HAL Id: hal-01114184
https://hal.sorbonne-universite.fr/hal-01114184
Submitted on 3 Nov 2015HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Polarizable Force Fields for Biomolecular Modeling
Yue Shi, Pengyu Ren, Michael Schnieders, Jean-Philip Piquemal
To cite this version:
Yue Shi, Pengyu Ren, Michael Schnieders, Jean-Philip Piquemal. Polarizable Force Fields for Biomolecular Modeling. Reviews in Computational Chemistry, 28 (51), Wiley, 2015, �10.1002/9781118889886.ch2�. �hal-01114184�
1
Polarizable Force Fields for Biomolecular Modeling
1
2
3
Yue Shi, Pengyu Rena 4
a. Department of Biomedical Engineering 5
The University of Texas at Austin 6 1 University Station, C0800 7 Austin, TX 78712 8 9 10 Michael Schniedersb 11
b. Department of Biomedical Engineering, College of Engineering and 12
Department of Biochemistry, Carver College of Medicine 13
The University of Iowa 14
Iowa City, Iowa, 52242 15
16 17
Jean-Philip Piquemalc 18
c. Laboratoire de Chimie Théorique (UMR 7616), 19
UPMC, Sorbonne Universités, 20
CC 137, 4 place Jussieu, 21
75252 Paris Cedex 05, France 22
2
1. Introduction
24
Molecular mechanics based modeling has been widely used in the study of chemical and 25
biological systems. The classical potential energy functions and their parameters are 26
referred to as force fields. Empirical force fields for biomolecules emerged in the early 27
1970's,1 followed by the first molecular dynamics simulations of the bovine pancreatic 28
trypsin inhibitors (BPTI).2-4 Over the past 30 years, a great number of empirical 29
molecular mechanics force fields, including AMBER,5 CHARMM,6 GROMOS,7 OPLS,8 30
and many others, have been developed. These force fields share similar functional forms, 31
including valence interactions represented by harmonic oscillators, point dispersion-32
repulsion for van der Waals (vdW) interactions, and an electrostatic contribution based 33
on fixed atomic partial charges. This generation of molecular mechanics force fields has 34
been widely used in the study of molecular structures, dynamics, interactions, design and 35
engineering. We refer interested readers to some recent reviews for detailed discussions.9, 36
10 37
Although the fixed charge force fields enjoyed great success in many areas, there remains 38
much room for improvement. In fixed charge based electrostatic models, the atomic 39
partial charges are meant to be “pre-polarized” for condensed phases in an averaged 40
fashion, typically achieved by the fortuitous overestimation of electrostatic charges by 41
low-level ab initio quantum mechanics. Such models thus lack the ability to describe the 42
variation in electrostatics due to many-body polarization effects, which have been shown 43
to be a significant component of intermolecular forces.10-12 With the rapid growth of 44
computational resources, there has been increasing effort to explicitly incorporate many-45
body induction into molecular mechanics to improve the accuracy of molecular modeling. 46
3
Classical electrostatics models that take into account polarization appeared as early as the 47
1950s. Barker in his 1953 paper “Statistical Mechanics of Interacting Dipoles” discussed 48
the electrostatic energy of molecules in terms of “permanent and induced dipoles”.13 49
Currently, polarizable models generally fall into three categories: those based on induced 50
point dipoles,9, 14-23 the classical Drude oscillators,24-26 and fluctuating charges.27-30 More 51
sophisticated force fields that are “electronic structure-based”31, 32 or use “machine 52
learning methods”33 also exist, but incur higher computational costs. Discussions of the 53
advantages and disadvantages of each model and their applications will be presented in 54
the following sections. 55
Compared to fixed charge models, the polarizable models are still in a relatively early 56
stage. Only in the past decade or so has there been a systematic effort to develop general 57
polarizable force fields for molecular modeling. A number of reviews have been 58
published to discuss various aspects of polarizable force fields and their development.9, 34-59
40
Here, we focus on the recent development and applications of different polarizable 60
force fields. We begin with a brief introduction to the basic principles and formulae 61
underlying alternative models. Next, the recent progress of several well-developed 62
polarizable force fields is reviewed. Finally, applications of polarizable models to a 63
range of molecular systems, including water and other small molecules, ion solvation, 64
peptides, proteins and lipid systems are presented. 65
1. Modeling Polarization Effects
66
1.1. Induced Dipole Models
4
To describe electrostatic interactions involving polarization, we consider a system 68
