3D Chromosome Structure and Chromatin Proteomics
By
Daniel Benjamin Dadon
B.S. Biology
UC San Diego, 2009
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL
FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
JUNE 2016
© Daniel Benjamin Dadon. All rights reserved The author hereby grants to MIT permission to reproduce
and to distribute publicly paper and electronic copies of this thesis document in whole or in part
in any medium now known or hereafter created.
Signature of Author: _____________________________________________________________________ Department of Biology May 12, 2016 Certified by: ___________________________________________________________________________ Richard A. Young Professor of Biology Thesis Supervisor Certified by: ___________________________________________________________________________ Rudolf Jaenisch Professor of Biology Thesis Supervisor Accepted by: ___________________________________________________________________________ Amy Keating Professor of Biology Chairman, Committee for Graduate Students
Dedication
Acknowledgments
I have many people to thank as this Ph.D. would not have been possible without their support.
First and foremost, I want to thank my mentors Rick and Rudolf for encouraging my scientific growth, teaching me how to be successful, and providing supportive guidance. It has been an honor to be a part of your labs.
I would like to thank my mentors from San Diego Armin Blesch, Andy McCammon, and Jacob Durrant for taking a chance with me and providing me with patient mentorship.
None of this work could have been possible without the loving support of my family. My parents always motivated me to press on to be the greatest in their own ways. Michael has been a wonderful listener and advisor during this work. Thank you to my brothers Jon and David, Mercy, Grandma Mercedes, Grandpa Leon, Uncle Albert, and the rest of my family for motivating me and telling me that I made them proud. A special thank you goes out to my love Erika whose patience with me is unmatched, whose 24-hour support has been unbelievable and whose compassion is angelic. I would also like to thank Erika’s family for treating me like family. I thank my grandfather Arthur A. Frias for inspiring me to do a Ph.D. at MIT.
I thank my talented scientific collaborators for their roles in my scientific and professional growth. My partner Ji Xiong has shown me how much is possible with hard work and determination. I thank my early mentors Haoyi Wang, Albert Wu Cheng, and John Cassady, who taught me a great deal about focus and laboratory technique. I thank Matt Guenther for sharing my dream of the impossible and actually trying. I thank Jacob
Lovén and Warren Whyte for lab and career advice. I thank Zi Peng Fan for his good attitude and discipline. Thanks to Diego Borges-Rivera for reminding me to think big.
I thank Katie, Nancy, and Gerry for always helping me to find my way around the Whitehead Institute and keeping the lab and me organized. Nancy has been like a lab mom to me. She was always available to listen and advise on life.
I would like to thank all of my colleagues present and past in the Young and Jaenisch labs for providing an environment that always challenges me to be a better scientist. Thank you to Jess Reddy for being an awesome bay mate who always made lab more fun. The Sequencing Core facilities and in particular Tom Volkert deserve thanks for supporting my genomic projects and leading the Biohazards to so many victories. Eric Spooner deserves tremendous thanks for working so hard with Ji Xiong and me to make the chromatin proteomic project work. A special thank you goes to Tony Lee for training me in the ways of the Jedi since I was a young Padawan. He has been an incredible mentor and buddy. Thank you to Frank Soldner for always providing an alternate perspective and wisdom. Thank you to Maya for always caring.
I would like to thank my committee members Phil Sharp and Laurie Boyer for providing world-class scientific guidance throughout this journey. Frank Solomon deserves acknowledgment for keeping me (and most of the MIT Biology Department) sane.
3D Chromosome Structure and Chromatin Proteomics
byDaniel Benjamin Dadon
Submitted to the Department of Biology
in partial fulfillment of the requirement for the degree of Doctor of Philosophy in Biology
Abstract
The selective interpretation of the genome through transcription enables the production of every cell type’s distinct gene expression program from a common genome. Transcription takes place within, and is controlled by, highly organized three-dimensional (3D) chromosome structures. The first part of the work presented here describes the generation of 3D chromosome regulatory landscape maps of human naive and primed embryonic stem cells. To create these 3D chromosome regulatory landscape maps, genome-wide enhancer and insulator locations were mapped and then placed into a 3D interaction framework formed by cohesin-mediated 3D chromosome structures. Enhancer (H3K27ac) and insulator (CTCF) locations were mapped using ChIP-sequencing, whereas 3D chromosome structures were detected by cohesin-ChIA-PET. 3D chromosome structures connecting insulators (CTCF-CTCF loops) were shown to form topologically associating domains (TADs) and insulated neighborhoods, which were mostly preserved in the transition between naive and primed states. Insulated neighborhoods are critical for proper gene expression, and their disruption leads to the improper regulation of local gene expression. Changes in enhancer-promoter loops occurred within preserved insulated neighborhoods during cell state transition. The CTCF anchors of CTCF-CTCF loops are conserved across species and are frequently mutated in cancer cells. These 3D chromosome regulatory landscapes provide a foundation for the future investigation of the relationship between chromosome structure and gene control in human development and disease. The work presented in the second part focuses on developing an approach called “chromatin proteomic profiling” to identify protein factors associated with various active and repressed portions of the genome marked by specific histone modifications. The histone modifications assayed by chromatin proteomic profiling are associated with genomic regions where specific transcriptional activities occur, thus implicating the identified proteins in these activities. This chromatin proteomic profiling study revealed a catalog of known, implicated, and novel proteins associated with these functionally characterized genomic regions.
Thesis Supervisor: Dr. Richard A. Young Title: Professor of Biology Thesis Supervisor: Dr. Rudolf Jaenisch Title: Professor of Biology
Table of Contents Title page 1 Dedication 2 Acknowledgments 3 Abstract 5 Table of Contents 6 Preface 7 Chapter 1: 10 Introduction Chapter 2: 65
3D Chromosome Regulatory Landscape of Human Pluripotent Cells
Chapter 3: 104
Chromatin Proteomic Profiling Reveals Novel Proteins Associated with Histone-marked Genomic Regions
Chapter 4: 135
Conclusions and Future Directions
Appendix A: 163
Supplementary Material for Chapter 2
Appendix B: 212
Preface
The mammalian body is composed of a large diversity of cell types, each characterized by specialized properties and functions. This leads us to ask: how can this diversity of cell types be achieved when every cell in the body shares a nearly identical genome? Much of this cellular diversity can be explained by the selective transcription of the genome to produce distinct gene expression programs that lead to defining cell identity. Accordingly, the precise transcriptional regulation of gene expression is critical to cell identity. Proper transcriptional regulation of gene expression occurs within, and is controlled by, a framework of three-dimensional (3D) chromosome structures.
In the first chapter of this work, I review the components and mechanisms known about 3D chromosome structures and their roles in gene control. I begin by providing an overview of the hierarchical organization of chromosome structure followed by a brief section on transcriptional control. The remainder of the introduction focuses on aspects of 3D chromosome structures such as the key proteins involved, their relationship to gene control, and their regulation.
In the second chapter of this thesis, I describe 3D chromosome regulatory landscape maps created in human embryonic stem cells (hESCs) that provide a foundation for understanding how the genome is transcriptionally regulated in the context of 3D chromosome structures. Most previous genomic studies have overlooked the 3D nature of the genome and have interpreted genomic data in a linear manner. This classic view of genomics is shifting because of new technologies that allow interrogation of the interactions underlying the 3D organization of the genome. The human genome is highly organized into complex 3D chromosome structures, which contribute to gene expression
by constraining and enabling interactions between regulatory elements and their target promoters. The maps described in chapter three provide a framework for the 3D interpretation of genomic data in hESCs. This work strongly supports a model where cohesin-associated chromosome loops are the molecular basis for many features of genome organization in mammalian cells. The work contributes to understanding how millions of potential enhancer regions find their proper target genes. Finally, the study concludes that the underlying genomic sequences that organize these 3D genome structures are highly conserved across vertebrates, but are often affected by mutations in cancer. The study of genomics from a 3D perspective can thus elucidate basic principles about gene regulation in human health and clues as to how it may malfunction in disease.
