinaemic.
(7) 1974. H. J. Willison et al.. neuropathies. These episodes may be misdiagnosed, for example as vascular or demyelinating brainstem events. Since the anti-ganglioside IgM paraproteins are likely to be persistently present at a stable level throughout the illness, additional factors such as intercurrent infections must influence the development of these acute craniobulbar relapses. It is possible that some cases may have CNS involvement due to antibody-mediated inflammatory injury to neuronal or glial components, but this has not been established. The ganglioside antigens are present within the CNS, and CNS involvement is known to occur in acute anti-GQ1b IgG antibody syndromes within the Miller Fisher syndrome spectrum (Yuki, 1998) and in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) (Thomas et al., 1987). Two of the current cases had multifocal MRI lesions in patterns suggestive of CNS demyelination in the absence of CSF oligoclonal bands, but interpretation is difficult as both were elderly. The electrophysiological features of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies include prominent sensory nerve abnormalities, most commonly an absence or a marked reduction in amplitude of sensory nerve action potentials. In the three cases with pure sensory involvement, it is difficult to decide whether the underlying pathology was a neuropathy or a sensory ganglionopathy. The relapsing course in one of them (Case 11) favours the former explanation. Motor conduction abnormalities are found in over half the cases, even in the absence of frank clinical motor involvement in the limbs. Denervation changes are variable. The three patients with axonal degeneration only showed the pattern of ‘abnormal median/normal sural’ sensory potentials that has been reported in CIDP (Notermans et al., 2000). In some patients, the features would fit diagnostic criteria for CIDP but the clinical picture is unlike that of CIDP and the syndrome should thus be readily distinguishable from CIDP on clinical and laboratory grounds, even in the presence of similar electrophysiology. Pathological analysis of tissues from cases of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies is so far limited. In a detailed post-mortem study, Obi and colleagues reported severe loss of myelinated nerve fibres in dorsal roots and sensory nerves, with relative preservation of anterior horn cells, ventral roots and motor nerves (Obi et al., 1999). There was some loss of dorsal root ganglion cells and pallor of the dorsal columns. No inflammatory changes or deposition of immunoglobulin or complement were observed. The present sural nerve biopsies showed severe loss of myelinated nerve fibres, particularly those of larger size, together with evidence of demyelination. A T5 dorsal root ganglion removed during the early stage of the disease in Case 10 was normal. This negative finding may be due to the fact that a relatively unaffected area was biopsied, compared with the cervical or lumbar dorsal root ganglia that subserve limb proprioception, or that the pathological changes were primarily in the peripheral nerves and not in sensory ganglia. No IgM deposition was shown either in the post-mortem material or the sural nerve biopsies but, in one recently reported. exceptional case, extensive IgM deposition was noted on myelin sheaths in a biopsied sural nerve (Mitsui et al., 1999). The presence of IgM paraproteins was demonstrated in almost all cases when sought using HRAGE coupled with IFE or using the very sensitive technique of IEF. Of considerable interest was the demonstration by IEF of at least two and possibly multiple paraproteins in four cases. Interpretation of banding patterns in IEF gels is in part subjective, and a conservative estimation was adopted in this study. Biclonal gammopathies have been reported previously in paraproteinaemic neuropathy (Ilyas et al., 1988). This may indicate the occurrence of clonal microheterogeneity within a single IgM B-cell clone producing anti-disialosyl antibodies, arising through the acquisition of additional somatic mutations within a parental B-cell clone that shift the isoelectric point of a monoclonal immunoglobulin (Willison et al., 1996). However, the