HAL Id: tel-02003501
https://tel.archives-ouvertes.fr/tel-02003501
Submitted on 1 Feb 2019HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Mécanismes de sensibilité/résistance des cellules
tumorales aux inhibiteurs de réparation de l’ADN Dbait.
Wael Jdey
To cite this version:
Wael Jdey. Mécanismes de sensibilité/résistance des cellules tumorales aux inhibiteurs de réparation de l’ADN Dbait.. Biologie cellulaire. Université Paris Saclay (COmUE), 2016. Français. �NNT : 2016SACLS463�. �tel-02003501�
THESE DE DOCTORAT
DEL’U
NIVERSITEP
ARIS-S
ACLAY PREPAREE AL’UNIVERSITE
PARIS-SUD
XI
ECOLE DOCTORALE N° 582 – CBMS Cancérologie : Biologie - Médecine - Santé Discipline : Aspects moléculaires et cellulaires de la biologie
Présentée et soutenue par
Wael Jdey
Le 25 Novembre 2016
MECANISMES DE SENSIBILITE/RESISTANCE DES
CELLULES TUMORALES AUX INHIBITEURS DE
REPARATION DE L’ADN DBAIT
Directeur de thèse : Dr. Marie DUTREIX
Composition du Jury :
Dr, Kannouche, Patricia Gustave Roussy Présidente du jury Dr. Hall, Janet Centre de recherche en cancérologie de Lyon Rapporteur Dr. Mes-Masson, Anne-Marie Institut du cancer de Montréal Rapporteur
Dr., Stern, Marc-Henri Institut Curie Examinateur
Dr., Lopez, Bernard Gustave Roussy Examinateur
Dr., Dutreix, Marie Institut Curie Directeur de thèse
Dr., Dixon, Graham Onxeo Invité
Dr., Sun, Jian-Sheng Muséum National des Histoires Naturelles Invité NNT : 2016SACLS463
CONFIDENTIEL 3 « Le monde est dangereux à vivre ! Non pas tant à cause de ceux qui font le mal, mais à cause de ceux qui regardent et laissent faire »
CONFIDENTIEL 5
PREFACE
Les travaux de recherche présentés dans ce manuscrit ont été menés dans le cadre d’une collaboration entre le laboratoire du Dr. Marie DUTREIX à l’Institut Curie et la société DNA THERAPEUTICS, aujourd’hui filiale d’ONXEO. L’équipe du Dr. Marie DUTREIX fait partie de l’unité de recherche U1021/UMR3347 dirigée par le Pr. Simon SAULE. La société DNA THERAPEUTICS a été crée en 2006 dans l’optique de développer un nouveau concept d’inhibiteurs, les molécules appâts, aussi appelées siDNA pour « short interfering DNA ». Plusieurs chercheurs, médecins, physiciens, ingénieurs et techniciens ont contribué à la création et au développement de cette société, tels que ma directrice de thèse, le Dr. Marie DUTREIX, et le précédent PDG de la société DNA THERAPEUTICS, le Pr. Jian-Sheng SUN. Ces travaux de thèse ont été financés par une bourse CIFRE (Conventions Industrielles de Formation par la Recherche en Entreprise) attribuée par l’ANRT (Association Nationale de la Recherche et de la Technologie) en Décembre 2013.
La majorité des traitements anticancéreux doivent leur efficacité à leur capacité d’endommager l’ADN. En effet, les cellules qui ne réparent pas efficacement ces dommages accumulent des lésions au niveau de leur ADN et meurent au moment de la division cellulaire. Les défauts dans les voies de réparation de l’ADN sont aujourd’hui largement exploités pour le traitement du cancer. En effet, la capacité des tumeurs à réparer les lésions induites par les traitements génotoxiques (chimio- et radiothérapie) leur confère une résistance intrinsèque ou acquise à ces traitements. Développer des inhibiteurs de réparation de l’ADN permettrait de contrecarrer cette résistance et de sensibiliser les tumeurs à ces thérapies conventionnelles. Cette problématique est devenue un enjeu majeur pour de nombreuses sociétés qui développent cette classe d’inhibiteurs. Malgré ces efforts, tous les inhibiteurs développés sont spécifiques d’une seule protéine ou d’une seule voie de réparation de l’ADN. Etant donné la redondance et la complémentarité de ces voies de réparation, inhiber une seule voie n’est pas suffisant pour tuer les cellules tumorales. Il est donc urgent de développer des agents plus efficaces qui permettraient de limiter cette problématique.
Le laboratoire du Dr. Marie Dutreix a développé une nouvelle classe d’inhibiteurs de réparation de l’ADN, les Dbait, consistant en une petite molécule d’ADN double-brin qui mime un dommage à l’ADN et agit ainsi comme un leurre qui perturbe plusieurs voies de réparation de l’ADN.
CONFIDENTIEL 6 Ces molécules « leurres » séquestrent plusieurs enzymes de réparation et perturbent la signalisation globale et la reconnaissance des dommages à l’ADN générés par les traitements génotoxiques. Les travaux précédents de l’équipe ont montré que les Dbait sensibilisent les tumeurs à la radiothérapie et la chimiothérapie.
Mes travaux de recherche au cours de cette thèse s’inscrivent dans la continuité de ces projets et portent sur deux questions complémentaires :
« MECANISMES DE SENSIBILITE/RESISTANCE DES CELLULES
TUMORALES AUX INHIBITEURS DE REPARATION DE L’ADN
DBAIT »
et
« EFFETS ET MECANISME D’ACTION DE LA COMBINAISON DES
DBAIT AVEC LES INHIBITEURS DE PARP »
CONFIDENTIEL 7
REMERCIEMENTS
Je tiens tout d’abord à remercier ma directrice de thèse, le Dr. Marie Dutreix, de m’avoir accueilli dans son laboratoire et offert la possibilité d’évoluer dans un très bon cadre de travail. Malgré certains moments et échanges difficiles, je sors de cette expérience plus mûr scientifiquement et humainement. Ses critiques scientifiques m’ont permis d’apprendre à me remettre en question, relativiser, réfléchir et trouver d’autres solutions, ce qui, à mon avis, représente des qualités essentielles pour devenir chercheur. Je la remercie de m’avoir fait confiance et laissé gérer de grands projets. Cette confiance et ce soutien ont été la clé de la réussite et de la concrétisation de ces projets. Je voudrais également la remercier de m’avoir « contraint » à un moment d’arrêter les expériences, afin de faire le bilan de mon travail. Il est toujours difficile pour un jeune chercheur de quitter sa paillasse, mais cela a été très utile pour approfondir ma formation doctorale et apprendre à rédiger des articles scientifiques. Je tiens à avouer que j’y ai pris goût.
Je souhaite remercier le Pr. Simon SAULE, directeur de l’unité « Signalisation Normale et Pathologique (UMR3347/U1021) : de l'embryon aux thérapies innovantes des cancers » à l’Institut Curie d’Orsay, de m’avoir accueilli au sein de son unité.
Je souhaite par ailleurs remercier mes rapporteurs, le Dr. Janet Hall et le Dr. Anne-Marie Mes-Masson d’avoir accepté de juger ce travail ; le Dr. Patricia Kannouche, le Dr. Marc-Henri Stern et le Dr. Bernard Lopez d’avoir accepté de faire partie du jury en temps qu’examinateurs, et les invités, le Dr. Graham Dixon et le Pr. Jian-Sheng SUN.
De plus, je souhaite remercier les membres de la société DNA Therapeutics, pour m’avoir apporté un support à la fois financier et scientifique. Sans le soutien de DNA Therapeutics, je n’aurai pu obtenir de bourse de financement et faire une thèse. Je remercie le Pr. Jian-Sheng SUN, ancien CEO de la société, pour sa confiance et son soutien. Un énorme merci à Nirmitha Herath (Nim pour les intimes), Flavien Devun, Aurélie Herbette, Céline Agrario (ou Misstress), Marjorie d’Agostino (Marjo) et Marie-Christine Lienafa (ou MC) pour vos conseils scientifiques, techniques et surtout votre grande humanité. J’en avais fortement besoin pour affronter cette aventure. Merci à Marjo pour tes cours de fitness, j’ai été ravi de t’apprendre en retour les secrets de la langue française! Merci à MC, à qui je dois toute mon expérience in vivo ! Tu m’as formé comme une chef. Merci MC pour les mesures des animaux pendant mon absence, ton sourire et ton réconfort dans les moments difficiles ! DNA Therapeutics est aujourd’hui une filiale de la société ONXEO. Je tiens à remercier mes nouveaux collaborateurs à Onxeo pour le parfait accueil qu’ils m’ont réservé. Je me suis senti rapidement intégré, utile et valorisé. Un grand merci en particulier à Graham Dixon, directeur R&D, Sandrine Demare, directrice analytique et laboratoire et Jean-Louis Labernardière, responsable laboratoire, pour leur soutien et leurs encouragements. Grâce à
CONFIDENTIEL 8 eux, je suis en train de m’imprégner de la « culture » de l’entreprise, qui me sera certainement très utile pour ma future carrière.