consisting of a collection of charge distribution sites located at lone-pair positions, atomic 69
centers and/or molecular centers, depending on the resolution of the model. The total 70
charge distribution at site i is the sum of permanent and induced charge 71
[1] 72
where M represents the charge distribution. This distribution can be a simple point charge, 73
a point multipole expansion with charge, dipole, quadrupole and/or higher order moments, 74
or a continuous charge distribution. While the principles described below are not limited 75
to any particular representation of charge distribution, we will use point multipoles for 76
convenience. 77
The electrostatic interaction energy between two charge sites i and j is given by 78
[2] 79
where T is the interaction operator and is a function of the distance between i and j. In the 80
case of point charge interactions, T is simply 1/r. The work (positive energy) needed to 81
polarize a charge distribution also has a quadratic dependence on the induced charge 82
distribution: 83
[3] 84
where α is the polarizability of site i that includes all orders of polarizability including 85
dipole polarizability.41 Although α is generally treated as an isotropic quantity, as in the 86
5
Applequist scheme 41, ab initio anisotropic polarizability tensors can be derived from 87
quantum mechanical calculations.42, 43 88
The total electrostatic energy is 89
[4] 90
The values of the induced moments minimize the total energy, by satisfying 91 [5] 92 As a result 93 [6] 94
Equation [6] can be solved iteratively to obtain the induced dipoles. The self-consistent 95
calculation is computationally expensive; however it can be accelerated with predictors 96
and non-stationary iterative methods.44 97
Substituting from Eq [5] into Eq [6], the final electrostatic energy becomes 98
[7]
99
where the first term is the permanent electrostatic energy and the second term is the 100
polarization energy. 101
1.2. Classic Drude Oscillators
102
In the Drude oscillator model, the polarization effect is described by a point charge (the 103
Drude oscillator) attached to each non-hydrogen atom via a harmonic spring. The point 104
6
charge can move relative to the attachment site in response to the electrostatic 105
environment. The electrostatic energy is the sum of the pairwise interactions between 106
atomic charges and the partial charge of the Drude particles 107
, 108
[8] 109
where ND and N are the number of Drude particles and non-hydrogen atoms, qD and qC
110
are the charges on the Drude particle and its parent atom, respectively, rD and rC are their
111
respective positions, and kD is the force constant of the harmonic spring between the
112
Drude oscillator and its parent atom. The last term in Equation [8] accounts for the cost 113
of polarizing the Drude particles. 114
The atomic polarizability (α) is a function of both the partial charge on the Drude particle 115
and the force constant of the spring 116
[9] 117
Both the induced-dipole and Drude oscillator approaches benefit from short-range Thole 118
damping to avoid a polarization catastrophe and to produce an anisotropic molecular 119
polarization response.45 120
1.3. Fluctuating Charges
7
The formalism of the fluctuating charge model is based on the charge equilibration 122
(CHEQ) method,46 in which the chemical potential is equilibrated via the redistribution of 123
charge density. The charge-dependent energy for a system of M molecules containing Ni
124
atoms per molecule is expressed as 125
126
(10) 127
where Qi is the partial charge on atomic site i. The χ describes the atomic
128
electronegativity controlling the directionality of electron flow, and J is the atomic 129
hardness that represents the resistance to electron flow to or from the atom. These 130
parameters are optimized to reproduce molecular dipoles and the molecular polarization 131
response. The charge degrees of freedom are typically propagated via an extended 132
Lagrangian formulation:47 133
[11] 134
where the first two terms represent the nuclear and charge kinetic energies, the third term 135
is the potential energy, and the fourth term is the molecular charge neutrality constraint 136
enforced on each molecule i via a Lagrange multiplier λi. The extended Lagrangian
137
approach can also be applied to the induced dipole and Drude oscillator models described 138
earlier. While the extended Lagrangian seems to be more efficient than the iterative 139
method, fictitious masses and smaller time-steps are required to minimize the coupling 140
8
between the polarization and atomic degrees of freedom, which can never be completely 141
eliminated.44 142
A few general force fields have been developed based on these formulas to explicitly 143
treat the polarization effect. We now discuss development highlights for some of the 144