presence of both κ and λ light chain-containing antibodies in a single serum clearly indicates that different parental B-cell clones must be contributing towards the totality of the IgM anti-disialosyl antibody pool in these cases. In a previous study on chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies, we have shown that the major paraprotein present in the serum produces the anti-ganglioside antibody activity (Willison et al., 1996). However, we should not necessarily assume this always to be the case in all sera, as exemplified in this study. When present, as an accompanying feature in eight out of 17 cases in this series, the cold agglutinating activity of sera from cases of chronic sensory ataxic neuropathy with antidisialosyl IgM antibodies is due to reactivity with red blood cell carbohydrate antigens of the rare anti-Pr specificity (Herron et al., 1994). Anti-ganglioside IgM antibodies in general, and antidisialosyl antibodies in particular, are well recognized for having greatly increased binding avidity at lower temperatures (Willison et al., 1993). Cold agglutinin disease is rarely symptomatic, although this has been reported (Herron et al., 1994). The cold-reactive properties of antidisialosyl antibodies are also reflected in anecdotal reports of marked deterioration in motor function or sensory symptoms reported by these patients when exposed to cold environments, which many patients take care to avoid. All cases of chronic sensory ataxic neuropathy with antidisialosyl IgM antibodies have, by definition, antibodies that react with gangliosides containing a NeuNAc(α2– 8)NeuNAc(α2–3)Gal epitope, on either the internal or terminal galactose of the core oligosaccharide. These include GD2, GD3, GD1b, GT1b, GT1a and GQ1b. Some CANOMAD sera also react with other gangliosides, particularly GD1a and GM3, which do not have this epitope, indicative of an additional degree of cross-reactivity. Which of these, if any, are the target ganglioside(s) in neural tissue responsible for the pathophysiological development of the disease is unknown. It is well established from clinical serological studies in Miller Fisher syndrome that anti-GQ1b IgG antibodies correlate highly with ophthalmoplegia (Yuki and Hirata, 1998). Miller Fisher syndrome antibodies invariably cross-react with GT1a,.
(8) Neuropathy with anti-disialosyl IgM antibodies and occasionally with GD1b, GD3 and GT1b. Acute oropharyngeal palsy is also associated with high titre IgG GT1a and GQ1b antibodies (O’Leary et al., 1996). Also, acute-phase IgG anti-GD1b antibodies have been identified in patients with acute sensory and ataxic neuropathies without limb weakness (Willison et al., 1994; Yuki and Hirata, 1998) and are statistically associated with sensory symptoms in Guillain– Barre´ syndrome (Wicklein et al., 1997). Thus different elements of the ganglioside binding profile of chronic sensory ataxic neuropathy sera may mediate different pathophysiological components leading to the clinical phenotype. The source of anti-disialosyl antibodies found in the sera of cases of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies is unknown. In Miller Fisher syndrome, with which this syndrome has clinical and serological parallels, they may arise through cross-reactivity with bacterial lipopolysaccharides, for example, as carried on certain sero strains of Campylobacter jejuni (Jacobs et al., 1997). This principle of molecular mimicry has been demonstrated experimentally (Goodyear et al., 1999). B cells encoding this antigen specificity may form part of the natural anti-carbohydrate antibody repertoire that acts as an early defence against encapsulated bacteria. The mechanism by which these B-lymphocyte antibody responses might make the transition from acute-phase immune responses to chronically stable clones of paraproteinsecreting B cells is unknown. Considerable experimental evidence indicates that the antiganglioside antibodies are the primary cause of the neuropathy in chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies. Many studies have shown mouse and human antiGD1b, GD3 and GQ1b antibody binding to a wide range of peripheral nerve structures including dorsal root ganglion neurones, peripheral