In the third chapter of this thesis, I describe the development and application of a mass spectrometry-based approach called, “chromatin proteomic profiling,” which was designed to identify proteins associated with functionally characterized regions of the genome. Although a tremendous amount of effort has been invested in the characterization of key protein factors that control gene expression and chromatin, we are still learning where most factors associate and function across the genome. Chapter two describes the use of chromatin proteomic profiling to construct a catalog of proteins that physically associate with various “active” and “repressed” genomic regions marked by specific histone modifications. These histone modifications mark regions of the genome where specific transcription-associated activities occur, thus implicating proteins associated with these histone modifications in these activities. The catalogs presented in chapter two not only confirm many of the proteins that were known to function in the
chromatin regions assayed, but also reveal additional proteins not previously known to interact with these genomic regions.
Chapter 1
INTRODUCTION
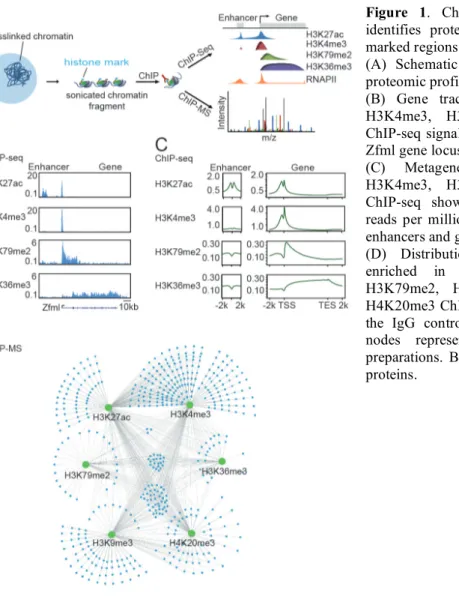
For many years, the study of chromosome structure has largely advanced independently of transcriptional studies. Simultaneously, the field of transcriptional control has developed with little influence from information about chromosome structure. However, transcription takes place within, and is controlled by, highly organized three-dimensional (3D) chromosome structures. The recent conceptual integration of these two fields of research is at the core of this thesis work (Figure 1). This introduction begins with a brief overview of chromosome structure presented as a hierarchical view of genome organization. The introduction continues with a short overview of transcriptional control that highlights the cis-regulatory elements involved. The remainder of the introduction focuses on conceptually uniting 3D chromosome structures with transcription, which is achieved by reviewing the functions of key proteins involved in chromosome structure, describing the relationship of 3D structures to gene regulation, and finally presenting what is known about the regulation of 3D chromosome structures.
Figure 1: 3D chromosome regulatory landscapes result from the conceptual integration of chromosome
Brief Overview of Chromosome Structure
At the highest level of nuclear organization, individual chromosomes are thought to occupy distinct zones of the nucleus called chromosome territories (CTs) (Figure 2). In 1909, Theodor Boveri proposed that interphase chromosomes occupied distinct regions of the nucleus that he termed chromosome territories. The first convincing evidence for CTs was collected by analyzing mutations made to a genome following ablation by a UV laser aimed at a small portion of the nucleus. It was found that only a limited number of chromosomes were affected with mutations. This finding confirmed the model where chromosomes did not thoroughly mix throughout the nucleus, but rather occupied distinct CTs (Cremer et al., 1996). Later, conclusive validations of the CT model were provided through the use of fluorescence in situ hybridization to visually label these territories and confirm their non-random organization in the genome (Cremer et al., 2006). Gross nuclear organization of chromosomes is determined by the size, number and activity of genes, as well as heterochromatin distribution (Bernardi, 2015; Guelen et al., 2008; Meaburn et al. 2016;
Figure 2: Chromosomes are organized into several tiers
of hierarchical structure. Chromosomes occupy distinct territories of the nucleus called chromosome territories. Within these territories, the chromosome organizes into domains that locally self-interact called Topologically Associating Domains (TADs) (~100 kb – 1000 kb). TADs are made of clusters of Insulated Neighborhoods (INs) (average size = ~200 kb). Enhancer-promoter loops are constrained within INs. Nucleosomes are the smallest unit of chromatin (~146 bp).
Pombo and Dillon, 2015). Other influences help to organize these nuclear chromosome structures, such as association with the lamina, nuclear envelop proteins and larger macromolecular structures like the nucleolus or transcription factories (Meaburn et al. 2016; Papantonis and Cook, 2013; Pombo and Dillon, 2015; van Steensel and Dekker, 2010). Within CTs, the core of these CTs is associated with compact chromatin, while other sub-domains of the CT that are associated with transcriptional activity loop out to the edges of the CTs (Cremer et al., 2006; Fraser et al., 2015; Fritz et al., 2015). Through microscopy, domains smaller than CTs called “CT domains” have been visualized between CTs and were suggested to be the unit building blocks of CTs (Albiez et al., 2006; Markaki et al., 2010). Microscopy-based approaches power an active area of research that continues to inform us about the organization of the nucleus.
High throughput sequencing studies have also greatly enhanced our understanding of genome organization. In the original genome-wide chromatin interaction mapping (Hi-C) paper, conducted at one megabase (Mb) resolution in two human cell types, it was found that there were two major pairwise interacting compartments of the nucleus termed compartments A and B (Lieberman-Aiden et al., 2009) (Box 1). Genomic regions of one compartment type tended to interact more frequently with other genomic regions of the same compartment type. Compartment A is more AT-poor and gene-rich, containing mostly active chromatin regions that tend to be more centrally located in the nucleus compared to the compartment B. Compartment B is AT-rich and gene poor, containing mostly repressed chromatin regions that are often associated with the peripheral nuclear lamina (Dixon et al., 2012; Lieberman-Aiden et al., 2009). Compartment B regions correlate well with lamina associating domains (LADs) and a late replication timing
consistent with the localization of this compartment at the nuclear periphery (Ryba et al., 2012). The emerging theme is that chromatin domains of similar chromatin state tend to associate through intrachromosomal interactions and interchromosomal interactions; however, mostly through intrachromosomal interactions (Grubert et al., 2015; Rao et al., 2014; Sexton and Cavalli, 2015). Further work has led to sub-divisions of this compartment model to account for other molecular features of chromatin (Rao et al., 2014), although the prevailing feature observed among interactions remains the homotypic organization of chromatin. The compartment model attained through DNA interaction sequencing studies is compatible with the CT model of nuclear organization.
Around the megabase size range, self-interacting domains called Topologically Associating Domains (TADs) have been identified as the basic unit of chromosome Box 1: Studying the 3D Genome Through Sequencing
Historically, fluorescent in situ hybridization (FISH) has been used to gain insight into genome structure, but more recently a suite of tools to assay genome structure using high-throughput sequencing have emerged. These sequencing efforts use crosslinking and intramolecular ligation to assay the contacts of distal genomic loci, and are considered part of the family of chromatin conformation capture “3C” assays. These methodologies work by either taking a one-vs-all approach—using a “bait” locus and assessing what genomic loci interact with the bait—or by assaying all-vs-all interactions (de Wit and de Laat, 2012). The original [perhaps ‘traditional’ or ‘classical’ instead of original]3C assay uses a “bait” region of the genome and checks for suspected interacting loci (Dekker et al., 2002), whereas the 4C assay chooses one bait and assays all interactions in an unbiased manner (Simonis et al., 2006). The 5C assay is a “many-vs.-many” method used for a given stretch of the genome acquired through massively tiled probes (Dostie et al., 2006), whereas the Hi-C method indeed performs an “all-vs-all” assay across the entire mappable genome (Lieberman-Aiden et al., 2009) with recently made protocol improvements (Hughes et al., 2014; Rao et al., 2014). The major issue hindering such Hi-C assays is that the depth of sequencing has a mathematically squared relationship relative to the acquisition of new data, making higher resolution maps very expensive to achieve.