representative force fields. 145
2. Recent Developments
146
2.1. AMOEBA
147
The AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applications) 148
force field, developed by Ponder, Ren and co-workers,15, 18, 37 utilizes atomic multipoles 149
to represent permanent electrostatics and induced atomic dipoles for many-body 150
polarization. The valence interactions include bond, angle, torsion and out-of-plane 151
contributions using typical molecular mechanics functional forms. The van der Waals 152
interaction is described by a buffered-14-7 function. The atomic multipole moments 153
consist of charge, dipole and quadrupole moments, which are derived from ab initio 154
quantum mechanical calculations using procedures such as Stone’s Distributed Multipole 155
Analysis (DMA).48-50 The higher order moments make possible anisotropic 156
representations of the electrostatic potential outside atoms and molecules. The 157
polarization effect is explicitly taken into account via atomic dipole induction. The 158
combination of permanent atomic multipoles and induced dipoles enables AMOEBA to 159
capture electrostatic interactions in both gas and condensed phase accurately. The vdW 160
parameters of AMOEBA are optimized simultaneously against both ab initio gas-phase 161
data and condensed-phase experimental properties. 162
9
In the past decade, AMOEBA has been applied to the study of water,15 monovalent and 163
divalent ions,51-53 small molecules,54, 55 peptides18, 56 and proteins.57-59 AMOEBA 164
demonstrated that a polarizable force field is able to perform well in both gas and 165
solution phases with a single set of parameters. In addition, AMOEBA is the first 166
general-purpose polarizable force field utilized in molecular dynamics simulations of 167
protein-ligand binding and calculation of absolute and relative binding free energies.58-62 168
The computed binding free energies between trypsin and benzamidine derivatives 169
suggests significant non-additive electrostatic interactions as the ligand desolvates from 170
water and enters the protein pocket (see Section 4.4 for further discussion). AMOEBA 171
has recently been extended to biomolecular X-ray crystallography refinement63, 64, and 172
consistently successful prediction of the structure, thermodynamic stability and solubility 173
of organic crystals65 are encouraging. 174
AMOEBA has been implemented in several widely used software packages including 175
TINKER,66 OpenMM,67 Amber,68 and Force Field X.69 The AMOEBA polarizable force 176
field was first implemented within the FORTRAN-based TINKER software package70 177
using Particle Mesh Ewald (PME) for long-range electrostatics. Implementation of the 178
polarizable-multipole Poisson-Boltzmann,71 which depends on the Adaptive Poisson-179
Boltzmann Solver (APBS),72 and generalized Kirkwood73 continuum electrostatics 180
models also exist in TINKER, which is now being parallelized using OpenMP. The 181
algorithms in TINKER are also available from within CHARMM using the MSCALE 182
interface.74, 75 Alternative FORTRAN implementations of AMOEBA using PME are 183
available in the Sander and PMEMD molecular dynamics engines of AMBER,68 with the 184
latter parallelized using MPI. The PME treatment of AMOEBA electrostatics has recently 185
10
been extended within the Java Runtime Environment (JRE) program Force Field X by 186
incorporating explicit support for crystal space group symmetry,63 parallelizing for 187
heterogeneous computer hardware environments63 and supporting advanced free energy 188
methods such as the Orthogonal Space Random Walk (OSRW) strategy.65, 76 These 189
advancements are critical for applications such as AMOEBA-assisted biomolecular X-ray 190
refinement,63, 77 efficient computation of protein-ligand binding affinity,57, 61 and 191
prediction of the structure, stability and solubility of organic crystals.65 Finally, the 192
OpenMM software is working toward a general implementation of AMOEBA using the 193
CUDA GPU programming language.78 194
2.2. SIBFA
195
The SIBFA (Sum of Interactions Between Fragments Ab initio computed) force field for 196
small molecules and flexible proteins, developed by Gresh, Piquemal et. al,79-83 is one of 197
the most sophisticated polarizable force fields because it incorporates polarization, 198
electrostatic penetration 84 and charge-transfer effects.85 199
The polarization is treated with an induced dipole model, in which the distributed 200
anisotropic polarizability tensors43 are placed on the bond centers and on the heteroatom 201
lone pairs. Quadrupolar polarizabilities are used to treat metal centers. The force field is 202
designed to enable the simultaneous and reliable computation of both intermolecular and 203
conformational energies governing the binding specificities of biologically and 204