nerve axons and myelin, nodes of Ranvier, muscle spindles and motor nerve terminals (Oka et al., 1996; Willison et al., 1996; Kusonoki et al., 1997; Mitsui et al., 1999; Willison and O’Hanlon, 1999). Rabbits immunized with GD1b develop an anti-GD1b response accompanied by an ataxic neuropathy (Kusonoki et al., 1996). Mouse motor nerve terminals can be paralysed by sera from cases of chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies, and also by sera containing related antibodies, including those from Miller Fisher syndrome cases. (Roberts et al., 1994; Buchwald et al., 1998; Goodyear et al., 1999; Plomp et al., 1999). Despite some deficiencies, the principal one being the lack of strongly supportive human pathological studies, the existing evidence points to a direct role for anti-disialosyl antibodies in causing the neural injury seen in chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies (Quarles and Dalakas, 1996). Although case ascertainment in this study was random through ad hoc referral, in many of these cases specialist serological investigation had been sought because the distinctive clinical phenotype was recognized by the referring clinician. In our view, it is probable that these cases represent a true clinicoserological syndrome and that the diagnosis is dependent on recognizing the phenotype and investigating. 1975. appropriately, i.e. testing for anti-ganglioside antibodies. Not all cases of chronic sensory ataxic neuropathy with antidisialosyl IgM antibodies present every feature of CANOMAD. For this reason, and the reluctance to accumulate arguably clumsy acronyms in the neurological literature, there is a good case for describing this neuropathy syndrome in its most simple form as it really is: a chronic sensory ataxic neuropathy with anti-disialosyl IgM antibodies. What remains to be determined is (i) the prevalence of antidisialosyl antibodies in the normal or disease control population, and (ii) whether the clinical phenotype of chronic sensory ataxic neuropathy with or without the additional motor features characteristic of CANOMAD occurs in the absence of anti-disialosyl antibodies. The former is likely to be very low; the latter could be investigated by prospective clinical studies of patients with sensory ataxic neuropathies (Pourmand and Maybury, 1996; Dalakas and Quarles, 1997; O’Leary and Willison, 1997; Wokke and van Dijk 1997). No clinical trials of treatment have been performed and no attempt was made in this study to assess treatment responses systematically. Data on protocols and responses are thus anecdotal and limited to single case descriptions. Both plasma exchange and intravenous immunoglobulin have been used with temporary benefit in some cases (Ponsford et al., 2000). Others have either failed to respond to these treatments or have deteriorated on treatment. The two cases reported by Ponsford and colleagues who responded to intravenous immunoglobulin both showed evidence of demyelination electrophysiologically (Ponsford et al., 2000). It is thus possible that it is such cases that are treatment responsive, whereas those in which the disorder is primarily an axonopathy are refractory, as has been suggested previously for paraproteinaemic neuropathy (Simmons et al., 1995; Di Troia et al., 1999). Plasma exchange may be complicated by agglutination in the extracorporeal arm of the system, and replacement/reinfusion fluid temperatures should be controlled carefully. The role of other treatments, including oral or intravenous corticosteroids, β-interferons and cytotoxic drugs, has not been evaluated systematically.. Acknowledgements We wish to thank Dr J. R. Ponsford for providing valuable clinical data. This study was supported by grants to H.J.W. from the Guillain–Barre´ Syndrome Support Group of Great Britain, European Community Biomed Programme BMH4CT96-0234 and The Wellcome Trust. References Ad hoc Subcommittee of the American Academy of Neurology AIDS Task Force. Research criteria for the diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). [Review]. Neurology 1991; 41: 617–8 Arai M, Yoshino H, Kusano Y, Yazaki Y, Ohnishi Y, Miyatake T. Ataxic polyneuropathy and anti-Pr2 IgMκ M proteinemia. J Neurol 1992; 239: 147–51..
