Cell-Intrinsic and Cell-Extrinsic Resistance to Classical
Chemotherapies
By
Simona Dalin
B.S. Biology and Mathematics Brandeis University, 2011
Submitted to the Department of Biology
in partial fulfillment of the requirements for the degree of: Doctorate of Philosophy in Biology
at the Massachusetts Institute of Technology February 2020
Ó 2020 Massachusetts Institute of Technology. All Rights Reserved.
Signature of Author ……… Department of Biology November 7th, 2019
Certified By ………. Michael T. Hemann Associate Professor of Biology Thesis Supervisor
Certified By ………. Douglas A. Lauffenburger Ford Professor of Biological Engineering, Chemical Engineering, and Biology Thesis Supervisor
Accepted By ………. Stephen Bell Uncas and Helen Whitaker Professor of Biology
Investigator, Howard Hughes Medical Institute Co-Director, Biology Graduate Committee
Cell-Intrinsic and Cell-Extrinsic Resistance to Classical
Chemotherapies
By:
Simona Dalin
Submitted to the Department of Biology on November 7th, 2019 in Partial Fulfillment of
the Requirements for the Degree of Doctor of Philosophy in Biology
Abstract
Resistance is a constant presence in chemotherapy treatment. The very action of these therapeutic compounds causes resistance to these drugs. There are two modes of resistance to chemotherapy: the cancer cells themselves can undergo modifications that result in resistance, known as cell-intrinsic resistance. Alternatively, the non-malignant cells surrounding the tumor can undergo modifications that result in chemo-protection of the nearby malignant cells, known as cell-extrinsic resistance. Both of these modes of resistance have been extensively studied in the lab, however our knowledge is not advanced enough to subvert chemotherapy resistance in the clinic.
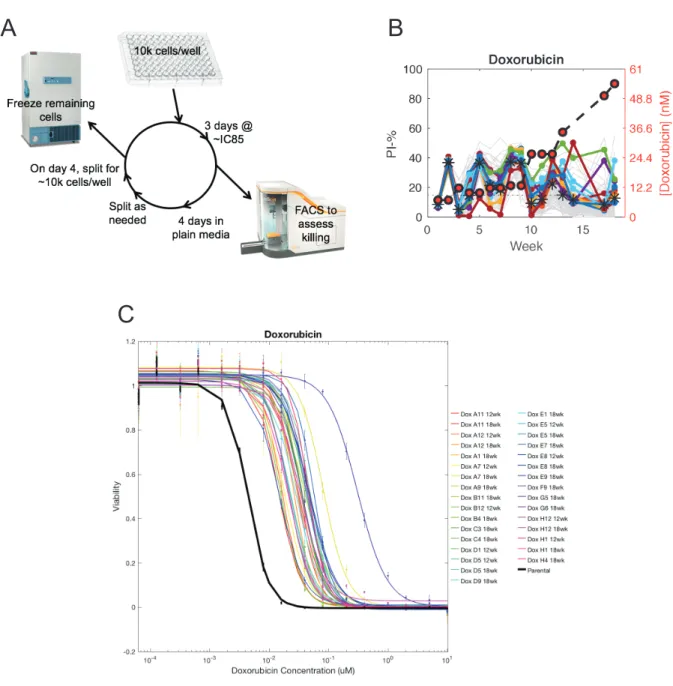
To further our knowledge of chemotherapy resistance with the ultimate goal of reducing or eradicating this public health challenge, I have studied mechanisms of cell-intrinsic and cell-extrinsic resistance. I first studied how to best choose the drugs in a combination regimen to reduce the emergence of chemotherapy resistance. I created over 100 cell lines resistant to front-line cytotoxic chemotherapies and surveyed collateral responses to acquisition of resistance. Previous research in bacteria and targeted cancer chemotherapeutics suggested that collateral effects of resistance to one drug can result in both resistance to a second drug, termed collateral resistance, as well as sensitivity to a second drug, termed collateral sensitivity. In contrast, I found that collateral sensitivity to resistance to classical chemotherapies is rare or nonexistent. Surprisingly, I also found that collateral responses to resistance to a single agent are not uniform, and are instead heterogeneous. Both of these results suggest that designing combination regimens with the objective of reducing resistance is not straightforward, and motivate more work to understand the forces at play.
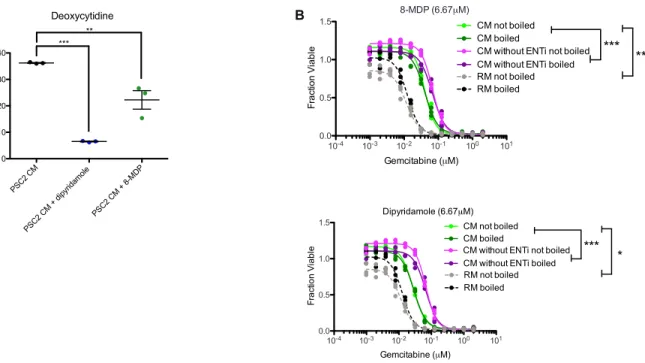
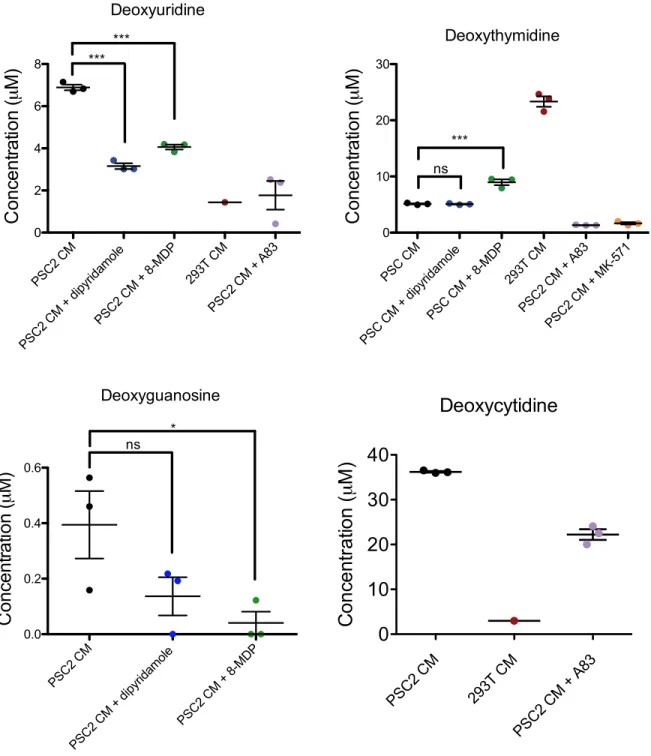
I next studied the role of the microenvironment on gemcitabine resistance in pancreatic cancer. Previously known mechanisms of microenvironment mediated resistance rely on paracrine signaling via heat-labile biomolecules such as cytokines and RNAs. In contrast, I found that pancreatic stromal cells secrete a heat-insensitive compound that confers gemcitabine resistance. I performed a series of biochemical experiments which identified this compound as deoxycytidine. This knowledge suggests that combining inhibition of deoxycytidine synthesis with gemcitabine treatment could increase gemcitabine efficacy.
Thesis supervisor: Michael T. Hemann Title: Associate Professor of Biology Thesis supervisor: Douglas Lauffenburger
Acknowledgements
I look back on the past six years with primarily positive memories – perhaps an unusual feeling for a graduating PhD student. A very significant contributing factor has been the people I have interacted with in this time. First and most importantly, I am incredibly fortunate that my parents have always supported me and enabled me to follow my dreams, even when those dreams involved using all the tape in the house to build paper creations.
The rest of my family is equally as supportive – my brother Barry Dalin has always been my biggest champion. When I meet his friends they always say “so you’re the genius older sister?”. His wife Belen Florez, my sister-in-law completes our family with her grace and strength and our lives have been happier since she entered them.
My new husband Honi Sanders (!!) has also helped me in innumerable ways throughout grad school, by reading my papers and applications, constantly telling me that I am excellent even when I feel otherwise, and picking me up and turning me upside down whenever I’m feeling too morose for his taste. Honi, I officially forgive you for not returning the favor of my making you coffee from 100 coffee teabags as you wrote your thesis several years ago.