Techniques have been developed to minimize this sequencing depth issue. One assay called chromatin interaction coupled to paired-end tag sequencing (ChIA-PET) reduces the assayed genomic space by adding what is known as a “capture step.” In the case of ChIA-PET, this “capture step” is a chromatin immunoprecipitation (ChIP) for an epitope of interest, which enriches for a subset of the genome associated with a target epitope (Fullwood et al., 2009). The initial reduction in the genomic space assayed leads to the acquisition of much higher-resolution maps with less sequencing depth effort. The ChIP step also enables the enrichment of biologically relevant regions of interest (Dowen et al., 2014; Fullwood et al., 2009; Handoko et al., 2011; Heidari et al., 2014; Li et al., 2010). The downside of such capture assays is the creation of mapping bias. An illustrative example of the comparison of Hi-C vs. ChIA-PET was recently made (Tang et al., 2015). One way to study the CTCF-CTCF associated loops in the genome is to perform ChIP-seq combined with deeply sequenced Hi-C (Rao et al., 2014). However, it was recently reported that a single round of CTCF ChIA-PET sequencing on an Illumina MiSeq could detect nearly the same interactions, plus 10-fold higher resolution (Tang et al., 2015). Another group utilized RNA probe sets associated with genome-wide promoter predictions to enrich C libraries for promoter regions in a method called capture Hi-C (Hi-Chi-Hi-C) (Mifsud et al., 2015). Optimizing sequencing methods through capture methods or deeper sequencing will help to inform our models of 3D genome structure. A more in-depth discussion regarding techniques used to ask questions about genome organization is provided in Chapter 4.
interaction. Through various Hi-C studies, TADs have been defined by intensified local interaction frequencies with sharp boundaries, across which interactions are infrequent (Dixon et al., 2012; Lieberman-Aiden et al., 2009). TADs can also be identified using cohesin-based chromatin interaction analysis using paired-end tag sequencing (ChIA-PET) data (Ji et al., 2015), but not by using RNA Polymerase II (RNA Pol II) ChIA-PET data suggesting that cohesin loops are the dominant molecular structures underlying TADs. Coincidently, the approximate size of TADs is about the same as that of the “CT domains” observed under the microscope, suggesting that these units are likely to be the same structures (Albiez et al., 2006; Gibcus and Dekker, 2013; Markaki et al., 2010). The transcriptional activity of genes within TADs tends to be coordinated and has been seen to change concordantly with cell identity (Dixon et al., 2015; Grubert et al., 2015; Heidari et al., 2014; Ji et al., 2015; Rao et al., 2014). TADs exhibit relatively similar boundaries across cell types (Dixon et al., 2015; Ji et al., 2015; Rao et al., 2014). Mammalian TAD boundaries are enriched for genes such as housekeeping genes (ubiquitously expressed), transfer RNA genes and short interspersed element (SINE) retrotransposons (Dixon et al., 2012; Hou et al., 2012; Sexton et al., 2012). Also, boundaries of TADs are relatively enriched for the binding of proteins such as CTCF, cohesin, condensin and TFIIIC (Ong and Corces, 2014). Species that have conserved the CTCF insulator protein match well with species that have studies reporting sharp TAD boundaries, suggesting that CTCF plays a central role in TAD organization (Heger et al., 2012; Sexton and Cavalli, 2013). Although other associated factors have been identified in Drosophila studies, it remains unclear if orthologous proteins exist in mammals (Ali et al., 2016; Ong and Corces,
2014). TADs have distinctive features and apparently play an import role in organizing the genome.
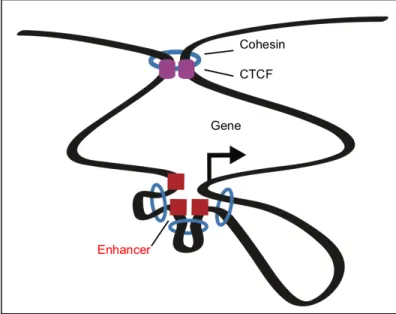
At the sub-TAD level, 3D chromosome structures exist to organize the genome into partitioned domains that constrain transcriptional activity called “insulated neighborhoods.” Such insulated neighborhoods are formed by cohesin-mediated CTCF-CTCF loops that bridge two distal CTCF-CTCF-bound insulator regions (Dowen et al., 2014) (Figure 3). Mechanistically, these CTCF-CTCF loops constrain internal enhancer looping towards target genes within the insulated neighborhood and prevent the spread of chromatin state beyond the insulated neighborhood boundaries (Dowen et al., 2014). Thus, some 3D chromosome structures appear to serve insulating functions, whereas others facilitate interactions among cis-acting regulatory elements.
At the molecular level, the genome is structured as a heterogeneous complex of DNA, RNA, and protein called chromatin. The structural state of chromatin can enable or restrict potential regions of transcriptional activity. Thus, the amount of transcription that occurs at a genomic locus can be correlated with the physical chromatin context of the locus. Overall, transcriptional activity across the genome can be
Figure 3: Insulated Neighborhood: Cohesin ring structures (blue)
and CTCF (purple) organize chromosome loop structures. Loops formed between CTCF sites create the insulated neighborhood domain in which cohesin loops enhancers (red) to the promoter (arrow) of the target gene.
thought of as the collaborative product of layers of regulation controlled by transcription factors, chromatin regulators and 3D chromosome structures.
Brief Overview of Transcriptional Control: Cis-Regulatory Elements
DNA binding transcription factors (also known as trans-acting factors) bind to cis-regulatory DNA sequences to activate or repress transcription at specific sites of the genome. The basic model of transcriptional regulation by transcription factors was revealed over half a century ago by studying the E. coli lac operon (Jacob and Monod, 1961). This work led to our basic understanding that protein-binding regulatory DNA sequences (also known as cis-regulatory elements) are associated with genes and that regulatory proteins bound to these sequences function to activate or repress transcription. In mammals, transcription factors can bind to promoter elements or distal enhancer elements to direct transcription to the promoter of a particular target gene. Once bound to DNA, transcription factors can recruit transcriptional machinery or trigger transcriptional pause-release (for further reading see (Adelman and Lis, 2012; Allen and Taatjes, 2015; Core et al., 2014; Kagey et al., 2010a; Kornberg, 2005; Kwak and Lis, 2013; Lee and Young, 2013; Malik and Roeder, 2010; Young, 2011)). Transcription factors can also affect chromatin state by recruiting chromatin regulators to these elements (for reviews see (Cavalli, 2002; Ernst et al., 2011; Fazzio and Panning, 2010; Flynn and Chang, 2012; Grewal and Jia, 2007; Gross et al., 2015; Jiang and Pugh, 2009; Lessard and Crabtree, 2010; Pirrotta, 2016; Smith et al., 2016; Zaret and Mango, 2016)). Some transcription factors apparently have a dominant role of binding to cis-regulatory elements called insulators that are responsible for partitioning the genome into regions of independent
transcriptional activity (Ali et al., 2016; Matharu and Ahanger, 2015; Ong and Corces, 2014; Phillips and Corces, 2009; Pombo and Dillon, 2015). The concerted activities of trans-acting factors associated with enhancers, promoters, and insulators govern the transcriptional program.
Enhancers and Promoters
Enhancers are distal cis-regulatory DNA elements that are involved in an increase of transcription at a target gene. Enhancers were first described as sequences that could distally activate recombinant reporter genes in an orientation-independent manner (Banerji et al., 1981; Moreau et al., 1981). Active enhancers are bound by transcription factors that recruit cofactors such as the Mediator complex, which can then recruit RNA Pol II to the core promoter (Allen and Taatjes, 2015; Kagey et al., 2010b; Kornberg, 2005; Malik and Roeder, 2005; Roeder, 1998). The collection of active enhancers in a cell type play roles in determining cell-type-specific gene expression across development (Buecker and Wysocka, 2012; de Laat and Duboule, 2013; Levine, 2010; Levine et al., 2014; Murakawa et al., 2016; Ong and Corces, 2012; Shlyueva et al., 2014).