(9) 1976. H. J. Willison et al.. Brindel I, Preud’homme JL, Vallat JM, Vincent D, Vasquez JL, Jauberteau MO. Monoclonal IgM reactive with several gangliosides in a chronic relapsing polyneuropathy. Neurosci Lett 1994; 181: 103–6.. Kusunoki S, Shimizu T, Chiba A, Ugawa Y, Hitoshi S, Kanazawa I. Experimental sensory neuropathy induced by sensitization with ganglioside GD1b. Ann Neurol 1996; 39: 424–31.. Buchwald B, Weishaupt A, Toyka KV, Dudel J. Pre- and postsynaptic blockade of neuromuscular transmission by Miller-Fisher syndrome IgG at mouse motor nerve terminals. Eur J Neurosci 1998; 10: 281–90.. Kusunoki S, Mashiko H, Mochizuki N, Chiba A, Arita M, Hitoshi S, et al. Binding of antibodies against GM1 and GD1b in human peripheral nerve. Muscle Nerve 1997; 20: 840–5.. Chassande B, Leger JM, Younes-Chennoufi AB, Bengoufa D, Maisonobe T, Bouche P, et al. Peripheral neuropathy associated with IgM monoclonal gammopathy: correlations between M-protein antibody activity and clinical/electrophysiological features in 40 cases. Muscle Nerve 1998; 21: 55–62.. Kyle RA. Monoclonal gammopathy of undetermined significance (MGUS). [Review]. Ballieres Clin Haematol 1995; 8: 761–81.. Chiba A, Kusunoki S, Shimizu T, Kanazawa I. Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller Fisher syndrome. Ann Neurol 1992; 31: 677–9. Dalakas MC, Quarles RH. Autoimmune ataxic neuropathies (sensory ganglionopathies): are glycolipids the responsible autoantigens? [editorial] Ann Neurol 1996, 39: 419–22. Di Troia A, Carpo M, Meucci N, Pellegrino C, Allaria S, Gemignani F, et al. Clinical features and anti-neural reactivity in neuropathy associated with IgG monoclonal gammopathy of undetermined significance. J Neurol Sci 1999; 164: 64–71. Duane GC, Farrer RG, Dalakas MC, Quarles RH. Sensory neuropathy associated with monoclonal immunoglobulin M to GD1b ganglioside. Ann Neurol 1992; 31: 683–5. Goodyear CS, O’Hanlon GM, Plomp JJ, Wagner ER, Morrison I, Veitch J, et al. Monoclonal antibodies raised against Guillain–Barre´ syndrome-associated Campylobacter jejuni lipopolysaccharides react with neuronal gangliosides and paralyse muscle–nerve preparations. J Clin Invest 1999; 104: 697–708. Herron B, Willison HJ, Veitch J, Roelcke D, Illis LS, Boulton FE. Monoclonal IgM cold agglutinins with anti-Pr1d specificity in a patient with peripheral neuropathy. Vox Sang 1994; 67: 58–63.. Latov N, Hays AP, Sherman WH. Peripheral neuropathy and antiMAG antibodies. [Review]. CRC Crit Rev Neurobiol 1988; 3: 301– 32. Leger JM, Chassande B, Musset L, Meininger V, Bouche P, Baumann N. Intravenous immunoglobulin therapy in multifocal motor neuropathy: a double-blind, placebo-controlled study. Brain 2001; 124: 145–53. Mitsui Y, Kusunoki S, Hiruma S, Akamatsu M, Kihara M, Hashimoto S, et al. Sensorimotor polyneuropathy associated with chronic lymphocytic leukemia, IgM antigangliosides antibody and human Tcell leukemia virus I infection. [Review]. Muscle Nerve 1999; 22: 1461–5. Notermans NC, Franssen H, Eurelings M, Van der Graaf Y, Wokke JH. Diagnostic criteria for demyelinating polyneuropathy associated with monoclonal gammopathy. Muscle Nerve 2000; 23:73–9. Obi T, Kusunoki S, Takatsu M, Mizoguchi K, Nishimura Y. IgM Mprotein in a patient with sensory-dominant neuropathy binds preferentially to polysialogangliosides. Acta Neurol Scand 1992; 86: 215–8. Obi T, Murakami T, Takatsu M, Kusunoki S, Serizawa M, Mizoguchi K, et al. Clinicopathological study of an autopsy case with sensorydominant polyradiculoneuropathy with