In addition to family, I have many academic supporters who were integral to my success in grad school. My advisors, Mike Hemann and Doug Lauffenburger have provided countless hours of advising throughout my time here. Mike’s goofiness and Doug’s sense of calm have been synergistic sources of support. Ernest Fraenkel and the members of my committee over the years (Piyush Gupta, Jackie Lees, Matt Vander Heiden, and Mike Yaffe) have also provided useful advice during my time in grad
school. All my labmates throughout the years have been sources of assistance, both technically and socially. In particular I thank Pete Bruno and Bo Zhao for training and excellent discussions. I also thank Emanuel Kreidl for starting the conditioned media project. My collaborators on the conditioned media project were huge sources of help – Mark Sullivan and Allison Lau provided both invaluable scientific support as well as camaraderie through both joyful and stressful times. Helen Mueller has had multiple roles in my life over the past several years, she performed essential experiments for the conditioned media project, we encouraged each other to learn R, and she has been a good friend, including dancing with me at my wedding two weeks ago!
My experience in grad school has been significantly influenced by the students I have mentored in the lab. I will always fondly remember doing RNA extractions on Christmas with Bouchra Benghomari, and remain impressed by her enthusiasm and continued success. Bea Grauman-Boss has played a critical role in my research during her time in the lab over the past year. Impressively, she had virtually no relevant lab skills when she started working with me last September, but has become almost completely independent in the lab this summer. This was critical as she completed the last experiments for the conditioned media project while I wrote this thesis and planned and celebrated my wedding. Without her help and skill, undoubtedly something crucial would not have been completed.
Finally, I need to thank my friends. My classmates in biograd2013 have been supportive and fun peers from the very beginning. I have had regular eat-lunch-and-support-each-other dates with Amelie Raz and Sonya Entova which were very helpful during several difficult periods of my PhD. In particular, Sonya and I have consumed a pint of ice cream together about once a month since 2nd year, a practice which has
played an integral role in keeping me sane, if not healthy.
I am forever indebted to my two best friends from college, Rachel Amouyal née Ellman, and Leah Mosenkis née Robsman. There is far far too much to write here, but I certainly would not have reached this day without the two of them. Without Cosco I likely would not have done this research, perhaps wouldn’t have even gone to grad school. The lessons and experiences from that time continue to reverberate in my life in ever sweeter ways ( בה ). I am so thankful we have managed to keep in touch as our ׳׳ friendship maintains me in many ways. I only wish we didn’t live spread over 9 hours of time zones. I am also grateful for the friendship of Ranit Patel, who I have known nearly since birth. Defenestrating ourselves and re-wiring her house as children were fun, but her presence during my 5th year of graduate school which began horribly and ended
monumentally (for both of us) was pivotal to my mental health and success. Lastly, I owe a big הברהדות to the Jewish community of Camberville. The community has (often unknowingly!) supported me through difficult times of my PhD, been a source of great friendship, and enriched my life with amazing kabbalat shabbat davening, simchat torah dancing, magical hag BBQs, and so much more…
TABLE OF CONTENTS
CHAPTER 1: INTRODUCTION ...6
HISTORY OF CHEMOTHERAPY RESISTANCE ...7
RESISTANCE TO SINGLE AGENTS ... 13
RESISTANCE TO COMBINATION THERAPIES ... 20
CHAPTER 2: COLLATERAL RESPONSES TO FRONT-LINE CYTOTOXIC CHEMOTHERAPIES ARE HETEROGENEOUS AND SENSITIVITIES ARE SPARSE 38 CHAPTER 3: DEOXYCYTIDINE RELEASE FROM PANCREATIC STELLATE CELLS PROMOTES GEMCITABINE RESISTANCE ... 64
CHAPTER 4: DISCUSSION ... 108
CELL-EXTRINSIC RESISTANCE ... 109
CELL-INTRINSIC RESISTANCE ... 112
CHANGING THE CURRENT STANDARD OF CARE ... 116
Chapter 1: Introduction
Chemotherapy resistance is both a new and old phenomenon: it was first observed relatively recently, in the 20th century, yet evidence suggests that resistant
clones exist prior to therapy. Thus, chemotherapy resistance likely was present in the first tumor to ever arise. This phenomenon is a major human health challenge – it prevents cancer treatments from working, and plays a role in many of the 8.2 million deaths worldwide due to cancer each year (1).
Efforts to combat chemotherapy resistance have ranged from research into resistance mechanisms, development of drugs targeting resistance mechanisms, using multiple drugs in combination, recruiting the immune system, and more. There have been some advances – some cancers now boast cure rates upwards of 90% –
however, resistance is still an issue for many cancer types, and no cancer patient can be unconcerned about chemotherapy resistance.
At the heart of this problem lies a lack of understanding of the basic problem – what is chemotherapy resistance? How does it arise? What factors contribute to it? And most importantly, how can it be prevented? In this thesis I set out to answer some of these questions by focusing on the classical, front-line chemotherapies that are still the mainstay of most chemotherapy regimens today. I first gained insight into the
consequences of resistance by studying broad phenotypic changes resulting from evolutionary acquisition of chemotherapy resistance. Then, I identified a
microenvironmental contribution to chemotherapy resistance in pancreatic cancer, and determined how that factor results in resistance.
History of Chemotherapy Resistance
Initial use of classical chemotherapies
Until the early 20th century, the only treatment for cancer was surgery and
radiotherapy. While these treatments were curative in some cases, most patients eventually succumbed to their disease (2). Curiously, the initial discovery that led to the development of the first chemotherapy was a direct result of war: the use of nitrogen mustards in WWI revealed that these compounds are toxic to lymphoid and myeloid cells (3). This observation led to tests of nitrogen mustards in mice and then humans with lymphoid tumors, revealing that these compounds effectively reduce the size of this type of tumor. While the initial tumor reduction was encouraging, this first success was accompanied by the first observation of chemotherapy resistance – patients’ tumors that initially responded eventually relapsed (4).
Shortly after the discovery that nitrogen mustards could shrink lymphoid tumors, Sydney Farber began investigating the role of folate metabolism in acute lymphoblastic leukemia (ALL). Folate was known to be important for bone marrow function so Farber hypothesized that that blocking the metabolism of folate-requiring compounds would be specifically toxic to ALL cells. Indeed, ALL patients treated with folate antagonists such as aminopterin achieved remissions. Again, these remissions were marred by the presence of chemotherapy resistance – the patients eventually succumbed to their disease (5, 6)
These two examples are widely considered to be the birth of chemotherapy and indeed the compounds discovered in those studies are still in wide use today. In fact,
the 1950s saw the genesis of many currently used anti-cancer compounds after the nitrogen mustard and anti-folate discoveries. For example, Actinomycin D, a
transcription inhibitor, was discovered through the screening of products of fermentation for various human-health related phenotypes (7). Encouragingly, the cure rate of
children with Wilms’ tumor rose from about 45% with surgery and radiation, to 80% with the addition of actinomycin D. Several nucleotide antimetabolites were also developed, including 6-mercaptopurine, 6-azathymidine, and 6-thioguanine (8, 9). Unfortunately, while these new antimetaboles were able to induce remissions, in most cases the remissions were short-lived (10, 11). 5-fluorouracil (5-FU), a third drug also developed in the 50s, was designed based on the observation that rat hepatomas metabolized more uracil than normal tissue. 5-FU targeted this pathway and was found to shrink tumors, however, again 5-FU eventually ceased being effective in these patients (12,
13). Interestingly, early use of these cytotoxic compounds was guided by patient toxicity
rather than drug mechanism of action.
More recent drug development has been guided by different principles. Targeted therapies, for example, are designed with a specific cellular target in mind. This is in contrast to earlier drug development which was guided by generally increasing cytotoxicity or interfering with a particular cellular process such as production of a particular metabolite. Importantly, although classical chemotherapies are often
discussed in contrast to targeted therapies, all drugs including classical chemotherapies bind to one or more cellular targets. The cellular targets of several classical
chemotherapies are discussed below. While targeted and classical chemotherapies do appear to be empirically different, those differences are not yet well understood.
Although the early work in the field of chemotherapy was unable to cure patients, remarkable progress was made in a very short time that laid the groundwork for modern cancer treatment like targeted therapies. The rapid advances were made possible in part by the close relationship between research and clinic. New compounds synthesized in labs were tested in humans after only a short time interval, a practice that is difficult or impossible today due to the current regulatory environment. The research/clinic crosstalk at the time allowed clinical practice to closely mirror the state of research and advance quickly, leading directly to the cancer treatments and cures we have today.