pharmacologically relevant molecules. Similar to AMOEBA, permanent multipoles are 205
used for permanent electrostatics in SIBFA. Flexible molecules are modeled by 206
combining the constitutive rigid fragments. SIBFA is formulated on the basis of quantum 207
11
chemistry and calibrated on energy decomposition analysis, as oppose to AMOEBA 208
which relies more on condensed-phase experimental data. It aims to produce accurate 209
interaction energy comparable with ab initio results. The development of SIBFA 210
emphasizes separability, anisotropy, nonadditivity and transferability. The analytical 211
gradients for charge-transfer energy and solvation contribution are not yet available in 212
SIBFA although molecular dynamics simulations with a simplified potential have been 213
attempted and will be reported in the near future. 214
SIBFA has been validated on a wide range of molecular systems from water clusters86 to 215
large complexes like metalloenzymes encompassing Zn(II).87-92 It has been used to 216
investigate molecular recognition problems including the binding of nucleic acids to 217
metal ions,93-95 the prediction of oligopeptide conformations,86, 96 and for ligand-protein 218
binding.97 Most of the SIBFA calculations reproduced closely the quantum chemistry 219
results, including both the interaction energy and the decomposed energy terms. At the 220
same time, electrostatic parameters are demonstrated to be transferable between similar 221
molecules. 222
,A Gaussian based electrostatic model (GEM) has been explored as an alternative to 223
distributed point multipole electrostatic representation.98 GEM computes the molecular 224
interaction energies using an approach similar to SIBFA but replacing distributed 225
multipoles by electron densities.99 GEM better captures the short-range effects on 226
intermolecular interaction energies, and it naturally includes the penetration effect. 227
Calculations on a few simple systems like water clusters99 have demonstrated GEM’s 228
capability to reproduce quantum chemistry results. Furthermore, implementating PME 229
for GEM in a PBC showed reasonable computational efficiency thanks to the use of 230
12
Hermite Gaussian functions.100 Therefore, replacing SIBFA’s distributed multipoles with 231
the GEM continuous electrostatic model will be a future direction of methodology 232
development.98 233
2.3. NEMO
234
NEMO (Non-Empirical Molecular Orbital) is a polarizable potential developed by 235
Karlström and co-workers.101-103 The NEMO potential energy function is composed of 236
electrostatics, induction, dispersion and repulsion terms. The induction component is 237
modeled using induced point–dipole moments with recent addition of induced point– 238
quadrupole moments.22 The electrostatics, previously represented by atomic charges and 239
dipoles, has also been extended to include atomic quadrupole moments leading to notable 240
improvement on formaldehyde. The atomic multipole moments are now obtained from ab 241
initio calculation using a LoProp procedure.104 The LoProp is claimed to provide atomic 242
multipoles and atomic polarizabilities that are less sensitive to basis sets than are other 243
methods such as Distributed Multipole Analysis (DMA). Also, NEMO is the only force 244
field that explores the possibility of including interactions between permanent multipoles 245
and higher-order induced multipoles involving higher-order hyperpolarizabilities.22 246
NEMO has demonstrated its ability to describe accurately both inter and intramolecular 247
interactions in small systems, including: glycine dipeptide conformation profiles,105 ion-248
water droplets,106 and urea transition from nonplanar to planar conformation in water.107 249
Its applicability to biomacromolecules is not yet known. 250
2.4. CHARMM-Drude
13
In addition to the induced dipole model, the classical Drude oscillator model is another 252
popular approach for modeling polarization effects.39, 108 Roux, MacKerell and their 253
colleagues have been developing a polarizable CHARMM force field based on this 254
approach. 25, 26, 109, 117 Unlike the induced dipole model, which treats the polarization 255
response using point dipoles, the Drude model represents the polarizable centers by a pair 256
of point charges. A point partial charge is tethered via a harmonic spring for each non-257
hydrogen atom. This point charge (the Drude oscillator) can react to the electrostatic 258
environment and cause the displacement of the local electron density. The atomic 259
polarizability depends on both the Drude particle charge and the harmonic force constant. 260
In MD simulations, the extended Lagrangian is used to evaluate the polarization response, 261