Toute cette aventure n’aurait pas été la même sans tous les membres de l’équipe Dutreix, ou les DUX (comme certains aiment nous appeler). Je les remercie infiniment pour leurs soutiens intellectuels, techniques et humains. Sans eux, je n’aurais pas eu le courage d’affronter certaines pistes qui me paraissaient bien compliquées. Merci à « mes DUX » pour les aventures autres que scientifiques que nous avons partagé ensemble. Merci à Nathalie Berthault, Maria Kozlak, Aurélie et Marie- Christine ; j’ai adoré notre sortie ski. C’était un joli weekend riche en émotions ! Merci à « maman » Nath pour les cours de ski qu’elle m’a donné et de m’avoir encouragé à affronter les pistes bleues. Merci Sofia Ferreira, ou « ma petite morue portugaise », pour ton sourire et ta bonne humeur. Je te rassure, tu apprendras le Français bien comme il faut si tu écoutes mes conseils Merci Maria Kozlak pour tes cours de Polonais, tes alcools un peu (beaucoup) forts, ton soutien et ta bonne humeur. Le cancer du « cyski » n’a plus aucun secret pour moi aujourd’hui. Merci à Wendy Philippon, la petite stagiaire en BTS qui est devenue une grande technicienne. Ton aide technique m’a été d’un grand support ! Merci à Sylvain Thierry, alias Sylvanus (non pas parce qu’il ressemble à un dieu Grec, mais pour une raison beaucoup moins glamour), pour tes conseils scientifiques, pour tous ces virus que l’on a produit ensemble (pardon, vecteurs lentiviraux ! ce n’est pas la même chose), et surtout pour ta grande humanité. Merci pour ton soutien pendant les moments difficiles. Je te souhaite bon courage pour la suite de ta carrière. Surtout n’oublie pas, garde tes valeurs et ton humanité, et préserve ta santé et ta famille avant tout. Tout le reste n’est pas aussi important… Merci aussi aux deux petits stagiaires que j’ai eu la chance de former au cours de cette thèse, Jordi Mano et Kim Cao, de m’avoir supporté. Vous m’avez appris beaucoup sur moi et j’espère vous en avoir appris beaucoup en retour. Continuez sur votre lancée, j’espère un jour venir vous voir à votre soutenance de thèse !
Malgré le développement de méthodes alternatives, l’utilisation de modèles animaux reste indispensable pour les thématiques de recherche de l’Institut Curie. Merci à la « plateforme d’expérimentation in vivo » de l’Institut Curie d’Orsay pour la qualité de leur travail. Le bien être des animaux a toujours été à son maximum. Merci au « Laboratoire d'investigation préclinique (Lip) » de l’Institut Curie Paris, pour leurs modèles de xénogreffes de cancer du sein. Je remercie par ailleurs la « plateforme d’histologie » de l’Institut Curie d’Orsay et plus particulièrement Sophie Dodier pour tout ce qu’elle m’a enseigné en histologie. J’ai pu, grâce à toi, acquérir de nouvelles compétences dans un domaine qui m’était jusque là inconnu. Je remercie aussi la « plateforme de Radiobiologie expérimentale » de l’Institut Curie d’Orsay : Frédérique Pouzoulet, Christophe Roulin, Sarah Bonnet-Boissinot, Dalila Labiod et Sophie Heinrich, pour leur aide et leur support scientifique. Pour terminer, je remercie Hamza Mameri et Roseline DELIC du laboratoire de Mounira AMOR-GUERET à l’Institut Curie d’Orsay pour tous vos conseils et aides concernant les lignées cellulaires de cancer du sein.
CONFIDENTIEL 9 Pour terminer, je souhaiterais remercier mes parents, sans qui je n’aurais pas été là où je suis aujourd’hui. Merci pour votre confiance, merci de m’avoir laissé partir faire ma vie où je le souhaitais, très loin de vous. Vous avez toujours été là pour me soutenir et m’encourager malgré la distance. Ressentir votre fierté a toujours été le plus grand moteur dans ma vie professionnelle et personnelle. J’espère vous rendre fiers de moi encore très longtemps. J’espère pouvoir aller vous voir plus souvent. Je resterai malgré vos craintes votre petit garçon, toujours très différent des autres, mais avec des valeurs, une sensibilité et une humanité que je tiens certainement de vous. Si vous voulez me remercier, BABA, OMMI, restez juste parmi nous encore très longtemps !! (et voilà, merci, vous avez réussi à me faire pleurer…).
On dit toujours que derrière un grand homme, il y a toujours une grande femme. Je ne sais pas si je suis un grand homme, mais je suis sûr d’une chose : derrière moi il y a une GRANDE (et très belle) femme. Ana (ma petite infirmière préférée)… il faudrait écrire une autre thèse rien que pour te remercier… Tu as toujours été là pour moi à mes côtés et tu as vécu avec moi les hauts et les bas d’une thèse. Voir ton sourire en rentrant d’une journée de travail très dure me faisait oublier toutes ces difficultés. Tes conseils, tes mots doux et ton grain de folie m’ont permis de supporter ces trois longues années chargées en émotions. « Je suis sûre que tu vas y arriver », « Ton travail est très bien », « Tiens bon, c’est ton avenir, le nôtre ! », « Tu peux te faire confiance, à toi, à ton travail et tes capacités», « Tu va relever le défi comme un pro », « Je suis très fière de toi » ; tous ces mots ont été les piliers de ma patience durant ces trois années. Tu as su me soutenir, et surtout me supporter (rien que pour ça, on devrait te couronner !). J’ai beaucoup appris avec toi lors de nos discussions scientifiques et médicales. J’espère que je t’ai aussi appris des choses, surtout en relisant cette thèse. Je ne te remercierais jamais assez d’ailleurs pour ces longues heures passées à la relire. Merci pour ces valeurs, ce grand cœur plein d’amour… tout ceci me rappelle tous les jours pourquoi je fais ce travail, et surtout pourquoi j’aime ce monde. MERCI !!!
J’espère n’avoir oublié personne … J’ai passé trois années de ma vie à apprendre sur moi et sur l’autre. Cette expérience m’a enrichi professionnellement et personnellement, et restera à jamais gravé dans ma mémoire.
CONFIDENTIEL 10
SOMMAIRE
PREFACE……….… 5
REMERCIEMENTS………...7
SOMMAIRE………....10
LISTE DES FIGURES, TABLES, ANNEXES………13
LISTE D’ABBREVIATIONS………15
CHAPITRE I – INTRODUCTION………...19
1. Les dommages à l’ADN………...21
1.1. Les dommages endogènes………..21
1.1.1. La réplication de l’ADN comme source de dommages ………....21
1.1.2. Le métabolisme cellulaire et les espèces réactives de l’oxygène (ROS)………...22
1.1.3. Les dommages récurrents de l’ADN………..23
1.2. Les dommages exogènes : l’environnement est hostile………..24
1.2.1. Les mutagènes alimentaires………..…..24
1.2.2. La consommation de cigarettes………..…24
1.2.3. Le soleil : ange ou démon ? Les rayonnements Ultra-Violets (UV)………..……25
1.3. Les dommages induits par les traitements anti-cancéreux………..25
1.3.1. Les agents alkylants………26
1.3.2. Les antimétabolites……….26
1.3.3. Les inhibiteurs des topoisomérases………26
1.3.4. Les radiations ionisantes………27
2. Signalisation des dommages à l’ADN……….……28
2.1. Les protéines kinases dans la signalisation des dommages à l’ADN……….28
2.1.1. La protéine ATM………...….30
2.1.2. La protéine ATR………...31
2.1.3. Interaction entre ATM et ATR………...31
2.1.4. La protéine kinase DNA-PK………..33
2.2. La protéine PARP dans la signalisation des dommages à l’ADN………..34
2.3. Modification de la structure chromatinienne en réponse aux dommages à l’ADN……35
2.3.1. La méthylation des histones………...36
CONFIDENTIEL 11
2.3.3. Acétylation et déacétylation des histones………...37
2.3.4. Le remodelage de la chromatine induit par la protéine PARP………...37
2.4. Défauts dans la signalisation des dommages à l’ADN et cancer………...40
3. Exploitation des voies de réparation de l’ADN en thérapeutique anti-tumorale 3.1. Les défauts intrinsèques dans les voies de réparation comme potentiel caractéristique pour stratifier les patients avant une chimiothérapie génotoxique………..42
3.1.1. Les défauts dans la voie NER………...42
3.1.2. La déficience dans la protéine MGMT………...…43
3.2. Les inhibiteurs des voies de réparation de l’ADN………..43
3.2.1. Association des inhibiteurs de réparation de l’ADN avec les traitements génotoxiques..43
3.2.1.1. Association avec les agents alkylants………...43
3.2.1.2. Association avec les sels de platine……….……….44
3.2.1.3. Association avec la radiothérapie……….45
3.2.1.4. Les inhibiteurs des points de contrôle du cycle cellulaire………...46
3.2.2. Utilisation des inhibiteurs de réparation de l’ADN en monothérapie………...48
3.2.2.1. Les inhibiteurs d’ATR en monothérapie………..49
3.2.2.2. Létalité synthétique avec les inhibiteurs de WEE1………..50
3.2.2.3. La déficience dans la voie de réparation par recombinaison homologue et les inhibiteurs de PARP……….50
4. Dbait : une nouvelle stratégie inhibant les voies de réparation de l’ADN……..56
4.1. Conception des molécules Dbait et leur mécanisme d’action………57
4.2. Applications………....59
4.2.1. Combinaison avec des traitements génotoxiques………...59
4.2.1.1. Combinaison avec la radiothérapie………...59
4.2.1.2. Combinaison avec la chimiothérapie………....62
5. Projet de thèse………..…65
CHAPITRE II – Résultats………...…67
1. Biomarqueurs prédictifs de la réponse au traitement par AsiDNA………69
1.1. Introduction ………..69
1.2. Article ...70
1.3. Conclusions et perspectives………....102
2. Effet de la combinaison AsiDNA et inhibiteurs de PARP ……….104
CONFIDENTIEL 12
2.2. Article ………...106
2.3. Conclusions et perspectives...…….140
3. Mécanismes impliqués dans la résistance à AsiDNA……….…….144
3.1. Introduction.…...144
3.2. Article en preparation………..145
3.3. Conclusions et perspectives………...163
CHAPITRE III – DISCUSSIONS ET PERSPECTIVES……….….165
ANNEXES……….….173
CONFIDENTIEL 13
LISTE DES FIGURES
Figure 1. Les agents génotoxiques, les dommages à l’ADN et les voies de réparation associées.