Early knowledge of resistance mechanisms
In response to the initial observations of chemotherapy resistance in the 1950s, research began into how cancers were able to relapse and grow in the presence of previously efficacious drugs. Knowledge of nucleic acid biosynthesis was particularly advanced at the time, so mechanisms of resistance to antimetabolites were the first to be discovered. 5-FU resistance was studied by serially transplanting tumors in mice treated with the drug and then assaying activity of uracil-metabolizing enzymes. This work revealed that 5-FU resistant lines lost the activity of enzymes that transform the uracil base into the riboside and deoxyriboside forms that are toxic to cells (14).
Resistance to methotrexate took longer to understand. Although it was known to work by inhibiting dihydrofolate reductase (DHFR), it took until the 1980s for
researchers to understand that this compound becomes polyglutamated in cells, and that this transformation is required for both its DHFR inhibitory effects and retention in cells (15). Similarly, in the late 1960s, resistance to nitrogen mustards was found to
result from decreased intracellular drug levels via reduced uptake in resistant vs. sensitive cells. Resistant cells were also found to have increased cellular levels of glutathione which can react with nitrogen mustards and reduce their ability to damage their cellular substrates (16, 17). More recently, glutathione conjugation has also been shown to allow the MRP1 transporter to export various drugs including chlorambucil, bringing the story full circle (18).
The main technique initially used to study chemotherapy resistance, of creating resistant lines in vitro or in vivo and characterizing differences to the sensitive parental line, is still in use today. Further, the initially discovered mechanisms of chemotherapy resistance – reduced activity of enzymes needed for toxification, detoxifying drug
modifications, and drug export - have all become well-established mechanisms that can lead to chemotherapy resistance in lab-based experiments. While these revelations answered the question of how some cancers can grow in the presence of some
cytotoxic drugs and laid a foundation for future research into chemotherapy resistance, they did not address the question of where and when these resistant cells originate.
Origin of resistant clones
The question of where resistance originates is not unique to cancer. It is relevant to all fields that use chemical compounds to kill biological entities including anti-viral therapies and antibiotic therapies. In all these cases, the central question asks if drug-resistance existed in the population prior to therapy, or if it was acquired as an
address this question in 1943 with a fluctuation test, asking if bacterial resistance to viral infection was pre-existing or caused by the infection.
To determine if viral resistance is pre-existing in a population of bacteria, Luria and Delbrück divided a culture of bacterial cells into two samples and further subdivided one of those into a large number of small aliquots. They grew all the cultures in the same conditions until, under the spontaneous mutation model, each would be likely to contain resistant cells. At this point they subdivided the previously undivided sample into aliquots, and added virus to all of the aliquots. If treatment with either a virus or drug causes resistance mutations, then the mean and variance of the number of resistant cells in all cultures from both the early-aliquoted and late-aliquoted samples will be equal to each other. If, on the other hand, resistance mutations arise prior to treatment, then the mean and variance of the number of resistant cells in the late-aliquoted sample will be equal to each other, but the mean will be less than the variance in the early-aliquoted sample. The data from the experiment fit the latter scenario, showing that viral resistance is pre-existing in bacterial populations.
Luria and Delbrück’s seminal experiment set off a wave of experimentalists searching for pre-existing vs. acquired resistance to a variety of stimuli in a variety of systems. In 1946, Evelyn Witkin showed that bacterial resistance to UV and X-ray radiation is pre-existing, and by 1948, Demerec showed that streptomycin and penicillin resistance is preexisting in bacteria (19, 20). In 1952, Law showed that resistance to folate antagonists in leukemia is also pre-existing (21). By 1956, he was convinced that the pre-existing nature of resistance in neoplasms required use of high doses of
Initial use of combination chemotherapy regimens to combat resistance
Despite reluctance to treat patients with multiple highly toxic drugs, evidence mounted in favor of high-dose combination chemotherapy. By the mid-50’s, the cancer-initiating potential of a single leukemia cell was well established (23). Further,
chemotherapies were shown to kill a constant fraction, rather than number, of tumor cells at a given dose, motivating the use of high doses to kill the largest fraction of cells. Additionally, in mice, combinations of chemotherapies were more effective than single agents (24). In a landmark effort, Frei and Freireich treated pediatric leukemia patients with a combination regimen termed “VAMP” which proved to be a large improvement over previous single-agent treatments (25, 26). Effective combinations for other malignancies soon followed, such as MOMP for Hodgkin’s lymphoma, C-MOPP for diffuse large B-cell lymphoma, and cyclophosphamide, methotrexate, and fluorouracil after mastectomy in breast cancer, (27–29). The rationale behind the choice of drugs for the combinations was to target different biochemical pathways within the cell to reduce the chance of cross-resistance. This methodology also reduced the overlap in side-effect profiles, allowing escalation of doses of each individual drug. By the mid 1970s, cancer was no longer seen as an invariably fatal disease. Due to the advances of high-dose combination chemotherapy, some cancers were curable, and there was hope for progress in other tumor types as well (30).
Since the 1970s, most of the progress in curing patients has been in the form of novel combinations, and more recently, new types of drugs like targeted therapies and immunotherapies. Despite these developments, the 5-year survival rate across all cancers has only increased from about 50% to about 68% in that time (31). Research in
the intervening decades has focused largely on mechanisms of action and resistance to chemotherapies. Today we have a greater understanding of how these drugs work and how cells evade their actions, but effort is needed to translate this increased
understanding to the clinic to further improve survival rates.
Resistance to Single Agents
Modes of resistance to classical chemotherapies
There are two modes by which tumors can resist chemotherapy. Modifications of the tumor cells themselves can facilitate their own survival in the presence of drug, termed “cell-intrinsic resistance”. Alternatively, modifications of non-malignant cells can result in inter-cell signaling that enables malignant cells to survive in the presence of the drug, termed “cell-extrinsic resistance”. Recognition of cell-intrinsic resistance dates back to the earliest days of chemotherapy, for example, Peter Reichard’s 1959 discovery that 5-FU resistant cells express less uridine and deoxyuridine
phosphorylases and thus cannot transform 5-FU to the toxic riboside or deoxyriboside forms (14). Cell-extrinsic resistance is a much more recent discovery – a role for non-malignant cells in resistance was first proposed in 1990 (32). In the section that follows, I will discuss aspects of these two modes of resistance relevant to my study of collateral effects of cell-intrinsic resistance detailed in chapter 2 of this thesis and my elucidation of a novel mechanism of cell-extrinsic resistance in pancreatic cancer discussed in chapter 3 of this thesis.
Cell-intrinsic resistance
There are many modifications of tumor cells that can result in resistance to chemotherapy. The drug’s target can be mutated such that the drug can no longer bind to it, signaling downstream of the target can be re-activated by increased signaling from parallel pathways, the amount of drug that accumulates in the cell can be decreased by reduced uptake or increased export, the drug can be metabolically inactivated, and the cell can become generally resistant to apoptosis (33–36). At least one of these
resistance mechanisms has been described for many classical chemotherapeutic agents. Here I will discuss known cell-intrinsic mechanisms of resistance to the drugs I studied in this thesis: doxorubicin, vincristine, paclitaxel, cisplatin, and gemcitabine.
Doxorubicin is an anthracycline that poisons the action of topoisomerase II. This enzyme manages the supercoiling and intertwining of DNA by inducing double-strand breaks (DSBs), passing one strand of DNA through the other then repairing the break (37). Doxorubicin interferes with this activity by preventing the re-ligation of the DSBs, resulting in DNA damage (38). Reduced expression of topoisomerase II decreases the number of DSBs caused by doxorubicin treatment, and thus causes resistance to this chemotherapy (39). Doxorubicin can also be pumped out of cells by several
transporters including P-gp, resulting in resistance (40). Finally, mutations in p53, signaling by several transcription factors such as NF-kB, FOXO3, and the activity of kinases such as AKT, MEK/ERK can allow cells to evade apoptosis following
doxorubicin treatment (41, 42). One common side-effect of doxorubicin treatment is cardiotoxicity associated with production of reactive oxygen and nitrogen species (ROS and RNS) (43). However, this mechanism of action is not generally discussed in terms
of doxorubicin’s anti-cancer activity, suggesting more binding partners and effects on tumor cell biology than is currently appreciated.
Vincristine is a natural product isolated from the Madagascar periwinkle plant that functions by destabilizing microtubules leading to mitotic spindle disintegration and eventually apoptosis (44). Similar to doxorubicin, vincristine can be effluxed from cells via transporters including P-gp (40). Additionally, mutations in vincristine’s targets, α- and β-tubulin, can stabilize microtubules and result in resistance to vincristine (45). Further, impairment of the mitotic spindle checkpoint can also cause resistance to this class of drug (46). Interestingly, despite the literature’s focus on vincristine’s role in inhibiting mitosis, this drug and others in its class have been shown to induce apoptosis regardless of cell-cycle stage, as well as to activate JNK signaling suggesting that this drug may have broader effects within the cell beyond inhibition of mitosis (47).