by allowing the Drude particles to move dynamically and experience nonzero forces. 262
Small fictitious masses are assigned to each Drude particle and independent low 263
temperature thermostats are applied to the Drude particle degrees of freedom.118 In case 264
of energy minimization, self-consistent iteration will be required to solve for the 265
polarization. 266
Determining electrostatic parameters for the Drude oscillator is not as straightforward as 267
for induced dipole models. Masses assigned to the Drude particles are chosen empirically. 268
The values for atomic charges and polarizabilities requires a series of calculations of 269
perturbed ESP maps. This force field has been parameterized for water25, 26, and for a 270
series of organic molecules including: alkanes,110 alcohols,111 aromatics,112 ethers,113, 114 271
amides,109 sulfurs,115 and ions.119, 120 An attempt has also been made to combine the 272
Drude-based polarizable force field with quantum mechanics in QM/MM methods.121 It 273
was noted that pair-specific vdW parameters are needed to obtain accurate hydration free 274
14
energies of small molecules using the polarizable force field. This is likely due to the 275
problematic combining rules used to compute the vdW interactions between unlike atoms. 276
The Drude model has been implemented in CHARMM74, 122 and in the NAMD 277
package,123 in which the computational cost is about 1.2 to 1.8 times greater than that of 278
fixed-charge CHARMM.124 279
2.5. CHARMM-FQ
280
The fluctuating charge model (FQ), also known as charge equilibration or 281
electronegativity equalization model, is an empirical approach for calculating charge 282
distributions in molecules. In this formalism, the partial charge on each atom is allowed 283
to change to adapt to different electrostatic environments. The variable partial charges are 284
computed by minimizing the electrostatic energy for a given molecular geometry. 285
Compared with the induced dipole and Drude models, the fluctuating charge models are 286
minimally parameterized and easier to implement because the polarizability is induced 287
without introducing new interactions beyond the point charges. Either extended 288
Lagrangian or self-consistent iteration can be used to compute the fluctuating charges in 289
MD simulations, with similar advantages and disadvantages as discussed above. 290
The CHARMM-FQ force field,125, 126 developed by Patel, Brooks, and their coworkers, 291
has been parameterized for small molecules,28 proteins,28, 127 lipids, lipid bilayers,113, 128 292
and carbohydrates.125 The force field has been applied to investigate liquid–vapor 293
interfaces in addition to biophysical studies.129 There are some known limitations for 294
fluctuating charge models, however, such models allow artificial charge transfer between 295
widely separated atoms but that can be controlled with additional constraints. Also the 296
15
intramolecular charge-flow is limited by the chemical connectivity. It is thus difficult to 297
capture the out-of-plane polarization in molecules such as aromatic benzenes with 298
additional charge sites. The CHARMM-FQ force field has been implemented in the 299
CHARMM software package.74 300
2.6. X-Pol
301
Gao and coworkers proposed the X-Pol framework by combining the fragment-based 302
electronic structure theory with a molecular mechanical force field.31, 32, 130 Unlike the 303
traditional force fields, X-Pol does not require bond stretching, angle, and torsion terms 304
because they are represented explicitly by quantum mechanics. The polarization and 305
charge transfer between fragments are also evaluated quantum mechanically.130 306
Furthermore, X-Pol can be used to model chemical reactions. 307
In X-Pol, large molecular systems are divided into small fragments. Electrostatic 308
interactions within the fragments are treated using the electronic structure theory. The 309
electrostatic interactions between fragments are described by the combined quantum 310
mechanical and molecular mechanical (QM/MM) approach. Also, a vdW term is added to 311
the interfragment interaction as a consequence of omitting electron correlation and 312
exchange repulsion. A double self-consistent-field (DSCF) procedure is used to converge 313
the total electronic energy of the system as well as the energy within the fragments (this 314
includes the mutual polarization effect). 315
The X-Pol potential has been applied to MD simulations of liquid water,131 liquid 316
hydrogen fluoride,132 and covalently bonded fragments.133, 134 This model was recently 317
used in a molecular dynamics simulation of a solvated protein.135 As expected the 318
16
computational efficiency of the X-Pol is in between that of a simple classical force field 319
and a full ab initio method. The solvated trypsin required 62.6 h to run a 5 ps simulation 320