Figure 2. Détection et réparation des cassures double-brins.
Figure 3. Rôles des protéines ATM et ATR dans la signalisation des dommages à l’ADN.
Figure 4. La signalisation ɣH2AX induite par une CDB.
Figure 5. Rôle de PARP1 dans la signalisation et la réparation des CSBs. Figure 6. Régulation des points de contrôle par la DDR.
Figure 7. Mécanismes de résistance aux inhibiteurs de PARP.
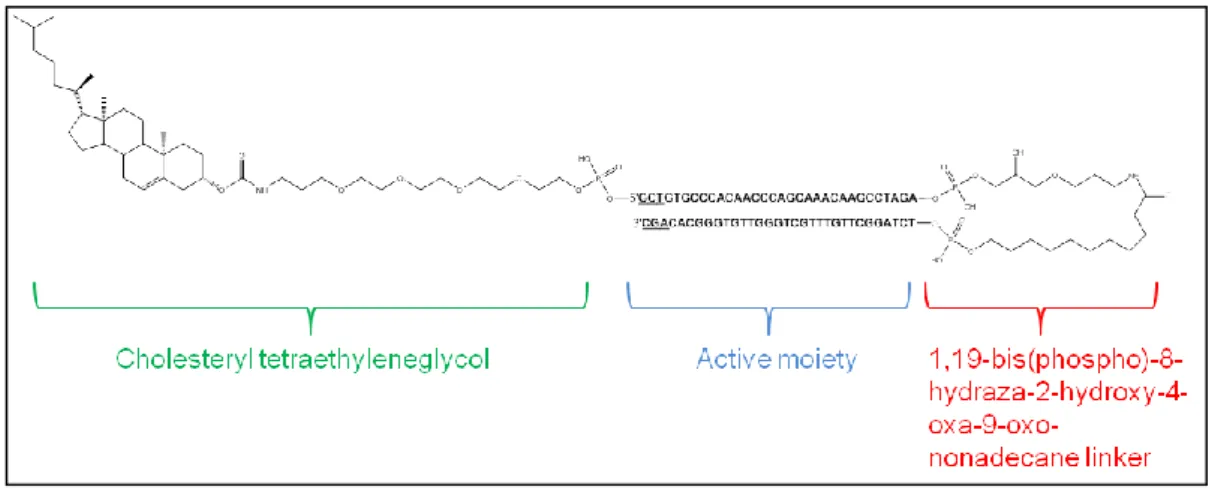
Figure 8. Représentation et formule d’AsiDNA, une molécule de la famille Dbait. Figure 9. Mécanisme d’action d’AsiDNA.
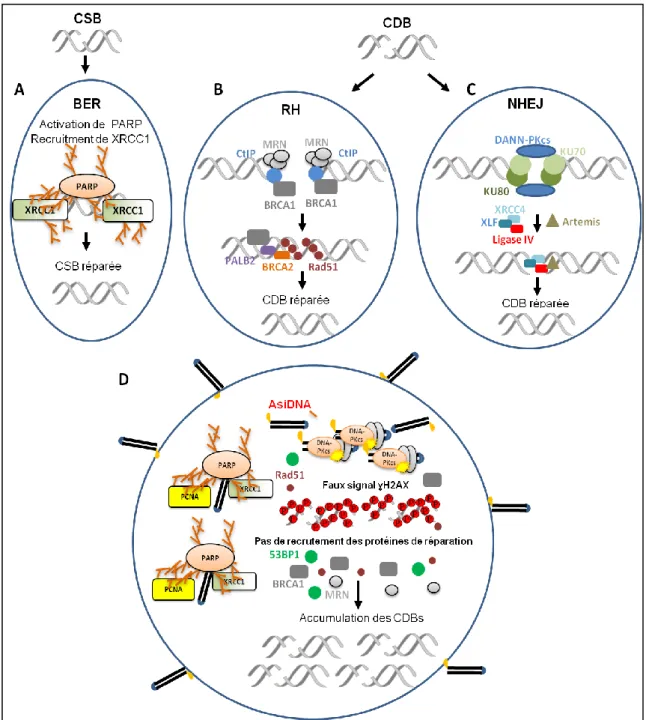
Figure 10. Effet du Dbait sur la réparation de l’ADN et la croissance tumorale.
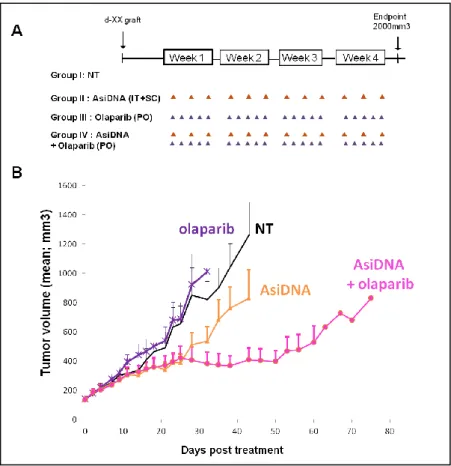
Figure 11. Réponse des patients de l’essai DRIIM à la combinaison AsiDNA + Radiothérapie. Figure 12. Effet de la combinaison d’AsiDNA et de l’olaparib sur la croissance tumorale.
Figure 13. Schéma du mécanisme de la létalité synthétique induite par l’olaparib et AsiDNA.
LISTE DES TABLES
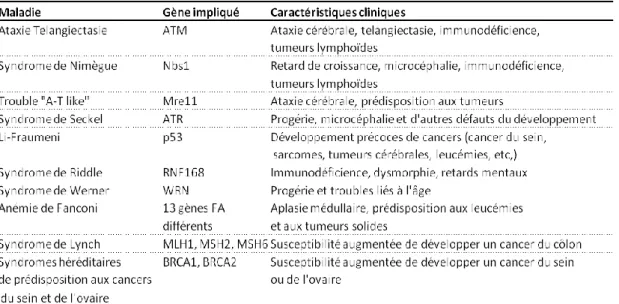
Table 1. Syndromes génétiques humains liés à la DDR. Table 2. Essais cliniques évaluant les inhibiteurs de la DDR.