Various molecular features are associated with active enhancers that aid in enhancer identification. Enhancers contain DNA sequences that can be bound by sequence-specific transcription factors. Enhancers can be bound by many transcription factors at a time, often binding in a biochemically cooperative manner (Hochschild and Ptashne, 1986; Siersbæk et al., 2014). Enhancer activity can be directly tested by its effect on transcriptional reporter assays (Babbitt et al., 2015). Active enhancer regions are flanked by nucleosomes with acetylated lysine 27 of N-terminal tail of histone H3
(H3K27ac), a mark that is deposited by histone acetyltransferases (HATs) such as the CBP/p300 complex (Creyghton et al., 2010; Hawkins et al., 2011; Neph et al., 2012; Rada-Iglesias et al., 2011). Enhancers are also marked with H3K4 mono-methylation (H3K4me1) (Calo and Wysocka, 2013; Hawkins et al., 2011; Smith and Shilatifard, 2014), although H3K4me1 is present at both active enhancers and inactive enhancers that are poised for activation later in development (Rada-Iglesias et al., 2011; Zentner et al., 2011). By using such epigenetic marks, many research groups have led efforts to map these enhancers finding approximately ~106 distinct enhancers across various human cell and tissue types (Andersson et al., 2014; Dunham et al., 2012; Hnisz et al., 2013; Thurman et al., 2012). These enhancer maps serve as a useful framework for studying gene regulation.
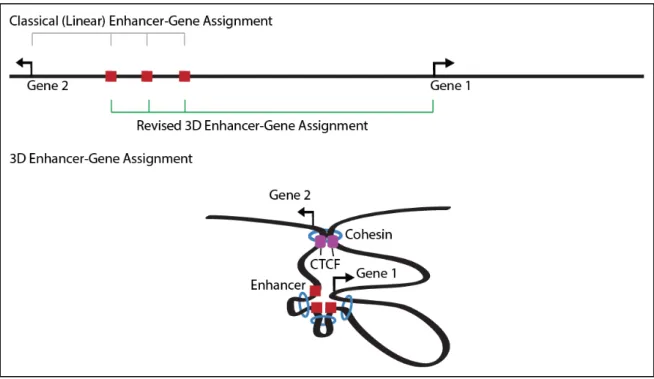
Once an enhancer is identified, it is often difficult to predict its target promoter. Conventionally, enhancers have been assigned to their nearest active gene. However, the linear proximity of enhancers to promoters is not always the best predictor of enhancer-promoter assignment (Blackwood and Kadonaga, 1998; Gheldof et al., 2010; Zhang et al., 2013). Enhancers commonly interact with the nearest active gene, but enhancers often operate over long linear distances to affect distal genes without having much effect on more proximal genes (Benko et al., 2009; Claussnitzer et al., 2015; Lettice et al., 2003; Montavon et al., 2011). This counterintuitive relationship is enabled by the 3D looping of the genome discussed later in this chapter (Figure 4). The looping mechanisms by which enhancers exert their effect on target genes are of fundamental interest to transcriptional biology.
Large clusters of enhancers, called “super-enhancers,” drive key genes associated with cell identity (Parker et al., 2013; Whyte et al., 2013). Hundreds to a few thousand super-enhancers can exist in a single cell type (Hnisz et al., 2013; Whyte et al., 2013). Super-enhancers are densely occupied by a disproportionate amount of transcription factors, cofactors, chromatin regulators, and transcription apparatus compared to typical enhancers (Hnisz et al., 2013). Genes associated with super-enhancers tend to be more highly expressed and individual super-enhancer constituents drive higher levels of transcription in enhancer reporter constructs than typical enhancer constituents (Hnisz et al., 2015; Loven et al., 2013; Whyte et al., 2013). Super-enhancers have been identified by analyzing ChIP-Seq data sets from enhancer-binding proteins (e.g. master transcription factors, mediator, BRD4, or H3K27ac) and applying a distance-based enhancer stitching algorithm to find regions of exceptional signal of these marks (Hnisz
Figure 4: Linear enhancer-gene assignment methods assign enhancers to the nearest gene on the linear
genome representation. Linear assignment leads to the assignment of these enhancers to Gene 2. 3D enhancer-gene assignment takes interaction data into account to identify the gene with the closest 3D proximity to the enhancers. 3D assignment leads to the proper assignment of these enhancers to Gene 1.
et al., 2013; Loven et al., 2013; Whyte et al., 2013). Individual enhancer constituents of multi-constituent super-enhancers were shown to respond to different signaling pathways to integrate multiple signals at an associated promoter (Hnisz et al., 2015). Super-enhancers are enriched for disease-associated genetic variation. The diseases whose associated variants fall in super-enhancers in a particular cell type are often known to involve that cell type (Hnisz et al., 2013). Oncogenic super-enhancers acquired by key oncogenes in cancer cells and these tumor-specific super-enhancers confer sensitivity to transcriptional inhibitors (Chipumuro et al., 2014; Kwiatkowski et al., 2014; Loven et al., 2013). The super-enhancer is a powerful construct that aids the discovery of cell-type-relevant transcriptional biology by focusing on genes of particular cell-type-specific interest.
Active enhancers are associated with the local production of RNAs. This phenomenon was first observed at the beta-globin locus control region (Tuan et al., 1992). These enhancer RNAs (eRNAs) were later found genome-wide in mouse cortical neurons at active enhancers (Kim et al., 2010). The established features of eRNAs are as follows: 1) eRNA expression can be detected at active enhancers and correlated with target gene expression, 2) eRNAs are 5’-capped and void of splicing, 3) most eRNAs are not polyadenylated and are bi-directionally transcribed, 4) other eRNAs are polyadenylated and generally unidirectionally transcribed, and 5) eRNAs are rapidly degraded by exosomes suggesting that these RNAs exert their function locally (Lai and Shiekhattar, 2014; Lam et al., 2014; Ren, 2010). It is not entirely clear whether the production of eRNAs directly contributes to transcription through RNA-mediated interactions (Lam et al., 2013) or if these eRNAs are produced as a transcriptional
by-product of enhancer-promoter interactions (Li et al., 2016). A recent study proposed that RNA at enhancers may function as transcription factor traps that increase the recruitment of transcription factors such as Yin-Yang 1 (YY1) to enhancers (Sigova et al., 2015). Further work will be necessary to dissect the roles of eRNA in transcription.
Promoters are cis-regulatory elements located at the 5’ end of genes that direct transcription to the transcription start site (TSS) through the binding of trans-acting factors that recruit and assemble the transcription initiation apparatus (Danino et al., 2015; Heintzman and Ren, 2007; Kadonaga, 2012; Muller and Tora, 2014). Promoters can be bound by general transcription factors (GTFs) or other transcription factors, which ultimately facilitate the recruitment of RNA Polymerase II (RNA Pol II), which in turn results in transcription. There are many types of promoters at different classes of genes associated with specific mechanisms (for reviews see (Danino et al., 2015; Kadonaga, 2012; Lenhard et al., 2012)). Active promoters are associated with nucleosome depletion, histone variant (H2A.Z, H3.3) incorporation, and characteristic histone modifications (Muller and Tora et al., 2014). Promoters are marked by the tri-methylation of lysine 4 of histone H3 (H3K4me3) by the Set1/Ash2L complex or by trithorax group proteins such as MLL (Barrera et al., 2008; Guenther et al., 2010; Ruthenburg et al., 2007). Promoters are also acetylated by the HAT complexes such as CBP/p300 at histone H3 at lysine 27 (H3K27ac) (Ji et al., 2015).