on a single 1.5 GHz IBM Power4 processor. A parallel version of X-Pol is being 321
developed. 322
2.7. PFF
323
Kaminski et al. developed a polarizable protein force field (PFF) based on ab initio 324
quantum theory.136, 137 The electrostatic interaction is modeled with induced dipoles and 325
permanent point charges. With the exception of a dispersion parameter, all other 326
parameters, including the electrostatic charges and polarizabilities, are obtained by fitting 327
to quantum chemical binding energy calculations for homodimers. The dispersion 328
parameters are later refined by fitting to the experimental densities of organic liquids.16 329
Gas-phase many-body effects, as well as conformational energies, are well reproduced,137 330
and MD simulations for real proteins are reasonably accurate at modest computational 331
costs.16, 138 332
To reduce the computational cost, a POSSIM (Polarizable Simulations with Second-order 333
Interaction Model) force field was later proposed, in which the calculation of induced 334
dipoles stops after one iteration.139, 140 The computational efficiency can be improved by 335
almost an order of magnitude by using this formalism. Because the analytical gradients 336
(forces) are unavailable, a Monte-Carlo technique is used in condensed-phase simulations. 337
POSSIM has been validated on selected small model systems, showing good agreement 338
with ab initio quantum mechanical and experimental data. Parameters for alanine and 339
protein backbone have been reported.141 340
17
Polarizable force fields for non-biological systems also exist. A many-body polarizable 341
force field by Smith and coworkers was developed and applied to the simulations of ion 342
conduction in polyethylene oxide (PEO).142-144 Cummings and coworkers developed an 343
interesting Gaussian charge polarizable force field for ions and in polyethylene oxide 344
(PEO).145-147 A polarizable force field for ionic liquids was also reported to provide 345
accurate thermodynamics and transport properties.148 346
3. Applications
347
3.1. Water Simulations
348
Due to its important role in life, water is a natural choice for polarizable force field 349
development. After the polarizable (and dissociable) water model of Stillinger and 350
David,149 more than a dozen polarizable water models have been reported.150 351
Similar to how the polarization models discussed previously, the polarizable water 352
models likewise fall into three major categories. Most belong to the first category, 353
including the Stillinger and David’s water model, SPCP,151 PTIP4P, 152 CKL,153 NCC,154 354
PROL,155 Dang-Chang156 and others. These models all adopted the induced dipole 355
framework to treat polarization, typically using a single polarizable site on water. TTM 356
models157-160 and the AMOEBA water model15 utilize an interactive, distributed atomic 357
polarizability with Thole’s damping scheme45 to treat electrostatics and polarization. The 358
Drude Oscillator-based water models include SWM4-DP,26 and SWM4-NDP,25 as well as 359
the Charge-On-Spring (COS) model,161 and its improved variation.162 The third group 360
includes the SPC-FQ and TIP4P-FQ163 water models that utilize the fluctuating charge 361
scheme to model polarization. The partial charges flow from one atom to another, and the 362
18
total charge of a water molecule need not be zero. Stern et al. proposed a unique water 363
model (POL5) by combining the fluctuating charge with the point induced dipole 364
scheme.164 Several more sophisticated polarizable water models based on quantum 365
mechanics were developed based on quantum mechanics, including QMPFF,165 DPP2,166 366
and Polarflex.167 For example, the charge penetration, induction, and charge transfer 367
effects have been incorporated into the DPP2 (Distributed Point Polarizable Model) 368
model which reproduces well the high-level ab initio energetics and structures for large 369
water clusters. 370
An advantage of a polarizable water model over most non-polarizable models is the 371
ability to describe the structure and energetics of water in both gas and condensed phases. 372
Water dimer interaction energies, the geometry of water clusters and the heat of 373
vaporization of neat water can be reproduced well by most polarizable models. Some 374
highly parameterized nonpolarizable force fields such as TIP5P, TIP4P-EW and 375
TIP4P/2005 actually perform as well or better than some polarizable force fields over a 376
range of liquid properties, including the density-temperature profile, radial distribution 377
function, and diffusion coefficient. However, for water molecules experiencing 378
significant changes in environment, e.g., from bulk water to the vicinity of ions or 379