Paclitaxel, another natural product that binds microtubules, is a taxane isolated from the bark of the Pacific yew tree. In contrast to vincristine, this compound stabilizes microtubules, causing a mitotic block and eventually cell death (48). Resistance to paclitaxel is very similar to vincristine resistance: destabilizing mutations in α- and β-tubulin can result in resistance (49). Similarly, paclitaxel can be effluxed by drug pumps including P-gp (50). Interestingly, due to their opposing mechanisms of action and resistance, resistance to vincristine can be co-occurrent with sensitivity to paclitaxel, and vice-versa (51). As with the other drugs discussed, there are hints in the literature for roles of paclitaxel beyond its canonical microtubule-stabilizing action. For example, it has been shown to induce expression of TNF-a (52). Further, one side effect of
axon-degrading microtubule destabilization of vincristine, on first consideration
stabilization of microtubules would not be expected to cause neuropathy. Instead, this effect may result from mitochondrial depolarization or disruption of axonal transport caused by paclitaxel (53). The role of these effects of paclitaxel treatment in malignant cells has not been investigated.
Cisplatin is a platinum-based DNA cross-linking agent. The discovery of this compound is particularly amusing: in the course of studying the effect of
electromagnetic radiation on cell growth, Barnett Rosenberg accidentally found that electrolysis products from his platinum electrodes, including cisplatin, inhibited bacterial cell division. Cisplatin kills cells by forming DNA adducts which activate the
DNA-damage response (54). In the laboratory setting, cisplatin resistance can be caused by reduced intracellular drug levels resulting from reduced drug uptake or increased export via MRP2 or other transporters, although the patient-relevance of this resistance
mechanism remains unclear (55, 56). Additionally, reduced levels of DNA damage repair (both nucleotide excision repair and mismatch repair) can also cause resistance to cisplatin. In this case, cells fail to detect DNA damage and therefore continue to divide (57, 58). Cells can also increase replicative bypass via translesion synthesis to continue DNA synthesis in the presence of cisplatin-induced DNA adducts (59). Curiously, there is a great deal of pre-clinical and clinical evidence that cisplatin and paclitaxel as combination therapy have greater than expected cytotoxic effects and reduced incidence of resistance (60). Given the distinct mechanisms of action and resistance of these two compounds, this relationship is unexpected and hints at additional roles for one or both compounds in cells.
Gemcitabine is a deoxycytidine (dC) analog which causes masked chain
termination via incorporation into DNA, and depletion of deoxynucleotides by inhibiting ribonucleotide reductase (RNR) (61). As with every other chemotherapeutic agent
discussed here, resistance to gemcitabine can result from decreased intracellular levels, in this case due to reduced expression of the nucleoside transporter, hENT1/2 that imports it into cells (62, 63). Overexpression of one of gemcitabine’s targets, RNR, can also result in resistance (64). Gemcitabine requires metabolic toxification, and changes in activity of toxifying enzymes like deoxycytidine kinase (dCK) or detoxifying enzymes like cytidine deaminase (CDA) can reduce the efficacy of this compound (65, 66). Beyond these more simple mechanisms of resistance, alteration in several signaling pathways such as NF-kB, WNT, and NOTCH can reduce cells’ propensity to undergo apoptosis in response to gemcitabine treatment (67).
All of the classical chemotherapies discussed above have several known
mechanisms of resistance, most often elucidated as single-agents in lab-based studies. Interestingly, in every case there are hints for more complex roles for the compounds within cells beyond the current state of knowledge, suggesting that much remains to be discovered in terms of clinically relevant mechanisms of action and resistance.
Recently, the drug discovery field has shifted from a “one drug one target” approach to a “one drug multiple targets” approach, termed polypharmacology, due to the
complexity of the disease and drug resistance. This approach has been most notable in the development of the poly-kinase inhibitor sorafenib (68). Considering the likelihood that classical chemotherapies bind multiple targets and have multiple currently
un-appreciated roles within cells, it is interesting to re-imagine these “tired and old” compounds as new and exciting polypharmacology agents.
Cell-extrinsic resistance
While cell-intrinsic resistance has been well established since nearly the dawn of chemotherapy, more recently researchers have come to appreciate the role of non-tumor cells in causing chemotherapy resistance (69). This phenomenon is particularly relevant in pancreatic ductal adenocarcinoma (PDAC), a malignancy with a striking accumulation of stromal tissue surrounding tumors (70). In fact, cancer cells make up as little as 10% of the mass of PDAC tumors, the large majority of these tumors is
comprised of stromal cells (71). This mode of resistance is termed “cell-extrinsic
resistance” and in this section I will discuss aspects of this phenomenon relevant to the work in chapter 3 of this thesis studying a novel mechanism of cell-extrinsic resistance in pancreatic cancer.
The first real evidence that factors other than the malignant cells themselves may contribute to chemotherapy resistance came from a 1990 study that showed differential sensitivity to several chemotherapeutic agents when assayed in vivo vs in vitro (32). More recently, gene expression based approaches have implicated the stroma in response to chemotherapy (72). Further, the amount of stroma accumulated around a tumor is predictive of patient clinical outcome in several cancer types. Greater stromal accumulation is prognostic of shorter disease-free survival, regardless of cancer type and clinical stage (73). There are many ways the tumor microenvironment can
fibroblasts, and even factors such as cytokines secreted by cells nearby or distal to the tumor.
Interactions with other cell types can even modify the way cells die. For example, some chemotherapeutic agents, such as anthracyclines like doxorubicin, can induce an “immunogenic cell death” that promotes antigen presentation on cancer cells as well as recruitment of antigen-presenting dendritic cells (74, 75). This type of cell death can essentially immunize mice against a particular cancer type under the right conditions (76). Other chemotherapies can also induce recruitment of various types of immune cells to the tumor, such as paclitaxel-induced macrophage infiltration. The presence of these macrophages causes a pro-tumorigenic effect, resulting in chemotherapy
resistance (77, 78). In another study, gemcitabine and 5-FU were shown to activate the inflammasome in myeloid-derived suppressor cells (MDSCs) which can cause T-cells to secrete the chemoprotective cytokine IL-1b (79). While the role of the immune system in the response to chemotherapy is complex, it is clear that these non-malignant cells can both enhance and blunt chemotherapy toxicity (80).
Factors secreted by many types of nearby cells can also play a role in malignant cells’ response to chemotherapy. For example, genotoxic therapies such as doxorubicin can induce senescence in both malignant and normal cells, resulting in secretion of a variety of cytokines and growth factors that can alter therapeutic response (81). IL-6, for example, is secreted from endothelial cells in the thymus after treatment with
doxorubicin. This pro-survival cytokine protects nearby malignant cells from cell death (82). Paracrine signaling of WNT16B from fibroblasts adjacent to tumors has also been shown to protect against anthracycline-induced cytotoxicity (83).
Tumor microenvironment-mediated chemotherapy resistance is particularly relevant in pancreatic cancer. In this malignancy, a variety of cytokines and extracellular matrix proteins have been implicated in therapeutic resistance. For example, b1-integrin signaling in pancreatic stellate cells (PSCs) surrounding pancreatic tumors causes malignant cells to resist radiotherapy-induced apoptosis (84). Similarly, PSCs can also secrete SDF-1a which activates CXCR4 signaling in pancreatic cancer cells, resulting in gemcitabine resistance (85). Another study found that IGFs secreted by various
pancreatic stromal components can activate pro-survival signaling in pancreatic cancer cells, blunting response to gemcitabine treatment (86). Non-soluble proteins can also play a role in resistance – the presence of various extracellular matrix proteins has been found to promote resistance to cytotoxic therapies in pancreatic cancer (87). In addition to proteins, exosomes have also been implicated in resistance to gemcitabine by
transferring a micro-RNA from macrophages to pancreatic cancer cells. The transferred micro-RNA, MiR-365, increased levels of dNTPs in cells as well as the activity of CDA, resulting in decreased gemcitabine efficacy (88).
Resistance to Combination Therapies Theories of combination regimen design
While the discoveries of chemotherapeutic agents starting in the 1950s heralded the start of a new age of cancer treatment options, it was only when these agents were used in combinations that cancer cures were seen (30, 89). One difficulty with
combination lies in choosing the specific drugs to use in the combination. Over the past 60 years there have been several different theories of how to best design combination
chemotherapy regimens. Importantly, none of these theories has been proven to be more effective than any others, so the design of efficacious combination chemotherapy regimens is still an open problem.