Promoters and enhancers share a number of characteristics, but can be distinguished at the molecular level (Kim and Shiekhattar, 2015). Although both enhancers and promoters produce bidirectional transcripts, the resulting transcripts have different molecular features. At enhancers, transcripts from either direction of
transcription are short-lived, experiencing exosome-mediated degradation (Andersson et al., 2014; Flynn et al., 2011). At promoters, however, transcripts in the sense direction can experience productive elongation and mRNA processing, whereas those from the antisense direction form short-lived noncoding RNAs called uaRNAs (Core et al., 2008; Preker et al., 2008; Seila et al., 2008), which experience exosome-mediated degradation (Andersson et al., 2014; Flynn et al., 2011). This asymmetric transcription is due to an enrichment of polyadenylation sites (PAS) and a depletion of U1 small nuclear ribonucleoprotein (snRNP) binding sites or 5’ splice like sequences (Almada et al., 2013; Core et al., 2014; Ntini et al., 2013). Additionally, enhancers and promoters are both marked with H3K27ac; however, H3K27ac can be a distinguishing feature between enhancers and promoters due to the stronger ChIP-seq signal enrichment for this mark at enhancers (Ji et al., 2015). Similarly, a higher ratio of H3K4me1/H3K4me3 signal can distinguish enhancers from promoters (Calo and Wysocka, 2013). Generally, the distinctive property that sets promoters apart from enhancers is the ability to direct the transcription of a spliced, polyadenylated transcript (Kim and Shiekhattar, 2015), but this definition is being blurred by functional assays that measure promoter or enhancer activities. By using chromatin signatures based on the above histone marks, it was recently reported that 15% of strong promoters in one cell type were marked as enhancers in another cell type (Leung et al., 2015). Upon cloning these promoter regions that were marked as enhancers into luciferase reporter vectors for enhancer activity, the predictions regarding the enhancer activity of these elements were largely validated (Leung et al., 2015). The potential overlapping roles of promoters and enhancers is an area of active research.
Insulators
Insulator elements help to form boundaries that partition the genome into chromatin domains of independent transcriptional activity. Insulators perform this duty in two classically described ways: insulator elements possess the abilities to block an enhancer from operating across the insulator and to prevent the spread of chromatin state beyond the insulator. These two functions of insulators were initially described separately, but are now becoming appreciated as the dual roles of insulators. In the following section, the studies that led to this model of insulator activity are reviewed.
The fundamental properties of insulators have been elucidated through decades of research. Insulators were first described in 1985 in Drosophila at the 87A7 cytogenic chromomere locus. At this locus containing two heat shock protein hsp70 genes, scientists discovered flanking chromatin structures that were proposed to serve as boundaries and anchor points for the higher-order organization of chromosome domains (Udvardy et al., 1985). Later, through genetic studies in Drosophila, others placed the Su(Hw) binding element from the gypsy retrotransposon between a transcriptional enhancer and its target yellow gene promoter, which led to repression of the target yellow gene. This observation led to a basic enhancer blocking model where an insulator element blocks an enhancer from activating a gene across the insulator element (Geyer and Corces, 1992). Other endogenous boundary elements were later proposed at the homeotic gene cluster in Drosophila. By integrating a lacZ-containing transposon into the iab-7 segment of the homeotic gene cluster, it was observed that the lacZ reporter had a specific expression pattern consistent with the transcriptional activity within the iab-7 cis-regulatory domain, and the reporter was protected from the activity of regulatory
elements in neighboring domains (Galloni et al., 1993). At the homeotic gene cluster locus, it was also observed that Polycomb-coated segments of this gene cluster appeared to have barriers that prevented the spreading of Polycomb to other segments of the gene cluster (Orlando and Paro, 1993). Cai and Levine later suggested that these insulators might not be restricted to the functional isolation of neighboring loci, but rather might function as flexible regulatory elements that coordinate complex promoters and genetic loci (Cai and Levine, 1995). In 1999, the eleven zinc-finger protein CTCF was found to bind to an upstream insulator element at the chicken beta-globin locus. It was concluded that CTCF binding prevented transcriptional enhancers from operating across such elements (Bell et al., 1999). CTCF was later shown to demarcate areas of active and repressed chromatin genome-wide (Cuddapah et al., 2007). These studies have contributed to a model where insulators function as enhancer blockers and barriers to the spread of chromatin state.
The constraints that cause enhancer elements to operate on their specific targets (Zabidi et al., 2015) and control the spreading of chromatin state are still not completely understood. Recent work suggests that insulators elements impose these constraints by organizing 3D genome structures (Dixon et al., 2012; Dowen et al., 2014; Handoko et al., 2011; Heidari et al., 2014; Nora et al., 2012; Phillips-Cremins et al., 2013; Rao et al., 2014). The following sections discuss some of the major proteins thought to be involved in human chromosome looping, lessons learned from studying model loci, a genome-wide view of chromosome looping, and the regulation of chromosome loops. For a more expansive list of chromosome structuring proteins and their putative regulators see Table 1.
3D Chromosome Structure Proteins Cohesin
The structural maintenance of chromosomes (SMC) protein complex cohesin is a ring-shaped complex made up of four protein subunits (SMC1, SMC3, RAD21, STAG) that can accommodate and link two nucleosome-bound DNA regions to form DNA loops (Dowen and Young, 2014; Hirano, 2006). The cohesin complex is highly conserved, having homologs from humans to bacteria (Gligoris et al., 2014), suggesting that cohesin has an indispensable role in the cell. Cohesin was first described for its vital role in sister
chromatid cohesion during G2 and mitosis (Nasmyth and Haering, 2009; Pombo and Dillon, 2015; Remeseiro et al., 2013; Seitan and Merkenschlager, 2012). Cohesin was later shown to have an important role in gene regulation (Dowen et al., 2013; Kagey et al., 2010a; Krantz et al., 2004; Rollins et al., 1999; Schaaf et al., 2013; Tonkin et al., 2004). Cohesin plays this role in gene regulation by connecting interacting DNA segments including enhancers, promoters, and insulators (Dowen et al., 2013, 2014; Ji et al., 2015).
Cohesin is loaded at enhancers and promoters of active genes by NIPBL (Dorsett, 2011; Hadjur et al., 2009; Nasmyth and Haering, 2009; Parelho et al., 2008; Phillips-Cremins and Corces, 2013; Schmidt et al., 2010; Seitan and Merkenschlager, 2012; Wendt et al., 2008). Genetic screens for repressors of a cut gene separated from its enhancer by a gypsy insulator in Drosophila, showed that cohesin contributed to gene regulation through mutations identified in its loading protein (Nipped-B) (Rollins et al., 1999). Cohesin was later identified in humans through detection of mutations in the loading protein ortholog (NIPBL) in a genetic disorder called Cornelia de Lange syndrome (Krantz et al., 2004; Tonkin et al., 2004). In yeast, the Nipped-B (NIPBL homolog) components SCC2 and SCC4 were shown to be necessary for the binding of cohesin to chromosomes leading to a model where NIPBL is the loader of cohesin (Ciosk et al., 2000). It was later found that knockdown of cohesin and Nipped-B lead to the disruption of the same set of genes (Schaaf et al., 2013). NIPBL is associated with the Mediator complex, which binds to transcription factors at enhancers and promoters to function as a co-activator in the recruitment RNA Pol II (Allen and Taatjes, 2015; Kornberg, 2005; Malik and Roeder, 2010; Roeder, 1998). ChIP-sequencing in murine
embryonic stem cells showed that sites occupied by the Mediator complex are co-occupied by cohesin (Kagey et al., 2010a). Thus, the cohesin loader NIPBL is thought to be recruited by Mediator to enhancers and promoters, which ultimately leads to enhancer-promoter looping via the formation of the cohesin ring at the base of the loop. In addition to enhancer and promoter sites, cohesin also functionally associates and co-occupies genomic sites with CTCF (Parelho et al., 2008), most sites of which appear to be mutually exclusive from Mediator-occupied sites, although many exceptions exist (Wendt et al. 2008; Parelho et al. 2008; Rubio et al. 2008; Kagey et al. 2010; Creighton et al. 2013; Faure et al. 2012; Hnisz et al. 2013). One model suggests that cohesin is loaded at enhancers and promoters by NIPBL and then it slides along the DNA loop forming an extrusion complex until cohesin runs into CTCF sites where the cohesin ring is stalled (Fudenberg et al., 2015). Cohesin could also conceivably be loaded at CTCF sites in a NIPBL-independent manner (Dorsett, 2011; Dowen and Young, 2014c). Once loaded onto the genome the loops mediated by cohesin for the basis for much of 3D genome organization. These 3D organizational functions are described in more detail in the following sections.