antiganglioside antibodies. Muscle Nerve 1999; 10: 1426–31. Oka N, Kusaka H, Kusunoki S, Tsuda H, Kaji R, Imai T, et al. IgM M-protein with antibody activity against gangliosides with disialosyl residue in sensory neuropathy binds to sensory neurons. Muscle Nerve 1996; 19: 528–30.. Hitoshi S, Kusunoki S, Chiba A, Takatsu R, Sunada Y, Nukina N, et al. Cerebellar ataxia and polyneuropathy in a patient with IgM M-protein specific to the Gal(beta-1–3)GalNAc epitope. J Neurol Sci 1994; 126: 219–24.. O’Leary C, Willison HJ. Autoimmune ataxic neuropathies (sensory ganglionopathies). [Review]. Curr Opin Neurol 1997; 10: 366–70.. Ilyas AA, Quarles RH, Dalakas MC, Fishman PH, Brady RO. Monoclonal IgM in a patient with paraproteinemic polyneuropathy binds to gangliosides containing disialosyl groups. Ann Neurol 1985; 18: 655–9.. O’Leary CP, Veitch J, Durward WF, Thomas AM, Rees JH, Willison HJ. Acute oropharyngeal palsy is associated with antibodies to GQ1b and GT1a gangliosides. J Neurol Neurosurg Psychiatry 1996; 61: 649–51.. Ilyas AA, Willison HJ, Dalakas MC, Whitaker JN, Quarles RH. Identification and characterization of gangliosides reacting with IgM paraproteins in three patients with neuropathy associated with biclonal gammopathy. J Neurochem 1988; 51: 851–8.. Pestronk A. Invited review: motor neuropathies, motor neuron disorders, and antiglycolipid antibodies. [Review]. Muscle Nerve 1991; 14: 927–36.. Jacobs BC, O’Hanlon GM, Breedland EG, Veitch J, van Doorn PA, Willison HJ. Human IgM paraproteins demonstrate shared reactivity between Campylobacter jejuni lipopolysaccharides and human peripheral nerve disialylated gangliosides. J Neuroimmunol 1997; 80: 23–30. Kahn SN, Riches PJ, Kohn J. Paraproteinaemia in neurological disease: incidence, associations, and classification of monoclonal immunoglobulins. J Clin Pathol 1980; 33: 617–21.. Plomp JJ, Molenaar PC, O’Hanlon GM, Jacobs BC, Veitch J, Daha MR, et al. Miller Fisher anti-GQ1b antibodies: α-latrotoxin-like effects on motor end plates. Ann Neurol 1999; 45: 189–99. Ponsford S, Willison HJ, Veitch J, Morris R, Thomas PK. Long-term clinical and neurophysiological follow-up of patients with peripheral neuropathy associated with benign monoclonal gammopathy. Muscle Nerve 2000; 23: 164–74. Pourmand R, Maybury BG. AAEM case report #31: paraneoplastic sensory neuronopathy. Muscle Nerve 1996, 19: 1517–22..
(10) Neuropathy with anti-disialosyl IgM antibodies Quarles RH. The spectrum and pathogenesis of antibody-mediated neuropathies. Neuroscientist 1997; 3: 195–204. Quarles RH, Dalakas MC. Do anti-ganglioside antibodies cause human peripheral neuropathies? J Clin Invest 1996; 97: 1136–7. Quarles RH, Weiss MD. Autoantibodies associated with peripheral neuropathy. [Review]. Muscle Nerve 1999; 22: 800–22. Roberts M, Willison H, Vincent A, Newsom-Davis J. Serum factor in Miller–Fisher variant of Guillain–Barre syndrome and neurotransmitter release. Lancet 1994; 343: 454–5. Simmons Z, Albers JW, Bromberg MB, Feldman EL. Long-term follow-up of patients with chronic inflammatory demyelinating polyradiculoneuropathy, without and with monoclonal gammopathy. Brain 1995; 118: 359–68. Thomas PK, Willison HJ. Paraproteinaemic neuropathy. Baillie`res Clin Neurol 1994; 4: 129–47. Thomas PK, Walker RW, Rudge P, Morgan-Hughes JA, King RH, Jacobs JM, et al. Chronic demyelinating peripheral neuropathy associated with multifocal central nervous system demyelination. Brain 1987; 110: 53–76. Wicklein EM, Pfeiffer G, Yuki N, Hartard C, Kunze K. Prominent sensory