In the earliest days of combination chemotherapy, two main principles guided regimen design. Researchers knew that cancers could be initiated by as little as one malignant cell, and that chemotherapies follow a logarithmic killing model – that is, they kill a constant fraction of tumor cells at a given dose, rather than a constant number of tumor cells at a given dose (23, 24). The conclusion from these facts was that to cure cancer, all tumor cells must be eradicated, and to eradicate all tumor cells, the highest possible dose of a chemotherapeutic agent must be administered. The obstacle is that all chemotherapeutic agents available at the time (and most chemotherapies to this day) have very significant toxicities. Therefore, one of the initial guiding principles behind combination chemotherapy design was to select drugs with non-overlapping toxicities, regardless of mechanism of action (30).
The second principle guiding early combination chemotherapy regimen design resulted from the early elucidation of the mechanism of action of many
chemotherapeutic agents. The existing knowledge of drug targets combined with the goal of eradicating all tumor cells led to the hypothesis that combining drugs with different targets would lead to greater overall killing. In the early days of chemotherapy, this was found to be true for targeting different steps in linear and branching metabolic pathways (90, 91). In the modern era, agents are chosen to target different pathways within a cell, and in fact most currently used combinations such as CHOP
cisplatin) follow this rule (92–94). Unfortunately, while these types of combinations can be more effective than empirically designed combinations, they are still curative in only a subset of cases.
The rational choice of drugs in a combination regimen based on the targets of the drugs heralded decades of research into how to design efficacious drug combinations. One theory is to search for drugs that kill more cells in combination than would be predicted based on adding their single-agent efficacy, termed drug synergy. While the concept of synergy seems simple intuitively, it is complicated to formalize
mathematically. For that reason, multiple mathematical definitions of synergy have been developed.
Bliss Independence, one of the earliest mathematical definitions of synergy, is still in wide use today. This definition of synergy assumes that each drug in a
combination acts independently of the other drugs. Therefore, for a given combination of drug A and drug B, the expected killing from the combination of drug A and drug B under the additive model would just be for the cells remaining alive after treatment with drug A to be killed by drug B in the same fraction as drug B kills on its own. More formally, if EA is the killing of drug A at a particular dose, and EB is the killing of drug B
at a particular dose, the expected killing of the combination will be simply:
𝐸
"#= 𝐸
"+ 𝐸
#(1 − 𝐸
")
(1)If the observed killing of drug A in combination with drug B is greater than EAB, the
combination is said to be synergistic. If the observed killing is less, the combination is said to be antagonistic (95, 96).
A second popular method of defining drug synergy, Lowe Additivity, considers the actual dose of a drug used to achieve a particular level of killing. In this case under the additive model, adding the same amount of drug A or drug B should result in the same increase in killing. To formalize that, if dose Ai of drug A results in killing E, a drug
pair is additive if the same dose of drug B, dose Bi, results in the same killing, E. This
intuition leads to the understanding that the doses of drug A and B required to achieve killing E when used in combination (dose Ac and dose Bc), are proportional to the
individual doses required to achieve the same killing E:
"+ ",
+
#+
#,
= 1
(2)When this is not the case, and equation (2) sums to less than 1, then AC and BC are less
than expected, and the combination is synergistic (96, 97).
Many studies have used either Loewe Additivity or Bliss Independence to
understand interactions between drugs. These two definitions of synergy form the basis of analysis of combination drug screens. For example, using Bliss Additivity (eq. 1) as a metric to define screen hits, Borisy et al found that the antipsychotic agent
chlorpromazine, and the antiprotozoal agent pentamidine, are synergistic when used together on A549 lung carcinoma cells (98). Additionally, Meco et al found that doxorubicin and the FUS-CHOP inhibitor ET-743 are weakly synergistic in a
rhabdomyosarcoma cell line using Lowe Additivity (eq. 2) as a synergy metric (99). In fact, further analysis of combination drug screens have shown that synergy tends to be more common between drugs targeting the same pathway rather than different
promising results based on synergistic combinations found in the lab have not made a major impact on clinical practice, perhaps in part due to changes that result when cells are grown in the lab (101, 102).
While the goal with synergistic combinations is to maximize malignant cell killing, any resistant cells that escape or emerge after therapy can grow and result in a
relapsed tumor. Therefore, a more recent theory of combination chemotherapy design, termed collateral sensitivity, aims to design combinations with the goal of reducing the emergence of resistance. The theory of collateral sensitivity actually originated in the antibiotic resistance field in the 1950s. Waclaw Szybalskia and Vernon Bryson surveyed the antibiotic sensitivities of a variety of resistant microbes to other antibiotics and found that chloromycetin resistance often results in polymyxin B and circulin sensitivity. They suggested that use of antibiotics in a rational sequence may enable the back-selection of selection of sensitivity (103). More recently this concept has gained traction as a way to prevent antibiotic resistance by using collaterally sensitive antibiotics either in
sequence, or in combination (104–106).
For many years, study of collateral sensitivity in chemotherapy resistance focused solely on collateral sensitivities due to efflux-pump mediated resistance (107). In the past few years, studies have begun to more broadly survey collateral responses to chemotherapies. In one study, a temporal collateral sensitivity between dasatinib, and the c-MET and VEGFR inhibitors crizotinib, foretinib, cabozantinib, and vandetinib was reported in Ph+ ALL cells (108). Another study found that cell lines resistant to first-line tyrosine kinase inhibitors (TKIs) are often sensitized to non-TKIs including the classical chemotherapies etoposide and pemetrexed (109). Further, review of the literature
suggests the existence of other unappreciated collateral sensitivities, such as reciprocal collateral sensitivities between microtubule destabilizers such as vinblastine and
microtubule stabilizers such as paclitaxel, as well as between cisplatin and paclitaxel (51, 110). Unfortunately, the field of collateral sensitivity in chemotherapy resistance is very young, and no results have yet been translated to the clinic. Further, the prediction of collateral sensitivity appears to be more complex than first imagined, as different lines resistant to the same therapy can result from different evolutionary trajectories and thus occupy different cell states and have different phenotypic drug sensitivities (109, 111). Finally, collateral sensitivities are currently discovered by evolving resistant lines in the lab and it is unclear how faithfully lab-based evolutionary pressures mimic those within patients undergoing chemotherapy treatment.
The concepts of collateral sensitivity, drug synergy and different drug targets all focus on the interactions between drugs in a combination. However, at the dawn of combination chemotherapy, researchers thought that perhaps the most effective combinations were comprised of drugs that did not interact at all. Specifically, this hypothesis states that tumors or cells that don’t respond at all to one drug will respond to a second drug – the drugs do not interact at all (112–115). After a long foray studying interacting combinations of drugs, the field has recently returned to the idea of
independent action of drugs in a combination. The first experimental evidence in favor of independent action came with the demonstration that the drugs in the CHOP regimen demonstrate less pairwise synergy than might be expected, and further that this
regimen homogenizes single-agent genetic dependencies both in cell lines and mouse models (116). More recently, Palmer and Sorger demonstrated that the efficacy of the
large majority of FDA approved drug combinations is explained by independent action – in other words, in any given patient, only one drug is active (117). This surprising result implies that, in theory, each patient could be treated effectively with only one drug. However, currently we are unable to determine which drug to use for each patient and so need to use combinations in order to maximize the chances of treating a patient with the “correct” drug for their tumor. This motivates two avenues of research: in the short-term, we need to learn how to predict drugs that act independently of each other. In the long-term, we need to find effective biomarkers to a priori determine the “best” drug for a given patient, in order to spare them the side effects of the other drugs in the
combination.
Extending knowledge of resistance to single agents to resistance to combination therapies
The causes of resistance to combination therapies are not well understood in spite of the surfeit of knowledge about resistance to single therapies. While
combinations are often touted as the solution to drug resistance, very little research has focused on resistance in a multi-drug setting. The known mechanisms of resistance to combinations of drugs often rely on general detoxification of the drugs in question. For example, expression of aldehyde dehydrogenase 1A1, an enzyme that detoxifies
aldehyde substrates via NAD+-dependent oxidation, is associated with poor response to
CHOP therapy (118). Another commonly studied multi-drug resistance mechanism is drug efflux via several channels, including P-gp (107, 119, 120). Interestingly, although P-gp expression is associated with drug resistance, and in a lab-based setting P-gp
inhibition can reverse drug resistance, P-gp inhibitors have not been successful in the clinic after three decades of research into P-gp inhibitor development (119, 121).