LISTE DES ANNEXES
CONFIDENTIEL 15
LISTE D’ABREVIATIONS
53BP1 53-Binding Protein 1 5-FU 5-FluoroUracile 8-oxoG 8-Oxo Guanine
ADN Acide DésoxyriboNucléique ADP-ribose Adénosine Di Phosphate-Ribose AP site Site Abasique
APE1 AP Endonucléase 1
ATM Ataxia-Telangiectasia-Mutated ATP Adénosine TriPhosphate
ATR Ataxia-Telangiectasia-and-Rad3-Related ATRIP ATR-Interacting Protein
BER Réparation Par Excision De Base (Base Excision Repair)
BLM Bloom Syndrome Protein
B-NHEJ Voie De Réparation Alternative Du NHEJ (Back-Up NHEJ) BRCA1 BReast Cancer Associated Protein 1
BRCA2 BReast Cancer Associated Protein 2
CDB Cassure Double-brin
Cdc25 Phosphatase Du Cycle De Division Cellulaire 25
CSB Cassure Simple-Brin
DDR Réponse Au Dommage à L'ADN (DNA Damage Response) DNA-PK Protéine Kinase ADN Dépendant
DNA-PKcs Sous-unité Catalytique De La Protéine Kinase ADN Dépendant DSB Cassure Double-brin (Double Strand Break)
FEN-1 Flap Endonuclease 1
hOGG1 Human 8-Hydroxyguanine DNA-Glycosylase HR Réparation Par Recombinaison Homologue
IT Intratumoral
CONFIDENTIEL 16
Lig Ligase
MDC1 Mediator Of DNA Damage Checkpoint Protein 1 MGMT Methylated-DNA-Protein-Cysteine Methyltransferase MMR Réparation Des Mésappariements De Bases
MN Micronoyaux
MRN MRE11-Rad50-NBS1
MYH Glycosylase Homologue Muty NAD Nicotinamide Adénine Di nucléotide NBS1 Nijmegen Breakage Syndrome-1
NER Réparation Par Excision De Nucléotides
NHEJ Réparation Par Recombinaison Non Homologue
P Phosphate
PAR Poly (ADP-Ribose) ou PARylation PARG Poly (ADP-Ribose) Glycohydrolase PARP Polymérase Poly (ADP-Ribose) PCNA Proliferating Cell Nuclear Antigen PI3K Phosphatidylinositol-3 Kinases
PIKK Phosphatidylinositol 3-Kinase-Related Kinases
PO Per Os
Pol ADN Polymérase
RFC Replication Factor C
ROS Espèces Réactives De l'Oxygène RPA Protéine De Réplication A siDNA short interfering DNA
SSB Cassure Simple-brin (Single Strand Break)
SSBR Réparation des Cassure Simple-brin (Single Strand Break Repair) TDP1 Tyrosyl-DNA Phosphodiesterase 1
TGD Tumor Growth Delay
TMZ Témozolomide
CONFIDENTIEL 17
UV Ultra-Violet
XPC Xeroderma Pigmentosum Group C
XRCC1 X-Ray Repair Cross-Complementing Protein 1 XRCC4 X-Ray Repair Cross-Complementing Protein 4 γH2AX Phosphorylation de H2AX
CONFIDENTIEL 19
CHAPITRE I
CONFIDENTIEL 20 Nos cellules sont des entités sensibles qui se remettent continuellement en question après avoir commis des erreurs regrettables. Elles subissent également une énorme pression de leur entourage ainsi que des agressions externes impitoyables. Malgré tous ces défis endogènes et exogènes, ces cellules résistent et arrivent à gérer tous ces problèmes. Elles ont appris au cours de leur évolution des techniques performantes pour se maintenir en forme, éviter de vieillir, de mourir ou pire encore de se transformer en un « monstre » ingérable qu’on appelle CANCER. En effet, la cellule lutte pour maintenir l’intégrité de son génome, une caractéristique essentielle pour continuer de proliférer dans de bonnes conditions et garder son statut de cellule normale. Cette intégrité génomique est primordiale pour le bon fonctionnement de la cellule et à plus grande échelle, de l’organisme qu’elle constitue. De ce fait, en réponse aux agents endogènes et exogènes qui endommagent l’ADN, la cellule a développé un système délicat de détection des lésions sur son ADN, induisant l’arrêt de sa prolifération pour permettre la réparation de l’ADN endommagé, ou pousser la cellule à se « suicider » si les dommages sont très sévères. Ce jeu de détection des lésions, arrêt de cycle cellulaire et réparation est orchestré par différentes voies complémentaires ou redondantes, constituant une roue de secours pour la cellule si jamais une des voies est surpassée. Plusieurs protéines œuvrent à différents niveaux dans cette cascade pour assurer une prise en charge et une réparation efficace des dommages à l’ADN. Ceci commence de la détection de la lésion, sa signalisation pour recruter d’autres protéines, le ralentissement du cycle cellulaire, jusqu'à sa réparation. Etant donnée l’importance de ces voies dans l’homéostasie cellulaire, il est essentiel de les étudier et de caractériser leurs fonctions et leurs régulations.
CONFIDENTIEL 21
1. Les dommages à l’ADN
Les cellules sont compartimentées en deux parties : le noyau contenant l’ADN génomique et les protéines nucléaires jouant un rôle dans la stabilité de l’ADN et sa réparation, et le cytoplasme contenant toute la machinerie nécessaire pour le bon fonctionnement de la cellule, allant de la production de l’énergie cellulaire à la synthèse protéique et l’adressage de ces protéines aux différents compartiments cellulaires.
L’ADN, codant pour chaque protéine jouant un rôle essentiel dans cette homéostasie cellulaire, est soumis à des agressions multiples d’origine intracellulaire et extracellulaire. La protection de l’ADN, son entretien et sa réparation sont primordiaux pour assurer l’intégrité de la cellule et sa survie. Différentes voies de réparation de l’ADN, redondantes ou complémentaires, sont utilisées par la cellule pour réparer son ADN et éviter la perte d’informations génétiques ou l’accumulation de mutations qui pourraient conduire à sa mort ou à sa transformation maligne (Roos, Thomas, & Kaina, 2016).
1.1. Les dommages endogènes
1.1.1. La réplication de l’ADN comme source de dommages
La réplication de l’ADN, qui se produit au cours de la phase S du cycle cellulaire, est un processus essentiel pour permettre la duplication fidèle de l’information génétique avant son transfert aux cellules filles au cours de la mitose. Cette réplication chromosomique entraine des risques tels que la possibilité d’introduire des mutations dans le génome suite à des erreurs de réplication non réparées (Wallace, Southard, Schimenti, & Schimenti, 2014). Le stress réplicatif (SR), défini par un ralentissement ou un blocage de la progression de la fourche de réplication et/ou de la synthèse de l’ADN (Zeman & Cimprich, 2014), peut engendrer des dommages à l’ADN. Ce stress peut être causé par un déficit en nucléotides, des lésions à l’ADN, l’incorporation de ribonucléotides qui ne sont pas reconnu par la machinerie de réplication, des structures secondaires de l’ADN, des éléments répétés, ainsi que des complexes de transcription et/ou des hybrides ADN-ARN (Muñoz & Méndez, 2016). En effet, des erreurs de mésappariement ou de délétion de nucléotides peuvent se produire au cours du SR. L’ADN polymérase, dont la fonction est d’ajouter des nucléotides à l’extrémité du brin d’ADN en cours de réplication, devient
CONFIDENTIEL 22 moins fidèle en rencontrant les problèmes à l’origine du SR. Elle introduit ainsi des bases non complémentaires au brin-codant, omet de copier un certain nombre de bases induisant la délétion d’une séquence donnée, ou re-recopie des bases conduisant à l’introduction d’une nouvelle séquence, tout ceci malgré sa capacité de relecture lui permettant de s’assurer de l’exactitude de la séquence ajoutée. Il existe cependant une voie de réparation de l’ADN, la voie de réparation des mésappariements des bases, qui permet de réparer la plupart de ces erreurs de réplication (Kunkel & Erie, 2015) (Figure 1).
Figure 1. Les agents génotoxiques, les dommages à l’ADN et les voies de réparation associées. L’ADN peut être endommagé par différents agents endogènes et exogènes (par exemple, l’irradiation ionisante, les agents alkylants ou les radicaux d’oxygène)(A), ce qui induit différents types de dommages (B), l’activation d’une cascade de signalisation et de réparation de ces dommages, orchestrée par une panoplie d’enzymes de détection, de recrutement et de réparation (C).UV, Ultra-violet ; MMR, Mismatch repair ; HR, Homologous recombination ; SSBR, Single-strand break repair ; NHEJ, Non-homologous end joining ; BER, Base excision repair ; NER, Nucleotide excision repair. (adapté de Quanz et al, 2011).
1.1.2. Le métabolisme cellulaire et les espèces réactives de l’oxygène (ROS)
Le métabolisme cellulaire est une source majeure de radicaux libres et d’espèces réactives de l’oxygène qui engendrent des modifications importantes sur les bases des nucléotides et de multiples lésions (Møller & Loft, 2004). Selon une étude, plus de 5000 lésions sont générées par cycle cellulaire à cause des ROS (Vilenchik MM, Knudson AG, 2003). La respiration cellulaire, par exemple, entraine la libération de ces radicaux qui interagissent avec les riboses des nucléotides induisant une rupture du squelette ribose-phosphate de
CONFIDENTIEL 23 l’ADN, et l’accumulation de cassures simple-brins de l’ADN (Tanaka, Halicka, Huang, Traganos, & Darzynkiewicz, 2006). D’autres lésions peuvent également être induites par les ROS, comme les sites apuriniques/apyrimidiniques, les bases oxydées 8-oxoguanine et certaines alkylations des bases (Barzilai & Yamamoto, 2004). Approximativement 1% de ces lésions sont transformées en cassures double-brins (CDB), essentiellement au moment de la réplication cellulaire, ce qui constitue en moyenne 50 CDBs « endogènes » par cycle cellulaire (Vilenchik MM, Knudson AG, 2003). La quasi-totalité de ces conversions se produisent au cours de la phase S du cycle cellulaire. Par conséquent, les cellules qui ne se divisent pas produisent des CDBs endogènes à un taux beaucoup plus faible. Il est également important de souligner que le taux calculé de ces lésions est approximative et ne réflète pas la spécificité tissulaire. Les simples modifications des bases sont généralement réparées par la cellule par la O6-méthylguanine-DNA méthyltransférase (MGMT) et la voie de réparation par excision de base (BER).
1.1.3. Les dommages récurrents de l’ADN
Malgré l’état très compacté et protégé de l’ADN, il est sujet à des lésions spontanées comme les désaminations, les méthylations et les dépurinations des bases, engendrant des cassures et des mésappariements. La désamination est une hydrolyse pendant laquelle une base nucléique perd son groupement amine, conduisant à des modifications comme: cytosine en uracile, adénine en hypoxanthine, guanine en xanthine et 5methyl-cytosine en thymine. La dépurination, quant à elle, est la perte d’une base purique (guanine ou adénine). Il est connu que 5000 bases puriques sont perdues par cycle cellulaire (Nakamura & Swenberg, 1999). Ces défaut sont généralement pris en charge dans la cellule par la voie de réparation BER, mais peuvent parfois laisser des sites abasiques. En l’absence de réparation, ils peuvent conduire à des cassures simple-brin, ou une mutation lors de la réplication (Obeid et al., 2010) (Figure 1).