ataxia in Guillain–Barre´ syndrome associated with IgG antiGD1b antibody. J Neurol Sci 1997; 151: 227–9. Willison HJ, O’Hanlon GM. The immunopathogenesis of Miller Fisher syndrome. [Review]. J Neuroimmunol 1999; 100:3–12. Willison HJ, Veitch J. Immunoglobulin subclass distribution and binding characteristics of anti-GQ1b antibodies in Miller Fisher syndrome. J Neuroimmunol 1994; 50: 159–65. Willison HJ, Paterson G, Veitch J, Inglis G, Barnett SC. Peripheral neuropathy associated with monoclonal IgM anti-Pr2 cold agglutinins. J Neurol Neurosurg Psychiatry 1993; 56: 1178–83.. 1977. Willison HJ, Almemar A, Veitch J, Thrush D. Acute ataxic neuropathy with cross-reactive antibodies to GD1b and GD3 gangliosides. Neurology 1994; 44: 2395–7 Willison HJ, O’Hanlon GM, Paterson G, Veitch J, Wilson G, Roberts M, et al. A somatically mutated human antiganglioside IgM antibody that induces experimental neuropathy in mice is encoded by the variable region heavy chain gene, V1–18. J Clin Invest 1996; 97: 1155–64. Willison HJ, Veitch J, Swan AV, Baumann N, Comi G, Gregson NA, et al. Inter-laboratory validation of an ELISA for the determination of serum anti-ganglioside antibodies. Eur J Neurol 1999; 6: 71–9. Wokke JH, van Dijk GW. Sensory neuropathies including painful and toxic neuropathies. [Review]. J Neurol 1997; 244: 209–21. Younes-Chennoufi AB, Leger JM, Hauw JJ, Preud’homme JL, Bouche P, Aucouturier P, et al. Ganglioside GD1b is the target antigen for a biclonal IgM in a case of sensory-motor axonal polyneuropathy: involvement of N-acetylneuraminic acid in the epitope. Ann Neurol 1992; 32: 18–23. Yuki N. Anti-ganglioside antibody and neuropathy: review of our research. [Review]. J Peripher Nerv Syst 1998; 3: 3–18. Yuki N, Hirata K. Postinfection sensory neuropathy associated with IgG anti-GD1b antibody [letter]. Ann Neurol 1998; 43: 685–7. Yuki N, Miyatani N, Sato S, Hirabayashi Y, Yamazaki M, Yoshimura N, et al. Acute relapsing sensory neuropathy asociated with IgM antibody against B-series gangliosides containing a GalNAcβ1– 4(Gal3–2αNeuAc8–2αNeuAc)β1 configuration. Neurology 1992; 42: 686–9.. Received August 10, 2000. Revised March 3, 2001. Accepted May 24, 2001.
(11)
Figure


Documents relatifs
Case presentation: We report the first case of CHIKV infection with chronic associated rheumatism in a patient who developed progressive erosive arthritis with expression
Antibodies against CASPR2 (Contactin-2 Associated Protein), a neuroglial cell-adhesion protein, have been described in at least three neurological syndromes:
Champ : entreprises de 10 salariés ou plus du secteur marchand non agricole pour l’enquête Acemo « Dialogue social en entreprise » ayant déclaré une négociation collective au
Compléter sans justifications les phrases ci-dessous en remplaçant les pointillés par une lettre3. On veut démontrer le même résultat en utilisant une
Brain and spinal cord MRIs follow-up (July 2012) showed an extension of T2 lesions without Gd enhancement (Figure 1F) and a stable extensive T2 lesion without Gd
À partir d’enquêtes réalisées par questionnaire, l’auteur montre que ceux qui ont le moins de connaissances numériques, notamment sur les décompositions additives et
The mAb also identified by western blot sCD14 (53 and 58 kDa) in milk and blood and sCD14 (47 kDa) in a lysate of macrophages obtained from involuted bovine mammary gland
Selon Madame G., sage-femme à l’hôpital de Sion, étant donné que la mère et l’enfant ne restent que quatre jours à l’hôpital, il est essentiel qu’un-e