While the study of general detoxification mechanisms causing multi-drug resistance has not resulted in clinical progress, most research is still conducted in a single-drug setting in order to isolate the effects of single drugs. For example, CRISPR and RNAi screens to identify genetic modulators of resistance to chemotherapies are done in the presence of one drug (122–124). However, since most patients are treated with multiple drugs, resistance in the single-drug setting may not be relevant to the clinic. Further, there has been no research studying the relationship between resistance to single drugs and resistance to multiple drugs. For example – if drug A has resistance mechanism 1, and drug B has resistance mechanism 2, no one has studied if resistance to drug A and B used together results from mechanism 1, 2, both, or neither. Resistance in the multi-drug setting will need to be addressed more critically to make an impact on clinical practice.
Conclusion
As described above, biomedical research has amassed a great deal of knowledge about resistance to single agents and theories of design of combination chemotherapy regimens. In spite of this, chemotherapy resistance is still a significant problem, leading to millions of deaths worldwide every year. In this thesis I present work to extend our knowledge in both of these areas. In chapter 2 of this thesis, I discuss work exploring collateral sensitivities to front-line chemotherapies to determine if this concept could be useful for rational design of new combinations of front-line
chemotherapies. In chapter 3 I present the discovery of a novel mode of stroma-mediated gemcitabine resistance. This work represents a step toward understanding and preventing chemotherapy resistance.
References
1. Cancer Research UK, Worldwide Cancer Mortality Statistics, (available at
https://www.cancerresearchuk.org/health-professional/cancer-statistics/worldwide-cancer/mortality).
2. V. T. DeVita, E. Chu, A history of cancer chemotherapy. Cancer Res. 68, 8643– 8653 (2008).
3. E. B. Krumbhaar, H. D. Krumbhaar, The Blood and Bone Marrow in Yelloe Cross Gas (Mustard Gas) Poisoning. J. Med. Res. 40, 497-508.3 (1919).
4. A. Gilman, F. S. Philips, The Biological Actions and Therapeutic Applications of the B-Chloroethyl Amines and Sulfides. Science. 103, 409–36 (1946).
5. S. Farber, L. K. Diamond, R. D. Mercer, R. F. Sylvester Jr, J. A. Wolff, Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (aminopterin). N. Engl. J. Med. 238, 787–793 (1948). 6. S. Farber, Some observations on the effect of folic acid antagonists on acute
leukemia and other forms of incurable cancer. Blood. 127, 271 (2016).
7. S. Farber, G. D. Angio, A. Evans, A. Mitus, Clinical Studies of Actinomycin D With Special Reference to Wilms’ Tumor in Children. J. Urol. 168, 2560–2561 (1954). 8. G. H. Hitchings, G. B. Elion, The chemistry and biochemistry of purine analogs.
Ann. N. Y. Acad. Sci. 60, 195–9 (1954).
9. G. B. Elion, S. Singer, G. H. Hitchings, Antagonists of nucleic acid derivatives. VIII. Synergism in combinations of biochemically related antimetabolites. J. Biol.
Chem. 208, 477–88 (1954).
10. J. H. Burchenal, M. L. Murphy, R. R. Ellison, M. P. Sykes, T. C. Tan, L. A. Leone,
et al., Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the
treatment of leukemia and allied diseases. Blood. 8, 965–99 (1953).
11. S. Farber, D. Pinkel, E. M. Sears, R. Toch, Advances in chemotherapy of cancer in man. Adv. Cancer Res. 4, 1–71 (1956).
12. C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, et al., Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 179, 663–6 (1957).
13. A. R. Curreri, F. J. Ansfield, F. A. Mciver, F. Ansfield, F. Mciver, Clinical Studies with 5-Fluorouracil Clinical Studies with 5-Fluorouracir. Cancer Res. 18, 478–484 (1958).
14. P. Reichard, O. Skold, G. Klein, Possible enzymic mechanism for the
development of resistance against fluorouracil in ascites tumours. Nature. 183, 939–41 (1959).
15. R. Zhao, I. D. Goldman, Resistance to antifolates. Oncogene. 22, 7431–7457 (2003).
resistance to alkylating agents in Ehrlich tumor cells. Biochem. Biophys. Res.
Commun. 32, 650–7 (1968).
17. G. J. Goldenberg, Properties of l5178y lymphoblasts highly resistant to nitrogen mustard. Ann. N. Y. Acad. Sci. 163, 936–953 (1969).
18. K. Barnouin, I. Leier, G. Jedlitschky, A. Pourtier-Manzanedo, J. König, W. D. Lehmann, et al., Multidrug resistance protein-mediated transport of chlorambucil and melphalan conjugated to glutathione. Br. J. Cancer. 77, 201–209 (1998). 19. E. M. Witkin, Genetics of Resistance to Radiation in ESCHERICHIA COLI.
Genetics. 32, 221–48 (1947).
20. M. Demerec, Origin of Bacterial Resistance to Antibiotics. J. Bacteriol. 56, 63–74 (1948).
21. L. W. Law, Origin of the Resistance of Leukæmic Cells to Folic Acid Antagonists.
Nature. 169, 628–629 (1952).
22. L. W. Law, Differences between Cancers in Terms of Evolution of Drug Resistance Differences between Cancers in Terms of Evolution of Drug Resistance, 698–716 (1956).
23. J. Furth, M. C. Kahn, The transmission of leukemia of mice with a single cell. Am.
J. Cancer. 31, 276–282 (1937).
24. H. E. Skipper, F. M. Schabel, W. S. Wilcox, Experimental evaluation of potential anticancer agents. Xiii. On the criteria and kinetics associated with “curability” of experimental leukemia. Cancer Chemother. reports. 35, 1–111 (1964).
25. E. J. FREIREICH, M. Karon, E. Frei, in Proc (1964).
26. E. Frei III, in Proc Am Assoc Cancer Res (1963), vol. 5, p. 20.
27. V. T. Devita, J. H. Moxley, K. Brace, E. Frei III, in Proc Am Assoc Cancer Res (1965), vol. 6, pp. 881–895.
28. V. T. DeVita, G. P. Canellos, B. Chabner, P. Schein, S. P. Hubbard, R. C. Young, Advanced diffuse histiocytic lymphoma, a potentially curable disease. Lancet
(London, England). 1, 248–50 (1975).
29. G. Bonadonna, E. Brusamolino, P. Valagussa, A. Rossi, L. Brugnatelli, C. Brambilla, et al., Combination Chemotherapy as an Adjuvant Treatment in Operable Breast Cancer. N. Engl. J. Med. 294, 405–410 (1976).
30. V. T. DeVita, P. S. Schein, The use of drugs in combination for the treatment of cancer: rationale and results. N. Engl. J. Med. 288, 998–1006 (1973).
31. C. R. Manrow R., Cancer fact sheet (National Institutes of Health). NIH Fact
Sheet, 1–2 (2010).
32. B. A. Teicher, T. S. Herman, S. A. Holden, Y. Wang, M. R. Pfeffer, J. W.
Crawford, et al., Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science (80-. ). 247, 1457–1461 (1990).
Pathol. 205, 275–292 (2005).
34. M. J. Niederst, J. A. Engelman, Bypass Mechanisms of Resistance to Receptor Tyrosine Kinase Inhibition in Lung Cancer. Sci. Signal. 6, re6–re6 (2013). 35. G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe, M. M. Gottesman,
Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 5, 219–234 (2006).
36. Okushi, R. M. Mohammad, I. Muqbil, L. Lowe, C. Yedjou, H.-Y. Hsu, et al., Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 35, S78–S103 (2015).
37. L. Liu, DNA Topoisomerase Poisons As Antitumor Drugs. Annu. Rev. Biochem.
58, 351–375 (1989).
38. J. L. Nitiss, Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev.
Cancer. 9, 338–350 (2009).
39. D. J. Burgess, J. Doles, L. Zender, W. Xue, B. Ma, W. R. McCombie, et al., Topoisomerase levels determine chemotherapy response in vitro and in vivo.
Proc. Natl. Acad. Sci. U. S. A. 105, 9053–9058 (2008).
40. J. M. Ford, W. N. Hait, Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol. Rev. 42, 155–99 (1990).
41. S. Fan, W. S. El-Deiry, I. Bae, J. Freeman, D. Jondle, K. Bhatia, et al., p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res. 54, 5824–30 (1994).