De façon contradictoire, certaines voies de réparation de l’ADN peuvent engendrer des dommages si elles ne sont pas conduites jusqu’à l’étape finale ou si elles rencontrent la machinerie de réplication ou de transcription. En effet, les voies de réparation par excision de bases (BER), la réparation des mésappariements de bases (MMR) et la réparation par excision de nucléotides (NER) génèrent des intermédiaires de réparation en forme de cassures simple-brin. Ces intermédiaires, s’ils ne sont pas réparés, peuvent se transformer en cassures double-brin lors de la réplication (Cox, 2013).
CONFIDENTIEL 24
1.2. Les dommages exogènes : l’environnement est hostile
1.2.1. Les mutagènes alimentaires
Les carcinogènes environnementaux peuvent être lié au régime et aux habitudes alimentaires et au mode de vie tels que l’activité physique. Plusieurs composés mutagènes ont été identifiés dans des aliments, et des études épidémiologiques ont confirmé l’implication de ces molécules dans la prévalence de certains cancers comme les cancers colorectaux, le cancer du sein et de la prostate (Wilson, Giovannucci, & Mucci, 2012). Les hydrocarbones aromatiques polycycliques (HAP), les amines hétérocycliques (AHC), les composés N-nitroso (CNO), les mycotoxines (aflatoxines) ainsi que l’acrylamide sont des mutagènes alimentaires très bien décrits et contribuant à la tumorigénèse. Les HAP par exemple induisent la formation des adduits intra-brins qui peuvent conduire à des mésappariements de bases (Raman et al., 2013) responsables des mutations activatrices de certains oncogènes ou la perte de suppresseurs de tumeurs. Etant donné que nous sommes tous continuellement exposés à ces mutagènes, une stratégie pourrait être de développer et de consommer des anti-mutagènes pour contrer ces composés carcinogènes. Des molécules naturelles présentes dans les fruits et les légumes, ainsi que des probiotiques sont connus pour leur fonction anti-mutagénique (Słoczyńska, Powroźnik, Pękala, & Waszkielewicz, 2014).
1.2.2. La consommation de cigarettes
Il est aujourd’hui reconnu qu’une sous-population de fumeurs développe un cancer (11-20% développent un cancer du poumon, par exemple), suggérant qu’il existe une variabilité interindividuelle en réponse aux carcinogènes du tabac. Une partie des composants du tabac induisent des adduits à l’ADN (essentiellement les aldéhydes et les CNO) ainsi que l’oxydation des bases (conduisant à des 8-oxoG par exemple). Ces altérations sont normalement réparées par les voies NER ou BER. Des études épidémiologiques ont montré un lien entre un risque élevé des cancers du poumon ou tête et cou et les niveaux des protéines impliquées dans ces voies de réparation de l’ADN chez les patients (Povey, Margison, & Santibáñez-Koref, 2007). Par exemple, une perte du locus du gène OGG1, une glycosylase responsable de l’élimination des 8-oxoguanines, est un évènement fréquent chez les patients avec un cancer du poumon (Wikman et al., 2000). Un lien causal a également été établi entre le niveau de la protéine
O(6)-Alkylguanine-CONFIDENTIEL 25 DNA alkyltransferase (MGMT), impliquée dans la réparation des adduits, et le risque du cancer du poumon (Povey et al., 2007).
1.2.3. Le soleil : ange ou démon ? Les rayonnements Ultra-Violets (UV)
Les rayonnements UV solaires sont un facteur environnemental important avec une variété d’effets physiologiques comme l’inflammation, le vieillissement cellulaire photo-induit et les dommages à l’ADN qui peuvent induire une carcinogénèse de la peau. L’exposition aux UV-A conduit à des modifications oxidatives de l’ADN telles que la formation de 8-hydroxyguanine, des sites abasiques, des cassures simple et double-brins et des adduits ADN-protéines. Les UV-B et -C quant à eux endommagent directement l’ADN en causant la formation de dimères de pyrimidines adjacents (de Gruijl, 2000). Ces dimères bloquent la machinerie de transcription ou la fourche de réplications de l’ADN et conduisent à des erreurs de réplication ou la formation de cassures. Les rayonnements UVs augmentent également la production des ROS qui créent des dommages à l’ADN, et activent certaines voies de transduction responsables de l’arrêt du cycle cellulaire, la sénescence ou la mort cellulaire (López-Camarillo et al., 2012).
1.3. Les dommages induits par les traitements anti-cancéreux
Le cancer est une maladie complexe et multifactorielle caractérisée par de nombreuses altérations. Parmi celles-ci on peut citer une prolifération accrue et une résistance à la mort cellulaire. La plupart des cellules cancéreuses se divisent plus souvent que les cellules normales, rendant ce processus de division cellulaire une cible thérapeutique pour traiter le cancer. L’objectif principal en ciblant la prolifération est d’arrêter le cycle cellulaire et/ou d’induire la mort des cellules cancéreuses en utilisant des molécules cytotoxiques (chimiothérapies) ou les irradiations ionisantes (radiothérapie). Etant donné que la réplication de l’ADN est une étape primordiale au cours de la division cellulaire, la principale cible de ces traitements est l’ADN. En effet, ces traitements causent des dommages à l’ADN, induisant l’activation de points de contrôle (checkpoints) du cycle cellulaire, l’arrêt du cycle et/ou la mort des cellules (Helleday, Petermann, Lundin, Hodgson, & Sharma, 2008) (Figure 1).
CONFIDENTIEL 26 1.3.1. Les agents alkylants
Les agents alkylants constituent une classe importante de chimiothérapies de première ligne qui infligent des dommages à l'ADN. Ces composés électrophiles transfèrent de façon covalente un groupe alkyle au niveau des bases de l’ADN (Helleday et al., 2008). Il existe différents groupes d’agents alkylants: les dérivés de moutarde azotés, les nitrosourés, les triazines et les sels de platine. Les dérivés de moutarde azotées sont des agents alkylants bi-fonctionnels dont les plus utilisés sont l’ifosfamide, le cyclophosfamide et le melphalan, qui forment des liaisons inter-brins guanine-guanine ou guanine-adénine. La mitomycine C et les sels de platine ont une action analogue à celle des moutardes azotés. Les triazines, comme la dacarbazine et le témozolomide, sont des agents alkylants mono- fonctionnels qui ajoutent un groupement méthyle sur les guanines, induisant ainsi des dommages de bases et des adduits (Fu, Calvo, & Samson, 2012). Tous ces adduits bloquent la machinerie de réplication et conduisent à des cassures de l’ADN.
1.3.2. Les antimétabolites
Les antimétabolites ont une structure similaire à celle des nucléotides et agissent soit en inhibant le processus de synthèse des nucléotides, soit en s’incorporant au niveau de l’ADN ou l’ARN en cours de synthèse (Longley, Harkin, & Johnston, 2003). L’inhibition des voies du métabolisme des désoxynucléotides triphosphate (dNTP) déplètent les cellules en nucléotides et inhibent ainsi la réplication de l’ADN. L’incorporation des antimétabolites dans l’ADN bloque l’avancement de la fourche de réplication. Le 5-Fluorouracil (5-FU) possède à la fois la capacité de s’incorporer dans l’ADN ou l’ARN en cours de synthèse à la place de la thymine ou l’uracile, et inhibe également la thymidilate synthase, enzyme responsable de la synthèse de deoxythimidine monophosphate. Il est largement utilisé pour le traitement du cancer colorectal. Le méthotrexate est un antimétabolite qui inhibe la dihydrofolate réductase nécessaire à la synthèse des purines et de la thymidine.
1.3.3. Les inhibiteurs des topoisomérases
Les topoisomérases sont des enzymes qui relaxent l’ADN en cours de réplication ou de transcription, en induisant des cassures transitoires pour diminuer la tension induite par la torsion de l’ADN. Il existe deux types de topoisomérases : les TOP1 qui coupent un seul brin de l’ADN et font passer l’autre brin à travers pour diminuer la tension, et les TOP2
CONFIDENTIEL 27 qui coupent les deux brins et détendent ainsi la structure de l’ADN (Pommier, 2006). Des inhibiteurs ciblant ces enzymes ou le complexe de clivage enzyme-ADN ont été développés et agissent en bloquant les topoisomérases au niveau du complexe de clivage, inhibant ainsi la ligature des cassures. Les inhibiteurs de TOP1 créent ainsi des cassures simple-brins qui se transforment en cassures double-brins au moment du passage de la fourche de réplication, alors que les inhibiteurs de TOP2 créent directement des cassures double-brins. La camptothécine et ses dérivés (le topotécan et l’irinotécan) constituent des inhibiteurs de TOP1 et sont largement utilisés pour le traitement du cancer colorectal, ovarien et du poumon (Pommier, 2006). L’étoposide est un inhibiteur de TOP2 utilisé dans le traitement de plusieurs cancers comme le cancer du poumon, des testicules ou le cancer de l’ovaire (Nitiss, 2009). La doxorubicine, en plus de sa capacité à s’intercaler dans l’ADN, est également un inhibiteur de TOP2 utilisé dans le traitement de certains lymphomes et leucémies, ainsi que les sarcomes osseux.