CTCF
CTCF is an essential eleven zinc-finger containing protein that binds the CCCTC DNA sequence motif to organize the genome through looping distal DNA elements. The CTCF homozygous knockout is early embryonic lethal in mouse (Heath et al., 2008; Splinter et al., 2006). A majority of CTCF binding regions are thought to be shared across cell types (Cuddapah et al., 2009; Heintzman et al., 2009; Kim et al., 2007). CTCF binds
~55,000 sites in any cell type (Wang et al., 2012) with approximately 70% binding site similarity between any two cell types, however in a study conducted across 19 cell types ~50,000 CTCF sites were determined to be variable while ~28,000 sites were constitutive. ChIP-seq analysis additionally revealed that CTCF is strongly enriched at the boundaries of TADs (Dixon et al., 2012; Hou et al., 2012; Sexton et al., 2012), LADs (Guelen et al., 2008), and frequently enriched at the boundaries of domains containing the histone modifications H3K27me3 and H2AK5ac (Cuddapah et al., 2009; Dowen et al., 2014; Handoko et al., 2011). The effect that these genome-wide CTCF-binding profiles have on gene expression across cell types remains incompletely understood.
CTCF bound to distal segments of DNA forms a CTCF-CTCF protein dimer that enables the looping of these DNA segments with the aid of the cohesin ring complex. Mass spectrometry experiments using an epitope-tagged version of CTCF and an in vitro yeast two-hybrid experiment supports a model where CTCF forms a homodimer (Yusufzai et al., 2004). Dimerization of DNA-bound CTCF proteins is dependent on the orientation of CTCF motifs. CTCF homodimers occur in vitro only when DNA probes have convergent sequences (Pant et al., 2004). In the genome, the formation of such loops almost always has the convergent orientation of the CTCF motifs. This relative orientation of motifs occurs in approximately 80-90% of the loops with a single CTCF motif at each end (Ji et al., 2015; Rao et al., 2014). However, using CTCF ChIA-PET data, the Ruan group has argued that CTCF participates in functional loops that do not require such convergently oriented motifs by forming loops with tandemly oriented motifs (Tang et al., 2015). Further studies regarding the relevance of CTCF motif orientation in looping are needed.
In addition to the general requirement for the convergent orientation of CTCF motifs participating in loops, other properties have been observed regarding the sites bound by CTCF. The Casellas group investigated the potential for the distinct usage of finger clusters among the eleven zinc-fingers contained in the CTCF protein. They found three general classes of finger usage, that the core zinc fingers 4-7 are bound in ~80% of binding events, and that flanking DNA sequences could modulate CTCF binding (Nakahashi et al., 2013). Thus, sequence specificities may indeed guide CTCF binding site selection. In addition to sequence-specific influences on CTCF binding, nucleosomes may also play a role. CTCF binds in between nucleosomes in what is called the nucleosome linker region and is flanked by 20 evenly spaced nucleosomes suggesting that CTCF has a major role in nucleosome positioning (Fu et al., 2008). The alternate model suggests that nucleosome positioning may have a global role in CTCF genome binding (Cuddapah et al., 2009). Methylation may also play a role in determining CTCF binding site selection by disrupting the binding interface between CTCF and the DNA (Filippova et al., 2001; Hark et al., 2000). The observation that CTCF prefers not to bind methylated binding sites was first observed in the context of the imprinted H19/Igf2 locus (Bell and Felsenfeld, 2000), discussed in detail in the next section. Across CTCF binding sites genome-wide in multiple cell types ~41% of differential CTCF binding is linked to differential methylation measured by reduced representation bisulfite sequencing (Wang et al., 2012). By including more datasets from other cell types, the same group later concluded that this relationship was limited to a small set of CTCF sites with highly variable binding across the samples (Maurano et al., 2015). This weak correlation suggests that perhaps other mechanisms are at play in the regulation of CTCF binding.
An additional mechanism regulating CTCF may be transcription across the CTCF binding site. At the X-chromosome, transcription of Jpx was found to evict a repressive CTCF molecule allowing Xist expression and ultimately X-chromosome inactivation (Sun et al., 2013). In another study from the same group, the authors described CTCF as an RNA-binding protein with high affinity for RNA. Thus, they proposed that CTCF is targeted to specific sites of the genome by cis-acting long noncoding RNAs (Kung et al., 2015). Some molecular features have been identified that characterize the CTCF protein and its DNA-binding, yet much work remains to better characterize this relationship such as the identification of the complete crystal structure of CTCF in complex with DNA and the determination of CTCF’s potential to oligomerize in vivo beyond a dimer.
The Relationship Between 3D Chromosome Structures and Transcription Lessons from Studying Model Loci
A small set of loci has served as models for our understanding of looping interactions in chromatin. Such models have helped to unveil the potential allele-specific behavior of loops, the roles loops play in development, and the relationships between loop formation and inducible gene expression. Various sequencing (see Box 1) and microscopy-based techniques (reviewed in Chapter 4) have enabled the interrogation of these looping interactions.
Studies at the H19/Igf2 locus have served as the foundation for the understanding of the allele-specific behavior of loops. At the H19/Igf2 locus, paternal allele imprinting of the imprinting control region (ICR) along with methylation of the multiple differentially methylated regions (DMRs) prevent CTCF binding and CTCF-mediated
loop formation, resulting in H19 repression and Igf2 activation by distal enhancers (Han et al., 2008; Hark et al., 2000; Murrell et al., 2004; Szabo et al., 2004). In contrast, at the maternal locus, demethylation allows for CTCF binding and CTCF-mediated loop formation, which prevents the activation of the Igf2 gene by blocking the enhancer and thus leads to the demethylation of the H19 promoter region, allowing activation of the H19 gene (Phillips-Cremins et al., 2013). Various groups have reported parent-of-origin interactions at this locus using allele-distinguishable transgenic mouse models (Kurukuti et al., 2006; Murrell et al., 2004). Using this model locus it was also found that CTCF binding to the ICR and multiple DMRs is required to maintain imprinting in somatic cells by preventing de novo methylation (Fedoriw et al., 2004; Kurukuti et al., 2006; Szabo et al., 2004) suggesting that CTCF may play a global role in the maintenance of imprinting through preventing methylation. In addition to CTCF-mediated prevention of methylation at the ICR and the DMR regions, CTCF-binding prevented methylation of the H19 promoter on the maternal allele, which permits H19 activation (Pant et al., 2003). Another lesson learned at this locus about CTCF’s role in the allelic control of gene expression comes from studies regarding the Polycomb complex. In these studies, it was discovered that CTCF was necessary for the recruitment of SUZ12 of the PRC2 complex to the ICR to deposit the H3K27me3 mark at the silenced maternal Igf2 promoter (Han et al., 2008; Li et al., 2008). The allelic nature of this imprinted locus has enabled many studies regarding the allelic influence on gene expression.
The murine beta-globin locus has been useful for studying the role of looping in the context of development. At this well-studied locus, a cluster of globin genes active during particular stages of erythroid development are contained between two CTCF
binding elements that form a loop domain including the beta-globin genes and excluding two flanking clusters of olfactory receptor genes (Farrell et al., 2002). One of the two CTCF sites that form the loop around the globin genes is located within a complex cis-regulatory element that is called the locus control region (LCR), because of its ability to control the expression patterns of the globin genes within the beta-globin cluster loop. These loops were initially reported to be absent in brain cells (Tolhuis et al., 2002), but later described to be present (Simonis et al., 2006), likely due to technical differences in the experiments. Inducing an ectopic interaction between the LCR and the promoter of the beta-globin gene in a cell line that does not usually express beta-globin or have any interaction between these two elements resulted in a large increase in beta-globin gene expression (Deng et al., 2012). This study suggests that the formation of the loop between the LCR and the gene target is necessary to allow for gene expression. The beta-globin locus is a useful model locus for studying chromatin structure during different developmental contexts.