42. J. Cox, S. Weinman, Mechanisms of doxorubicin resistance in hepatocellular carcinoma. 3, 57–69 (2016).
43. A. De Angelis, D. Cappetta, L. Berrino, K. Urbanek, in Cardiotoxicity (InTech, 2018; http://www.intechopen.com/books/cardiotoxicity/doxorubicin-cardiotoxicity-multiple-targets-and-translational-perspectives).
44. E. Martino, G. Casamassima, S. Castiglione, E. Cellupica, S. Pantalone, F. Papagni, et al., Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorganic Med. Chem. Lett. 28, 2816–2826 (2018). 45. H. L. Cells, M. Kavallaris, A. S. Tait, B. J. Walsh, L. He, S. B. Horwitz, et al.,
Multiple Microtubule Alterations Are Associated with Vinca Alkaloid Resistance in, 5803–5809 (2001).
46. A. Masuda, K. Maeno, T. Nakagawa, Association between Mitotic Spindle Checkpoint Impairment and Susceptibility to the Induction of Apoptosis by Anti-Microtubule Agents in Human Lung Cancers. Am. J. Pathol. 163, 1109–1116 (2003).
47. D. Bates, A. Eastman, Microtubule destabilising agents : far more than just antimitotic anticancer drugs, 255–268 (2017).
48. M. A. N. N. Jordan, R. J. Toso, D. Thrower, L. Wilson, Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. 90, 9552–9556
(1993).
49. F. Cabral, L. Wlble, S. Brenner, R. Brinkley, Taxol-requiring Mutant of Chinese Hamster Ovary Cells with Impaired Mitotic Spindle Assembly. 97 (1983). 50. S. B. Horwitz, D. Cohen, S. Rao, I. Ringel, H. J. Shen, C. P. Yang, Taxol:
mechanisms of action and resistance. J. Natl. Cancer Inst. Monogr., 55–61 (1993).
51. M. Hari, Y. Wang, S. Veeraraghavan, F. Cabral, Mutations in alpha- and beta-tubulin that stabilize microtubules and confer resistance to colcemid and vinblastine. Mol. Cancer Ther. 2, 597–605 (2003).
52. Y. Huang, Y. Fang, J. Wu, J. M. Dziadyk, X. Zhu, M. Sui, et al., Regulation of Vinca alkaloid-induced apoptosis by NF- K B / I K B pathway in human tumor cells, 271–278 (2004).
53. E. Gornstein, T. L. Schwarz, Neuropharmacology The paradox of paclitaxel neurotoxicity : Mechanisms and unanswered questions. NP. 76, 175–183 (2014). 54. L. Kelland, The resurgence of platinum-based cancer chemotherapy. Nat. Rev.
Cancer. 7, 573–584 (2007).
55. P. Copper, K. Katano, A. Kondo, R. Safaei, A. Holzer, G. Samimi, et al.,
Acquisition of Resistance to Cisplatin Is Accompanied by Changes in the Cellular, 6559–6565 (2008).
56. H. Thomas, H. M. Coley, Overcoming Multidrug Resistance in Cancer : An Update on the Clinical Strategy of Inhibiting P-Glycoprotein (2003),
doi:10.1177/107327480301000207.
57. D. Fink, S. Aebi, S. B. Howell, The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 4, 1–6 (1998).
58. I.-Y. Chang, M.-H. Kim, H. B. Kim, D. Y. Lee, S.-H. Kim, H. Kim, et al., Small interfering RNA-induced suppression of ERCC1 enhances sensitivity of human cancer cells to cisplatin. Biochem. Biophys. Res. Commun. 327, 225–33 (2005). 59. J. Doles, T. G. Oliver, E. R. Cameron, G. Hsu, T. Jacks, G. C. Walker,
Suppression of Rev3 , the catalytic subunit of Pol ζ , sensitizes drug-resistant lung tumors to chemotherapy, 1–6 (2010).
60. B. Stordal, N. Pavlakis, R. Davey, A systematic review of platinum and taxane resistance from bench to clinic: an inverse relationship. Cancer Treat. Rev. 33, 688–703 (2007).
61. B. A. Chabner, D. L. Longo, Eds., Cancer Chemotherapy, Immunotherapy, and
Biotherapy. Principles and Practice (Wolters Kluwer, Philadelphia, ed. 6, 2019).
62. J. Spratlin, R. Sangha, D. Glubrecht, L. Dabbagh, J. D. Young, C. Dumontet, et
al., The Absence of Human Equilibrative Nucleoside Transporter 1 Is Associated
with Reduced Survival in Patients With Gemcitabine- Treated Pancreas Adenocarcinoma. 10, 6956–6961 (2004).
Transcription Analysis of Human Equilibrative Nucleoside Transporter-1 Predicts Survival in Pancreas Cancer Patients Treated with Gemcitabine, 3928–3936 (2006).
64. J. D. Davidson, L. Ma, M. Flagella, S. Geeganage, L. M. Gelbert, C. A. Slapak, An increase in the expression of ribonucleotide reductase large subunit 1 is
associated with gemcitabine resistance in non-small cell lung cancer cell lines.
Cancer Res. 64, 3761–6 (2004).
65. J. R. Kroep, W. J. P. Loves, C. L. Van Der Wilt, E. Alvarez, I. Talianidis, E. Boven,
et al., Pretreatment Deoxycytidine Kinase Levels Predict in Vivo Gemcitabine
Sensitivity 1. 1, 371–376 (2002).
66. T. Neff, C. A. Blau, Forced expression of cytidine deaminase confers resistance to cytosine arabinoside and gemcitabine. Exp. Hematol. 24, 1340–6 (1996).
67. Y. Jia, J. Xie, Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes Dis. 2, 299–306 (2015).
68. Z. A. Knight, H. Lin, K. M. Shokat, Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer. 10, 130–137 (2010).
69. F. Klemm, J. A. Joyce, Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 25, 198–213 (2015).
70. J. Kleeff, M. Korc, M. Apte, C. La Vecchia, C. D. Johnson, A. V. Biankin, et al., Pancreatic cancer. Nat. Rev. Dis. Prim. 2, 1–23 (2016).
71. M. Erkan, S. Hausmann, C. W. Michalski, A. A. Fingerle, M. Dobritz, J. Kleeff, et
al., The role of stroma in pancreatic cancer: Diagnostic and therapeutic
implications. Nat. Rev. Gastroenterol. Hepatol. 9, 454–467 (2012).
72. P. Farmer, H. Bonnefoi, P. Anderle, D. Cameron, P. Wirapati, V. Becette, et al., A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 15, 68–74 (2009).
73. J. Wu, C. Liang, M. Chen, W. Su, Association between tumor-stroma ratio and prognosis in solid tumor patients : a systematic review and meta-analysis. 7 (2016).
74. L. Zitvogel, L. Galluzzi, M. J. Smyth, G. Kroemer, Mechanism of Action of Conventional and Targeted Anticancer Therapies: Reinstating
Immunosurveillance. Immunity. 39, 74–88 (2013).
75. L. Galluzzi, A. Buqué, O. Kepp, L. Zitvogel, G. Kroemer, Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell. 28, 690–714 (2015).
76. O. Kepp, L. Senovilla, I. Vitale, E. Vacchelli, S. Adjemian, P. Agostinis, et al., Consensus guidelines for the detection of immunogenic cell death (2014), doi:10.4161/21624011.2014.955691.
77. W. M. Gallagher, N. Wadhwani, S. D. Keil, S. A. Junaid, H. S. Rugo, E. Shelley, Leukocyte Complexity Predicts Breast Cancer Survival and Functionally
Regulates Response to Chemotherapy (2011), doi:10.1158/2159-8274.CD-10-0028.
78. T. Shree, O. C. Olson, B. T. Elie, J. C. Kester, A. L. Garfall, K. Simpson, et al., Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer, 2465–2479 (2011).
79. M. Bruchard, G. Mignot, V. Derangère, F. Chalmin, A. Chevriaux, F. Végran, et
al., Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor
cells activates the Nlrp3 inflammasome and promotes tumor growth. 19 (2013), doi:10.1038/nm.2999.
80. D. W. Mcmillin, J. M. Negri, C. S. Mitsiades, The role of tumour–stromal
interactions in modifying drug response: challenges and opportunities. 12 (2013), doi:10.1038/nrd3870.
81. Y. Sun, P. S. Nelson, Molecular Pathways : Involving Microenvironment Damage Responses in Cancer Therapy Resistance, 4019–4026 (2012).