1.3.4. Les radiations ionisantes
L’irradiation ionisante est un agent physique qui crée des dommages à l’ADN. Il est estimé que 50% des patients ayant un cancer reçoivent une radiothérapie à un certain moment au cours de leur traitement (Baskar, Lee, Yeo, & Yeoh, 2012). Les rayonnements ionisants ont à la fois une action directe et indirecte sur l’ADN. Ils peuvent induire de façon directe des cassures au niveau de l’ADN en ionisant ou excitant certains de ses atomes (Jackson & Bartek, 2009). L’effet indirect est causé par les ROS produites au cours de l’irradiation (Hoeijmakers, 2001). Ces ROS sont responsables de plusieurs altérations comme l’oxydation des bases, la formation d’adduits et des sites abasiques ainsi que les cassures simple et double-brins (Jackson & Bartek, 2009). Il est connu que l’irradiation induit environ 1000 cassures simple-brins et 40 cassures double-brins par Gray (Friedberg, 1995). Les rayonnements ionisants induisent essentiellement une mort cellulaire par catastrophe mitotique (Jonathan, Bernhard, & McKenna, 1999).
CONFIDENTIEL 28
2. Signalisation des dommages à l’ADN
Le maintien de l’intégrité du génome est essentiel pour prévenir l’instabilité génétique responsable de plusieurs anomalies incluant le cancer, des défauts de développement et des désordres neuro-dégénératifs et d’immunodéficience (Lord & Ashworth, 2012). Comme discuté dans le chapitre précédent, l’ADN cellulaire est constamment soumis à des agressions endogènes et environnementales tels que les ROS et les UVs. Pour protéger l’intégrité génomique après un dommage à l’ADN, la cellule a développé une machinerie complexe et très bien coordonnée pour détecter et contrecarrer ces altérations, appelée la réponse aux dommages à l’ADN (DDR, pour « DNA Damage Response »). La DDR peut être divisée en plusieurs voies distinctes mais interconnectées, qui sont définies par le type de la lésion à réparer. En effet, les cassures double-brins (DSBs) sont réparées soit par la voie « fidèle » de recombinaison homologue (HR, pour Homologous Recombination) ou par la voie « non fidèle » par jonction d’extrémités non homologues (NHEJ, pour Non Homologous End Joining). Les lésions induites par les UVs sont réparées par la voie d’excision de nucléotides (NER, pour Nucleotide Excision Repair). Les lésions localisées au niveau des bases sont réparées par la voie d’excision de bases (BER, pour Base Excision Repair) et les mésappariements des bases sont réparés par la voie MMR (MisMatch Repair). La voie Fanconi Anemia (FA) prend en charge les adduits inter-brins à l’ADN comme ceux induits par les sels de platine par exemple (Pearl, Schierz, Ward, Al-Lazikani, & Pearl, 2015). Toutes ces voies de DDR englobent une série similaire de processus très bien coordonnés : la détection de la lésion, le recrutement et l’accumulation des protéines de réparation au site du dommage et enfin la réparation de la lésion. Toutes les protéines dans ces voies de signalisation peuvent être considérées comme des protéines détectrices du dommage, transductrices du signal ou effectrices, et communiquent ensemble dans une hiérarchie très bien organisée.
2.1. Les protéines kinases dans la signalisation des dommages à l’ADN
Afin que la cellule réponde de façon efficace aux dommages à l’ADN, ce dommage doit d’abord être détecté. Le complexe MRE11-Rad50-NBS1 (MRN) et les protéines KU70/KU80 sont des protéines détectrices des cassures double-brins, qui recrutent à leur tour les protéines transductrices. Les kinases reliées aux phosphatidylinositol 3-kinasesCONFIDENTIEL 29 (PIKKs), incluant les protéines ATM (pour Ataxia Telangiectasia Mutated), ATR (pour ATM and Rad3 related) et DNA-PKcs (la sous unité catalytique du complexe DNA-PK, pour DNA-dependant Protein Kinase), sont des protéines transductrices agissant précocement dans la cascade de signalisation des dommages à l’ADN (McKinnon, 2012)(Cimprich & Cortez, 2008) (Figure 2). Ces kinases transmettent le signal aux protéines effectrices qui, en agissant sur leurs cibles, induisent l’arrêt du cycle cellulaire pour permettre la réparation de la lésion, ou la mort cellulaire si les dommages sont très importants ou non réparables.
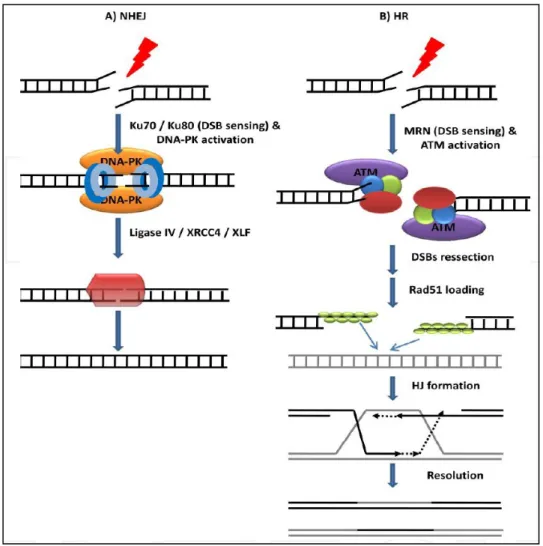
Figure 2. Détection et réparation des cassures brins. (A) NHEJ. Les cassures double-brins (CDBs) sont détectées par l’hétérodimère Ku70/Ku80 qui stabilise les deux extrémités libres de l’ADN endommagé et recrute la sous-unité catalytique DNA-PKcs (pour former le complexe DNA-PK). La DNA-PK s’auto-phosphoryle et s’active et active les effecteurs de la voie NHEJ (Ligase IV, XRC44, XLF) qui permettent de réparer la CDB. (B) HR. La kinase ATM est recrutée au niveau du site du dommage via son interaction avec le complexe MRN, permettant son activation, nécessaire pour la phosphorylation de multiples substrats. Dans une réaction qui
CONFIDENTIEL 30 dépend de multiples endo- et exo-nucléases (incluant Mre11, Exo1 et CtIP), les extrémités de la CDB sont réséquées formant ainsi des extrémités simple-brins, qui fixent Rad51 et d’autres protéines. Les brins recouverts de Rad51 envahissent la chromatide sœur non endommagée, permettant aux polymérases de recopier de façon fidèle les brins de cette chromatide. (Chapitre 8 « signaling DNA damage », Lopez-Contreras et Fernandez-Capetillo, 2012. http://dx.doi.org/10.5772/50863).
2.1.1. La protéine ATM
Les cassures double-brins (CDBs) sont réparées dans les cellules de mammifères par la voie de jonction d'extrémités NHEJ et par la voie de recombinaison homologue HR qui prédominent à différents stades du cycle cellulaire. Le complexe Mre11-Rad50-Nbs1 (MRN) est le premier senseur des CDBs, et il est prouvé qu'elle participe à ces deux voies de réparation (Xie, Kwok, & Scully, 2009)(Rass et al., 2009). Des mutations dans les membres de ce complexe donnent lieu à des troubles caractérisés par une sensibilité aux agents qui génèrent les CDBs, des défauts dans les points de contrôle du cycle cellulaire, une instabilité génétique et des anomalies neurologiques (Waltes et al., 2009). Ces syndromes se chevauchent dans leurs phénotypes cliniques et cellulaires avec l’ataxie télangiectasie (AT), une anomalie caractérisée par un défaut de la kinase ATM. Cette kinase, membre de la famille des PIKKs, est activée par les CDBs et agit directement sur la machinerie de réparation de l’ADN ou les points de contrôle du cycle cellulaire (Lavin, 2008). ATM est constitutivement présente sous forme de dimères qui se dissocient permettant l’activation d’ATM par auto-phosphorylation et acétylation. Une activation complète d’ATM nécessite à la fois la présence d’une CDB et une interaction avec le complexe MRN (Dupré, Boyer-Chatenet, & Gautier, 2006). Une fois activée, ATM peut alors phosphoryler une multitude de substrats qui participent à divers processus cellulaires, y compris la réparation de l'ADN et le contrôle du cycle cellulaire. Une cascade de réactions est donc initiée, débutant par la phosphorylation de l’histone H2AX (pour donner la forme phosphorylée ɣH2AX), induisant une modification de la structure chromatinienne qui joue un rôle important dans la signalisation et la réparation des dommages à l’ADN (van Attikum & Gasser, 2009). En effet, la phosphorylation de H2AX est essentielle pour le recrutement des protéines impliquées dans la DDR. ATM phosphoryle ainsi et active certaines de ces protéines, comme MDC1, 53BP1 et BRCA1.
CONFIDENTIEL 31 Tous ces processus régulent la réparation de la CDB et les points de contrôle du cycle, permettant d’éviter l’instabilité génétique (Figure 3-b).