The MHC-II (HLA) locus has been useful for studying the relationship between looping and signal induction, and how it leads to transcription. The two MHC-II genes (HLA-DRB1 and HLA-DQA1) are co-regulated by an intergenic element termed XL9. Upon stimulation with the cytokine interferon-gamma, cells that do not normally express these receptors can transcribe these genes. Additionally, induction in a human epithelial cell line leads to the formation of loops between the central XL9 element with both of the HLA genes (Majumder et al., 2008). The XL9 element displayed enhancer blocking effects in a reporter assay (Majumder et al., 2006); however, CTCF knockdown leads to reduced loop formation with the central CTCF and the two promoters as well as a
decrease in expression, suggesting that CTCF can contribute to specific transcriptionally functional looping. The authors went on to show that the promoter-associated portions of these loops were mediated by the factors RFX (in complex with CREB and NF-Y) and CIITA and did not report direct binding of CTCF to either of the promoters. Knockdown of CTCF, RFX or CIITA all led to reductions in looping (Majumder et al., 2008). This locus represents a model where loops can be formed in an inducible manner leading to gene expression. It is important to note that the loops in the model described here were not reported to be formed by CTCF-CTCF dimers but rather by a heterogeneous complex of proteins containing CTCF. More induction studies will be needed to tease out the molecular determinants for the proposed property of induced structure. Given that these loops are already pre-formed in MHC-II expressing cells, the concurrent induction of loop formation and gene expression may be a phenomenon that occurred because the experiment was performed in cells that do not express MHC-II receptors and are not normally exposed to interferon gamma, thus loop rearrangements are necessary. Other structural studies of induction will be needed to determine the prevalence of this phenomenon of concurrent loop formation with gene expression.
Loop formation may not necessarily occur simultaneously with gene activation. Other studies have found examples where loop structures already exist prior to the induction of gene expression (Eijkelenboom et al., 2013; Hakim et al., 2011; Jin et al., 2013). Treatment of IMR90 cells with TNF-alpha leads to the activation of hundreds of genes. The interactions of these genes with their enhancers were found to be already present before stimulation (Jin et al., 2013). Similarly, in-depth investigation of the glucocorticoid receptor responsive loci did not exhibit major rearrangements in genome
organization upon stimulation (Hakim et al., 2011). In both of these studies, exposure to potent molecular stimulation did not result in major looping rearrangements concordant with gene expression changes. These results suggest that these cells may already be structurally primed to respond these signals. In addition to assessing structural changes in response to signaling molecules, another group evaluated structural changes in response to the FOXO3 transcription factor. They found that FOXO3-mediated gene activation occurred through enhancers that had previously established loops to their target genes (Eijkelenboom et al., 2013). These models suggest that 3D structures may already be structurally poised to respond to certain environmental or developmental cues. Thus, loop formation alone between regulatory elements and their target genes is not sufficient for gene expression.
Cohesin-Mediated Chromosome Loops
Two major classes of loops formed by the cohesin ring complex have emerged from cohesin ChIA-PET studies: those connecting cis-regulatory elements with genes and those connecting CTCF-bound regions with other CTCF-bound regions. A developing model suggests that CTCF loops mediated by cohesin (from here on called CTCF-CTCF loops) form the molecular basis for the formation of TADs and that enhancer- and promoter-associated loops occur within these CTCF-CTCF loops. Although these TADs are relatively similar between cell types, many of the interactions within these domains can be strikingly dynamic (Dixon et al. 2015; Rao et al. 2014; Ji, Dadon. DB., Powell, BE., Fan, ZF., Borges-Rivera, D. et al. 2015). The variability of the interactions within TADs between cell types can largely be attributed to the subset of the cohesin-mediated
interactions associated with enhancers and promoters. This section discusses the key findings regarding cohesin-mediated CTCF-CTCF loops, enhancer-promoter loops, and loop networks.
CTCF-CTCF loops are of critical importance to the organization of TAD structure and the formation of insulated neighborhoods responsible for the coordination of proper interactions between enhancers and their target promoters. In a human cell line, the knockdown of CTCF led to an increase in interactions that crossed TAD borders, but the overall organization of the TADs did not change (Zuin et al., 2014), whereas another study reported weak but significant effects on the function of TAD boundaries (Sofueva et al., 2013). However, the knockdown approaches used in these studies suffer from an inability to achieve complete knockdown efficiencies possibly explaining why larger effects were not observed. Potentially, other more efficient protein degradation systems may reveal a clearer answer (Yu et al., 2015). In murine embryonic stem cells, the CTCF-CTCF loops forming insulated neighborhood domains were shown to insulate enhancer-associated loops at key pluripotency-enhancer-associated loci containing transcriptional activity and polycomb-silencing. Perturbation of insulated neighborhood boundaries containing the CTCF motif through CRISPR/Cas9-mediated deletion led to the inappropriate expression of genes within and outside of insulated neighborhoods. Additionally, new contacts could be detected between regions inside of the insulated neighborhoods and a promoter outside as measured by 3C when the CTCF boundary deletion was made (Dowen et al., 2014). This study provided direct evidence that the integrity of CTCF-CTCF loop structure is critical for the proper expression of genes within and directly adjacent to the loop. Another study elegantly showed that CRISPR/Cas9 inversion of the
orientation of CTCF binding sites at the Pcdh locus and the beta-globin locus led to changes in CTCF looping direction directly responsible for changes in gene expression and chromatin topology (Guo et al., 2015). However, others point out that not all CTCF sites have insulating behavior towards enhancer-promoter contacts (Sanyal et al., 2012), suggesting that other factors influence the degree of insulation conferred by CTCF. In light of knowledge regarding CTCF’s participation in 3D looping, the degree of insulation may be directly related to the 3D interactions made at the CTCF site and the presence of other local loop structures.
Disruption of CTCF-CTCF loops can lead to disease. A recent study conducted in humans, discovered that limb malformation syndromes were caused by genetic mutations of certain TAD boundaries that contained multiple CTCF binding sites (Lupianez et al., 2015). In the study, the authors found that the observed human limb phenotypes could be reproduced in mice by recapitulating these genetic mutations using CRISPR-Cas9 genome editing. Using 4C interactions sequencing, it was observed that these mutations led to long-range structural changes enabling ectopic interactions between genes within the CTCF-bound TAD boundary and regulatory elements outside of the TAD, and vice-versa. This work confirmed the idea that disruption of CTCF sites involved in topological organization could lead to ectopic interactions and changes in gene expression, and provided evidence that this mechanism was involved in disease. Recent work from the Young laboratory proposed a model in which proto-oncogenes can become activated by genetic mutations that disrupt looping structures because of a loss of enhancer insulation. The work identified common genetic disruptions of such CTCF-CTCF structures in T-ALL patients. Through CRISPR/Cas9-mediated genome editing they could recapitulate
the loss of the loop and the upregulation of the proto-oncogene (Hnisz et al. 2016). Other work from the Bernstein laboratory has provided evidence consistent with this model, where a gain of function mutation in the IDH2 enzyme leads to the productions of an onco-metabolite that interferes with DNA demethylation catalyzed by TET enzymes. Inactivated TET enzymes lead to hypermethylation of a CTCF binding site, which causes a loss of an insulating CTCF loop structure permitting the aberrant interaction of an enhancer with a glioma oncogene. CRISPR-mediated deletion of the CTCF site in WT gliomaspheres phenocopies the aberrant enhancer-promoter interaction and subsequent expression of the oncogene seen in the IDH2 mutant cancer cells (Flavahan et al., 2015). Evidence is mounting to support a role for loop disruption in cancer.