82. L. A. Gilbert, M. T. Hemann, DNA Damage-Mediated Induction of a Chemoresistant Niche. Cell. 143, 355–366 (2010).
83. Y. Sun, J. Campisi, C. Higano, T. M. Beer, P. Porter, I. Coleman, et al.,
Treatment-induced damage to the tumor micro - environment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 18, 1359–1368 (2012). 84. T. S. Mantoni, S. Lunardi, O. Al-Assar, A. Masamune, T. B. Brunner, Pancreatic
stellate cells radioprotect pancreatic cancer cells through β1-integrin signaling.
Cancer Res. 71, 3453–3458 (2011).
85. H. Zhang, H. Wu, J. Guan, L. Wang, X. Ren, X. Shi, et al., Paracrine SDF-1α signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget. 6, 3085– 3097 (2015).
86. L. Ireland, A. Santos, M. S. Ahmed, C. Rainer, S. R. Nielsen, V. Quaranta, et al., Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 76, 6851–6863 (2016).
87. S. Müerköster, K. Wegehenkel, A. Arlt, M. Witt, B. Sipos, M. L. Kruse, et al., Tumor Stroma Interactions Induce Chemoresistance in Pancreatic Ductal Carcinoma Cells Involving Increased Secretion and Paracrine Effects of Nitric Oxide and Interleukin-1β. Cancer Res. 64, 1331–1337 (2004).
88. Y. Binenbaum, E. Fridman, Z. Yaari, N. Milman, A. Schroeder, G. Ben David, et
al., Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug
Resistance in Pancreatic Adenocarcinoma, 5287–5300 (2018).
89. J. F. Holland, Hopes for tomorrow versus realities of today: therapy and prognosis in acute lymphocytic leukemia of childhood. Pediatrics. 45, 191–3 (1970).
90. H. E. SKIPPER, J. R. THOMSON, M. BELL, Attempts at dual blocking of biochemical events in cancer chemotherapy. Cancer Res. 14, 503–7 (1954).
91. V. R. Potter, H. Simonson, Sequential blocking of metabolic pathways in vivo.
Proc. Soc. Exp. Biol. Med. 76, 41–6 (1951).
92. T. A. Yap, A. Omlin, J. S. De Bono, Development of Therapeutic Combinations Targeting Major Cancer Signaling Pathways. 31 (2019),
doi:10.1200/JCO.2011.37.6418.
93. B. E. K. Rowinsky, M. R. Gilbert, W. P. Mcguire, D. A. Noe, L. B. Grochow, A. A. Forastiere, et al., Sequences of Taxol and Cisplatin : A Phase I and
Pharmacologic Study. 9, 1692–1703 (2019).
94. A. J. J. Wood, J. O. Armitage, Treatment of Non-Hodgkin’s Lymphoma. N. Engl.
J. Med. 328, 1023–1030 (1993).
95. C. I. Bliss, The toxicity of poisons applied jointly. Ann. Appl. Biol. 26, 585–615 (1939).
96. J. Foucquier, M. Guedj, Analysis of drug combinations: current methodological landscape. Pharmacol. Res. Perspect. 3 (2015), doi:10.1002/prp2.149.
97. S. Loewe, H. Muischnek, Über Kombinationswirkungen. Arch. für Exp. Pathol.
und Pharmakologie. 114, 313–326 (1926).
98. E. R. Price, G. Serbedzija, A. A. Borisy, P. J. Elliott, N. W. Hurst, M. S. Lee, et al., Systematic discovery of multicomponent therapeutics. Proc. Natl. Acad. Sci. 100, 7977–7982 (2003).
99. D. Meco, T. Colombo, P. Ubezio, M. Zucchetti, M. Zaffaroni, A. Riccardi, et al., Effective combination of ET-743 and doxorubicin in sarcoma: Preclinical studies.
Cancer Chemother. Pharmacol. 52, 131–138 (2003).
100. J. Lehár, G. R. Zimmermann, A. S. Krueger, R. A. Molnar, J. T. Ledell, A. M. Heilbut, et al., Chemical combination effects predict connectivity in biological systems. Mol. Syst. Biol. 3, 1–14 (2007).
101. A. Tsherniak, K. Hinohara, R. Beroukhim, G. Ha, G. Getz, F. Vazquez, et al., Genetic and transcriptional evolution alters cancer cell line drug response. Nature.
560, 325–330 (2018).
102. G. Ha, R. Beroukhim, B. Wong, N. F. Greenwald, J. Shih, C. Oh, et al., Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 49, 1567–1575 (2017).
103. W. Szybalski, V. Bryson, Genetic Studies on Microbial Cross Resistance To Toxic Agents I. ,. J. Bacteriol. 64, 489–499 (1952).
104. C. Munck, H. K. Gumpert, a. I. N. Wallin, H. H. Wang, M. O. a. Sommer, Prediction of resistance development against drug combinations by collateral responses to component drugs. Sci. Transl. Med. 6, 262ra156-262ra156 (2014). 105. L. Imamovic, M. O. a Sommer, Use of collateral sensitivity networks to design
drug cycling protocols that avoid resistance development. Sci. Transl. Med. 5, 204ra132 (2013).
evolution of antibiotic resistance. Nat. Rev. Microbiol. 7, 460–466 (2009).
107. K. M. Pluchino, M. D. Hall, A. S. Goldsborough, R. Callaghan, M. M. Gottesman, Collateral sensitivity as a strategy against cancer multidrug resistance. 15, 98– 105 (2013).
108. B. Zhao, J. C. Sedlak, R. Srinivas, B. Tidor, D. A. Lauffenburger, M. T. Hemann,
et al., Exploiting Temporal Collateral Sensitivity in Tumor Clonal Evolution
Exploiting Temporal Collateral Sensitivity in Tumor Clonal Evolution. Cell, 1–13 (2016).
109. A. Dhawan, D. Nichol, F. Kinose, M. E. Abazeed, A. Marusyk, E. B. Haura, et al., Collateral sensitivity networks reveal evolutionary instability and novel treatment strategies in ALK mutated non-small cell lung cancer. Sci. Rep. 7, 1232 (2017). 110. B. Stordal, N. Pavlakis, R. Davey, A systematic review of platinum and taxane
resistance from bench to clinic: An inverse relationship. Cancer Treat. Rev. 33, 688–703 (2007).
111. D. Nichol, J. Rutter, C. Bryant, A. M. Hujer, S. Lek, M. D. Adams, et al., Antibiotic collateral sensitivity is contingent on the repeatability of evolution. Nat. Commun.
10 (2019), doi:10.1038/s41467-018-08098-6.
112. J. . Gaddum, Pharmacology (Oxford University Press, First Edit., 1940). 113. E. Frei, E. J. Freireich, E. Gehan, D. Pinkel, J. F. Holland, O. Selawry, et al.,
Studies of Sequential and Combination Antimetabolite Therapy in Acute Leukemia: 6-Mercaptopurine and Methotrexate. Blood. 18, 431–454 (1961). 114. E. Frei, M. Karon, R. H. Levin, E. J. Freireich, R. J. Taylor, J. Hananian, et al.,
The Effectiveness of Combinations of Antileukemic Agents in Inducing and Maintaining Remission in Children with Acute Leukemia. Blood. 26, 642–656 (1965).
115. E. J. Freireich, E. Gehan, E. Frei III, L. R. Schroeder, I. J. Wolman, R. Anbari, et
al., The Effect of 6-Mercaptopurine on the Duration of Steorid-induced
Remissions in Acute Leukemia: A Model for Evaluation of Other Potentially Useful Therapy. Blood. 21, 699–716 (1963).
116. J. R. Pritchard, P. M. Bruno, L. a Gilbert, K. L. Capron, D. a Lauffenburger, M. T. Hemann, Defining principles of combination drug mechanisms of action. Proc.
Natl. Acad. Sci. U. S. A. 110, E170-9 (2013).
117. A. C. Palmer, P. K. Sorger, Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell. 171, 1678-1691.e13 (2017).
118. J. Jiang, Y. Liu, Y. Tang, L. Li, R. Zeng, S. Zeng, et al., ALDH1A1 induces resistance to CHOP in diffuse large B-cell lymphoma through activation of the JAK2/STAT3 pathway. Onco. Targets. Ther. 9, 5349–5360 (2016).
119. Z. Binkhathlan, A. Lavasanifar, P-glycoprotein inhibition as a therapeutic
approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr. Cancer Drug Targets. 13, 326–46 (2013).