2.1.2. La protéine ATR
Comme ATM, ATR est une protéine centrale dans la signalisation des dommages à l’ADN. Si ATM est essentiellement impliquée dans la signalisation des CDBs, ATR est activée par des structures d’ADN simple-brins, qui peuvent survenir suite à différents types de dommages, comme les CDBs, les adduits de bases, les ponts inter-brins, les cassures simple-brin (CSBs) ou les fourches de réplication bloquées. Le recrutement d’ATR au niveau de ces sites de dommages ou de stress réplicatif est médié par l’interaction directe de la protéine ATRIP avec la protéine RPA qui couvre ces structures d’ADN simple-brins (Zou & Elledge, 2003). Une fois activée, ATR agit à travers ses cibles pour promouvoir la réparation de l’ADN, l’arrêt du cycle cellulaire ou la stabilisation de la fourche de réplication bloquée. Ces fonctions sont assurées par la cible directe d’ATR, CHK1, qui joue un rôle important dans l’activation du point de contrôle de la phase S du cycle cellulaire en réponse à un dommage à l’ADN. CHK1 inhibe l’activation de nouvelles origines de réplication en induisant la dégradation de Cdc25A, permettant le ralentissement de la réplication de l’ADN et fournissant le temps nécessaire pour résoudre la source du stress (Sørensen et al., 2003). ATR joue également un rôle important dans le point de contrôle G2/M pour éviter une entrée précoce de la cellule en mitose avant que la réplication de l’ADN ne soit terminée ou en présence de dommages (Xiao et al., 2003)(Graves, Lovly, Uy, & Piwnica-Worms, 2001) (Figure 3-a).
2.1.3. Interaction entre ATM et ATR
Les kinases ATM et ATR sont activées par différents types de lésions, et agissent dans des voies distinctes. Néanmoins, elles ont en commun une partie de leurs cibles comme l’histone H2AX et p53. ATM et ATR coopèrent à travers ces cibles communes pour induire les réponses cellulaires adéquates après les différents stresses génotoxiques en coordonnant la progression du cycle cellulaire et la réparation de l’ADN. En effet, des études ont montré que ATM pourrait contribuer à l’activation des checkpoints en phase S et G2/M du cycle cellulaire, suggérant ainsi un chevauchement entre la signalisation ATM
CONFIDENTIEL 32 et ATR dans le contrôle du cycle (Abraham, 2001). Ces deux kinases coopèrent également pour assurer le maintien de la stabilité des télomères en évitant l’entrée des cellules avec des télomères non protégés en mitose, en induisant la dégradation de la phosphatase Cdc25C (Thanasoula, Escandell, Suwaki, & Tarsounas, 2012). Il existe également une interaction entre ATM et ATR dans la protection et le maintien de la stabilité des sites fragiles (Ozeri-Galai, Schwartz, Rahat, & Kerem, 2008). Ce chevauchement partiel entre la signalisation par ATM et ATR, même si les voies en aval ne sont pas redondantes, permet une compensation en cas de défaut dans l’une des deux voies (Figure 3-c).
Une déficience en ATM ou ATR est souvent associée à une hypersensibilité aux agents génotoxiques, due à une dérégulation des points de contrôle du cycle cellulaire. Ce phénotype a sollicité l’intérêt de développer des inhibiteurs d’ATM ou d’ATR pour sensibiliser les cellules tumorales à la chimio- ou radiothérapie, ou en monothérapie dans des cellules particulièrement dépendantes de certaines voies de réparation.
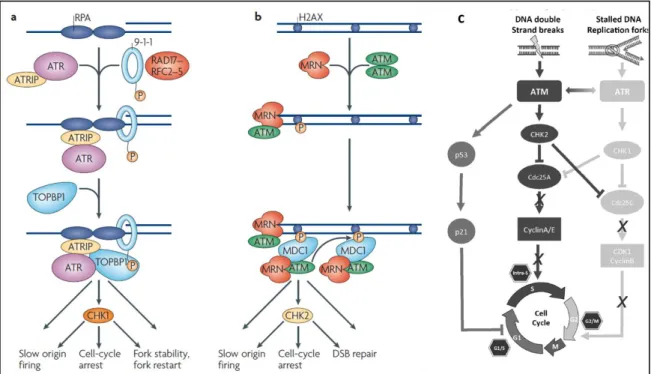
Figure 3. Rôles des protéines ATM et ATR dans la signalisation des dommages à l’ADN. (a) les deux complexes ATR-ATRIP et 9-1-1 sont recrutés indépendamment au niveau de la structure simple-brin en se liant à la protéine RPA. La protéine TOPBP1 est ensuite recrutée et active ATR, qui phosphoryle et active à son tour CHK1 et ses autres cibles effectrices. Cette cascade conduit au ralentissement des origines de réplication, à l’arrêt du cycle cellulaire et la stabilisation des fourches de réplication. (b) La formation d’une cassure double-brins conduit au recrutement du
CONFIDENTIEL 33 complexe MRN et l’activation de la protéine ATM. La protéine ATM activée phosphoryle l’histone H2AX (ɣH2AX) qui fixe et recrute MDC1, conduisant au recrutement de complexes MRN-ATM supplémentaires et l’amplification du signal ɣH2AX. La protéine ATM activée permet également l’activation d’autres cibles comme CHK2. Toute cette signalisation induit l’arrêt du cycle cellulaire et la réparation de la CDB. (c) Il existe des chevauchements entre les cascades de signalisation initiées par ATM ou ATR. Notamment, les deux enzymes CHK1 et CHK2 peuvent phosphoryler Cdc25A et Cdc25C, en fonction de la source du stress génotoxique. (adapté de (Weber et al., 2015) et (Cimprich & Cortez, 2008)).
2.1.4. La protéine kinase DNA-PK
La voie NHEJ (pour « Non-Homologous End Joining ») est une voie essentielle dans la réparation des CDBs, connue pour prendre en charge près de 80% des CDBs dans les cellules en prolifération (Mao, Bozzella, Seluanov, & Gorbunova, 2008a). Cette voie peut opérer pendant toutes les phases du cycle cellulaire, mais rentre en compétition avec la voie de réparation par recombinaison homologue pendant la phase S et G2 du cycle, quand la chromatide sœur est disponible (Mao, Bozzella, Seluanov, & Gorbunova, 2008b). La voie NHEJ comprend 3 étapes : une étape de détection de la CDB, une étape de « processing » des extrémités de la cassure et une étape de ligature. En effet, la CDB est reconnue par le complexe KU70/KU80, qui recrute à son tour la kinase DNA-PKcs, formant ainsi le complexe DNA-PK, une sérine/thréonine kinase active composée de l’hétérodimère KU70/KU80 et de la sous-unité catalytique DNA-PKcs (Figure 2). Les premières indications sur l’implication de DNA-PK dans la réparation des CDBs viennent du fait de l’hypersensibilité des cellules déficientes en DNA-PKcs ou KU à l’irradiation ionisante ou à d’autres traitements induisant des CDBs (Lees-Miller et al., 1995)(Taccioli et al., 1994). L’immunodéficience induite par une déficience en DNA-PKcs ou KU indique également l’implication de DNA-PK dans la réparation des CDBs « programmées » durant la recombinaison V(D)J, un processus essentiel qui permet de créer une grande diversité de récepteurs des cellules T et d’immunoglobulines qui sont nécessaires à la reconnaissance de l’immense variété des antigènes étrangers (Radhakrishnan, Jette, & Lees-Miller, 2014). En réponse aux CDBs, DNA-PK s’autophosphoryle et phosphoryle plusieurs protéines comme l’histone H2AX, signalant la CDB. Une expression élevée de DNA-PK accélère la réparation des CDBs induites par l’irradiation ou par des agents génotoxiques comme l’étoposide ou la doxorubicine, et
CONFIDENTIEL 34 confère ainsi une résistance à ces agents (Hansen et al., 2003). Des études cliniques rétrospectives ont également montré une corrélation entre le niveau d’expression de DNA-PK et la réponse des patients avec une leucémie lymphoblastique chronique à l’étoposide (Eriksson, Lewensoh, Larsson, & Nilsson, 2002). L’inhibition de DNA-PK semble ainsi être une voie intéressante pour sensibiliser les tumeurs aux agents thérapeutiques induisant des CDBs. Des inhibiteurs spécifiques de DNA-PK, comme le NU7026 ou le NU7441, retardent la réparation des CDBs et montrent une radio-sensibilisation et une augmentation de la cytotoxicité des inhibiteurs des TOP2 (Willmore et al., 2004).