Cohesin also mediates enhancer- and promoter-associated loops that are important in controlling cell state. Such loops ultimately function to bring distal enhancers into close three-dimensional proximity of promoters to allow for gene activation. Enhancers must efficiently coordinate looping interactions with other enhancers and promoters to target and locally concentrate transcriptional machinery in the vicinity of their target promoters (van Berkum and Dekker, 2009; Dowen and Young, 2014; Levine et al., 2014; Smallwood and Ren, 2013; Young, 2011). These cohesin-mediated loops are of critical importance to cell state because cell identity relies on a specific gene expression program orchestrated by enhancer- and promoter-associated genomic interactions (Cubenas-Potts and Corces, 2015; Dowen et al., 2013). Enhancer-promoter loops mediated by cohesin are generally constrained within insulated neighborhood boundaries formed by CTCF-CTCF loops (Dowen et al., 2014; Ji et al., 2015). Enhancer-promoter loops appear to be a
key cell-type-specific class of structures that exist within the framework of insulated neighborhoods demarcated by CTCF-CTCF loops.
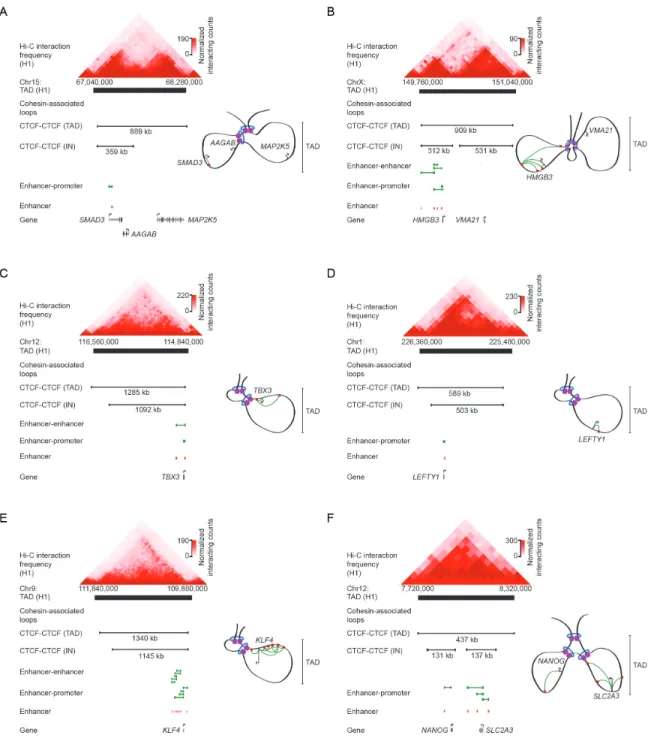
Enhancer-promoter associated loops may serve to integrate signaling pathways at promoters. Super-enhancer regions have been shown to reside inside of insulated neighborhoods with the TSS of their target gene (Dowen et al., 2014; Ji et al., 2015) (Figure 5). These super-enhancers have been shown to integrate multiple signaling pathways at their target promoter (Hnisz et al., 2015). These super-enhancers also exhibit high densities of looping between enhancer constituents and their target promoter. An intriguing possibility is that these 3D super-enhancer structures exist to physically integrate multiple signaling pathways at their target promoters. Little is known about the transience of such loops involved in the integration of signals, but at some examples, the formation of loops associated with cis-regulatory elements have been observed to pre-exist before transcriptional signals arrive or co-occur as a response to signaling cues (Eijkelenboom et al., 2013; Hakim et al., 2011; Jin et al., 2013; Majumder et al., 2008). More studies regarding enhancer-promoter loops will be needed to dissect these mechanisms.
Networks of cohesin-associated loops can form to connect multiple cis-regulatory elements and genes. Promoters can form networks of looping interactions with other promoters and enhancers, forming active transcriptional hubs (Grubert et al., 2015; Heidari et al., 2014) and fitting a model seen under the microscope called transcription factories. It is estimated that thousands of transcription factories exist in a given nucleus (Edelman and Fraser, 2012; Papantonis and Cook, 2013). A single-molecule live imaging study found that the organizational dynamics of single transcription factories appeared to exist transiently in a cycle of assembly, transcriptional bursting, and disassembly. It was observed that the temporal frequencies of such transcription factory cycles correlated with higher levels of expression (Cisse et al., 2013). Looking at such higher-order networks of looping interactions and the temporal dynamics of such loops may reveal further insights into the relationship between gene regulation and looping. Studying the networks and subnetworks of cohesin interactions along with their network dynamics may reveal features of cell state regulation. However, it is important to remember that the
Figure 5: Schematic of KLF4 TAD in naive hESCs. The TAD contains an insulated neighborhood that
constrains enhancer interactions to the region within the insulated neighborhood. Cohesin-associated loop data (black lines) for a TAD-spanning CTCF loop, insulated neighborhood-spanning CTCF-CTCF loop, enhancer-enhancer loops, and enhancer-promoter loops are displayed along with enhancer locations (red).
extent to which multiple loops simultaneously occur in a cell at one allele remains mostly unknown (discussed in Chapter 4).
Regulation of CTCF-CTCF Loops
The basic model of CTCF-CTCF loop formation requires the sequence-specific binding of CTCF to two distant genomic sites at either end of a cohesin-mediated loop (Dowen and Young, 2014c). CTCF functionality can depend on sterically incompatible modifications to CTCF binding sites and post-translational modifications to CTCF. Specifically, CTCF functionality can be perturbed through methylation of the DNA at the CTCF binding site, through post-translational modification of CTCF by poly(ADP-ribose)-associated proteins, and through phosphorylation of CTCF by kinases.
It is well appreciated that DNA methylation of the CTCF motif and the vicinity of a putative CTCF binding site can abrogate CTCF binding (Cubenas-Potts and Corces, 2015; De La Rosa-Velazquez et al., 2007; Phillips and Corces, 2009; Pombo and Dillon, 2015; Sexton and Cavalli, 2015). DNA methylation is controlled by writers such as DNA methyltransferases (DNMT1, 3A, 3B) and is erased by ten-eleven translocation methylcytosine dioxygenases (TET1, 2, 3) (Dawlaty et al., 2014; Leung et al., 2014; Pastor et al., 2013). CTCF binds to hypomethylated sites as methylation of CTCF binding sites obstructs the CTCF-DNA interface, thus, CTCF-CTCF loops tend not to form at methylated DNA sites. Upon investigation of the CTCF binding sites present at CTCF loop anchors of human embryonic stem cells, it was found that these sites were also generally hypomethylated across a panel of 37 human cell/tissue types (Ji et al., 2015). However, analysis of individual sites shows that a large number of differentially
methylated sites exist across cell types spanning the human body (Schultz et al., 2015). Some have suggested that this differential methylation across cell types poorly correlates with CTCF binding (Maurano et al., 2015; Wang et al., 2012), but these studies made conclusions without allelic information, which is required to make these claims definitively. Conversely to CTCF binding, it has been suggested that methylated CTCF binding sites can be bound by the methylation binding domain protein (MBD) Kaiso (also known as ZBTB33) (De La Rosa-Velazquez et al., 2007). Kaiso binds to a methylated CTCF motif upstream of retinoblastoma tumor suppressor gene (De La Rosa-Velazquez et al., 2007) along with methylated CTCF consensus motifs genome-wide (Defossez et al., 2005). This evidence suggests that a methylation-dependent competition of CTCF and Kaiso exists. Kaiso is ubiquitously expressed across the body suggesting that it may play an important role in the regulation of CTCF-CTCF loops. Further allele-biased studies will be needed to reveal the direct relationship between CTCF binding and DNA methylation.
Another regulatory mechanism of CTCF activity is the post-translational modification of CTCF by poly(ADP-ribose) associated proteins. Post-translational PARylation is the addition of Poly(ADP-ribose) (PAR) polymers to a protein. PARylated CTCF, modified by poly(ADP-ribosyl)ation polymerase 1 (PARP1), is more stably bound to DNA, which prevents DNA methylation by DMNT1 (Guastafierro et al., 2013; Yusufzai et al., 2004). Specifically, DNMT1 is inactivated by binding to activated PARP1 associated with CTCF in a complex, sterically preventing DNMT1-mediated DNA methylation, thus stabilizing CTCF binding (Zampieri et al., 2011). In another study, it was shown that the depletion of PAR leads to the accumulation of CTCF and