2.2. La protéine PARP1 dans la signalisation des dommages à l’ADN
La diversité des types de dommages à l’ADN nécessite de multiples mécanismes de réparation. Malgré ces différentes réponses, la plupart se produisent à travers l’activation de voies de signalisation impliquant différentes modification post-traductionnelles comme la phosphorylation, l’ubiquitination, l’acétylation et Poly(ADP-Ribosy)lation (ou PARylation). Comme les kinases PIKKs (ATM, ATR et DNA-PK), la protéine Poly(Adénosine diphosphate (ADP)-Ribose) Polymérase (PARP) est un acteur majeur dans l’initiation des ces cascades de réponse aux lésions à l’ADN. Même si elles sont moins dramatiques que les CDBs, les CSBs sont également toxiques. Ces lésions sont induites par l’action oxidative des ROS, les inhibiteurs de TOP1 ainsi que l’irradiation ionisante (il est admis que l’irradiation induit 25 fois plus de CSBs que de CDBs). Les CSBs sont détectées par la protéine PARP1, qui s’active en utilisant des NAD+ comme source d’ADP ribose pour synthétiser des homopolymères de Poly-ADP ribosyles (PAR) sur elle-même ainsi que sur d’autres protéines cibles (Panzeter & Althaus, 1994). L’enzyme poly(ADP-ribose) glycohydrolase (PARG) est responsable de la dégradation des ces polymères de PAR et agit de concert avec PARP pour assurer une présence transitoire de concentrations élevées de PAR dans la cellule (Braun, Panzeter, Collinge, & Althaus, 1994). Une inhibition ou une déficience en PARP1 ou PARP2, ainsi qu’une sous expression de PARG est associée à une réparation altérée de l’ADN, une hypersensibilité aux agents génotoxiques et une instabilité génétique élevée (Yang, Cortes, Patnaik, Jasin, & Wang, 2004)(Cortes et al., 2004). En effet, PARP1/2 participe dans la voie de réparation par excision de base (BER) ainsi que dans la signalisation des dommages (Schreiber et al., 2002)(Bouchard, Rouleau, & Poirier, 2003). L’auto-modification deCONFIDENTIEL 35 PARP1/2 permet le recrutement de tous les acteurs de la voie BER comme première réponse au dommage, ainsi que les effecteurs en aval responsables de l’arrêt du cycle ou de la mort cellulaire. En effet, en plus de son rôle dans la réparation de l’ADN, PARP interagit avec et régule les protéines des points de contrôle du cycle. La protéine p53 par exemple a une grande affinité pour la PARP auto-modifiée, qui permet son activation induisant ainsi l’arrêt du cycle (Lane & Hupp, 2003). De plus, PARP1 altère localement l’architecture chromatinienne, permettant ainsi de faciliter l’accès des protéines de réparation sur le site du dommage (Althaus, 2005).
2.3.
Modification de la structure chromatinienne en réponse aux
dommages à l’ADN
Après une CDB, le complexe MRN se fixe sur les extrémités de l’ADN endommagé, et recrute les PIKKs kinases ATM ou ATR, initiant une cascade de réponse au dommage (encore appelée DDR). Alternativement, c’est l’hétérodimère Ku70/Ku80 qui se fixe sur ces extrémités et recrute la kinase DNA-PKcs. Ces kinases phosphorylent H2AX au niveau du site du dommage, pour donner la forme phosphorylée ɣH2AX, initiant ainsi la réponse de la chromatine au dommage à l’ADN (Ataian & Krebs, 2006). ɣH2AX est la modification chromatinienne la plus documentée en réponse à une CDB. Cette modification est une étape précoce qui initie une série de remodelage de la chromatine nécessaire au recrutement d’autres protéines impliquées dans la modification de la chromatine ou la réparation des dommages. En plus de ɣH2AX, d’autres modifications d’histones sont requises pour une réparation efficace de la lésion, comme l’acétylation, la méthylation et l’ubiquitination (Figure 4).
CONFIDENTIEL 36 Figure 4. La signalisation ɣH2AX induite par une CDB. H2AX est phosphorylée par les membres de la famille PI3K (ATM, ATR et DNA-PK). La protéine impliquée dépend de l’origine du stress génotoxique. Les foci ɣH2AX sont impliquées dans le recrutement des protéines de la DDR comme le complexe MRN, MDC1, 53BP1, BRCA1 et ATM. Ils sont également nécessaires pour le remodelage de la chromatine en recrutant les complexes de remodelage, ainsi que pour la transduction des signaux de la DDR (amplification de l’activation d’ATM, point de contrôle du cycle G2/M). De plus, à travers le recrutement des cohésines et du complexe MRN, les foci ɣH2AX sont impliqués dans la liaison et l’attachement des extrémités de la CDB, empêchant ainsi la dissociation de ces extrémités. (Redon et al., 2010).
2.3.1. La méthylation des histones
Un défaut dans la méthylation de l’histone H3-K79 ou H3-K36 est associé à une hypersensibilité à l’irradiation (Game & Chernikova, 2009), suggérant un rôle important de ces modifications dans la réparation de l’ADN. La méthylation H3-K79 et la deméthylation H4-K20 sont reconnues par la protéine de réparation des CDBs 53BP1 au niveau de la chromatine relaxée. D’autre part, la méthylation H3-K9me3 stimule l’activité de l’histone acetyl-transférase (HAT) TIP60 au niveau du dommage, résultant en une acétylation des histones et de la protéine ATM ce qui l’active et permet une amplification du signal ɣH2AX (Xu & Price, 2011).
2.3.2. L’ubiquitination des histones
L’ubiquitination des histones joue un rôle important à différentes étapes de la réparation de l’ADN. Par exemple, l’ubiquitination de H2AX-K119 est induite après l’irradiation
CONFIDENTIEL 37 (Ikura et al., 2007). Il a également été montré que l’ubiquitibation de H2AX par RNF8 et RNF168 est essentielle pour le recrutement et la rétention des protéines de réparation BRCA1 et 53BP1 au niveau de la CDB (Doil et al., 2009). L’ubiquitination des histones H3 et H4 facilite également le recrutement des protéines de réparation au niveau des CDBs (Moyal et al., 2011).
2.3.3. Acétylation et déacétylation des histones
Plusieurs études ont montré que l’acétylation des histones par les histones acétyl-transférases (HATs) au niveau des CDBs facilite le remodelage de la chromatine vers un état moins compacté ou « ouvert », permettant ainsi l’accès des protéines de réparation au niveau de la lésion. Une acétylation des histones H3 et H4 par les HATs CBP et p300 facilite le recrutement des hétérodimères Ku70/Ku80 (H Ogiwara et al., 2011). CBP et p300 joue également un rôle dans la voie de réparation par recombinaison homologue, en régulant la transcription des gènes BRCA1 et Rad51 (Hideaki Ogiwara & Kohno, 2012). Etant donné que les modifications des histones sont requises pour décompacter la chromatine et faciliter la réparation de l’ADN, d’autres modifications sont nécessaires pour rétablir la conformation initiale de la chromatine une fois que la réparation est achevée. Les histones déacétylases (HDACs) sont recrutées pour reverser l’acétylation des histones et restaurer la structure de la chromatine. Cependant, ces HDACs pourraient également jouer un rôle plus direct dans le processus de réparation de l’ADN, car elles ont été détectées au cours des étapes précoces de la réparation (Bao, 2011). D’autre part, plusieurs études ont montré que l’inhibition des HDACs radio-sensibilise différents types de cancers comme le cancer de la prostate, le myélome multiple, l’ostéosarcome et le rhabdomyosarcome (Blattmann et al., 2010). Plusieurs essais cliniques sont en cours avec des inhibiteurs de HATs ou de HDACs, notamment dans les cancers de la thyroïde ou du cerveau et les cancers gastro-intestinaux (Ree et al., 2010)(Masoudi et al., 2008).
2.3.4. Le remodelage de la chromatine induit par la protéine PARP
Parmi les cibles de la protéine PARP, suite à la détection d’une lésion, il y a la protéine PARP elle-même, les protéines impliquées dans la même voie de réparation BER et également des histones avoisinant la lésion. En effet, le screening des séquences
CONFIDENTIEL 38 protéiques avec un site consensus de PARylation a permis l’identification de plusieurs partenaires. Parmi ces protéines, plusieurs sont impliquées directement dans les réponses cellulaires au dommage à l’ADN, au niveau de la reconnaissance des dommages et de la transformation des extrémités (XPA, XRCC1, MSH6, DNA ligase III, DNA polymérase ɛ), ou plus tardivement au cours des évènements responsables de la régulation du cycle cellulaire (p53, p21, …). Les ribonucléoprotéines, les DNA méthyltransférases, les DNA topoisomérases, et les protéines centromériques CENPA et CENPB possèdent également des motifs de PARylation dans leurs séquences (Gagné, Hunter, Labrecque, Chabot, & Poirier, 2003)(Reale, Matteis, Galleazzi, Zampieri, & Caiafa, 2005). La PARylation régule le remodelage de la chromatine à travers tous ces partenaires. La CHD4 par exemple, une sous-unité catalytique de la HDAC NuRD, est recruté par les polymères PAR en réponse à un dommage à l’ADN et facilite le chargement du complexe NuRD essentiel pour le remodelage de la chromatine (Polo, Kaidi, Baskcomb, Galanty, & Jackson, 2010). L’hélicase ALC1 se fixe également sur les PAR, s’active et permet de faire glisser les nucléosomes loin des sites de dommages pour faciliter l’accès des acteurs de réparation à la lésion (Ahel et al., 2009)(Gottschalk et al., 2009). Ainsi, les polymères PAR permettent le remodelage de la chromatine au niveau du dommage en interagissant directement avec la chromatine et à travers ses partenaires (Figure 5).
En conclusion, il est maintenant admis que la réparation de l’ADN est un domaine complexe régulé par différents processus. En effet, toute la machinerie impliquée dans la DDR ne travaille pas seule mais est plutôt coordonnée avec un ensemble de mécanismes complémentaires qui sont également essentiels pour le maintien de l’intégrité du génome. Par exemple, les protéines de remodelage de la chromatine permettent à la machinerie de réparation de l’ADN d’avoir accès facilement au site du dommage. Les composants de la DDR interagissent également avec les machineries du contrôle du cycle cellulaire et de la ségrégation des chromosomes. Toutes ces interactions permettent une réparation efficace de l’ADN avant la mitose et assurent la transmission d’une information génétique correcte à la cellule fille.