nonpolar molecules, only the polarizable models can capture the change of water dipole, 380
structure and energetics.168 381
Polarization water models are being extended and applied to other phases as well as to 382
the interface between different phases. Rick et al recently incorporated charge transfer 383
into their polarizable water model that was then used to study ice/water coexistence 384
properties and properties of the ice Ih phase.169 The POL3 water model14, 170 was used to 385
19
study the ice-vapor interface, and to calculate the melting point of ice Ih. Bauer and Patel 386
used the TIP4P-QP model to study the liquid-vapor coexistence.171 387
3.2. Ion Solvation
388
Ions are an important component in many chemical and biological systems. Nearly half 389
of all proteins contain metal ions, and they play essential roles in many fundamental 390
biological functions. Some metal ions are critical for both protein structure and function. 391
In enzymes, ions can bind and orient the substrates through electrostatic interactions at 392
the active sites, thus controlling catalytic reaction. Divalent ions are vital in nucleic acid 393
structures. Modeling ion-water and ion-biomolecule interactions accurately is very 394
important. 395
Due to the high electron density and small sizes of ions, the non-polarizable models fail 396
to capture the structural details adequately and do not or to reproduce the atomic dipole 397
of water around the ions.172-176 Several studies of ion solvation have been reported using 398
different polarizable models51-53, 116, 120, 177-187 with analyses focused on solvation 399
structures, charge distribution, and binding energies. Noted that no straightforward 400
experimental measurement of hydration free energy data exist because the macroscopic 401
system must be neutral. Different assumptions are used to decompose the experimental 402
hydration free energy into single ion contributions. The hydration free energy of some 403
monovalent ions such as Na+ and K+ from different sources can vary by as much as 10 404
kcal/mol. It is more reliable to compare the hydration free energy of the whole salt and 405
the relative energy between cations or anions. 406
20
The AMOEBA polarizable force field has been used to model a number of anions and 407
cations, including Na+, K+, Mg++, Ca++, Zn++, Cl-, Br-, and I-.51-53, 188 Parameters for these 408
ions, including the vdW parameters and polarization damping coefficients (for divalent 409
ions only), were obtained by fitting to the ab initio QM interaction energy profiles of ion-410
water pairs. Molecular dynamics simulations were then performed to evaluate the ion-411
cluster solvation enthalpies and solvation free energies.51-53, 188 The excellent agreement 412
between calculated and experimental hydration free energy, often within 1%, demonstrate 413
that polarizable force fields are transferable between phases. Ab initio energy 414
decomposition using, e.g., the Constrained Space Orbital Variations (CSOV) method,99, 415
189
have also been applied to examine the polarization component of the ion-water 416
interaction energy and to guide the force field parameterization.53, 190 More recently, the 417
AMOEBA force field was used to model the hydration of high valent Th(IV)94 and 418
studies on open-shell actinides are underway. 419
The SIBFA model was used to examine Pb(II),191 lanthanides (La(III) and Lu(III)) and 420
actinides (Th(IV)) in water.94 SIBFA-predicted interaction energies generally matched 421
well with the ab initio results, including the energy decompositions. Lamoureux and 422
Roux developed the CHARMM polarizable force field for alkali and halide ions based on 423
the Drude Oscillator.177 Hydration free energies, calculated via thermodynamic 424
integration,192 showed an encouraging agreement with experiment. 425
3.3. Small Molecules
426
Small molecules are building blocks of biomolecules and serve as substrates and 427
inhibitors. Abundant experimental measurements on various physical and chemical 428
21
properties exist for common organic molecules which in turn are used in the 429
parameterization of the force fields. Polarizable and non-polarizable force fields can 430
usually produce reasonable estimations of physical properties of neat liquids.193-196 431
Extensive studies using polarizable force fields, covering major functional group, 432
including alkanes, alcohols, aldehydes, ketones, ethers, acids, aromatic compounds, 433
amines, amides, and some halogen compounds have been reported.28, 36, 55, 110, 112, 126, 197-434
199
Calculations of structure, dipole moment, heterodimer binding energy, liquid diffusion 435
constant, density, heat of vaporization, and hydration free energy are usually performed 436
to assess the quality of force field parameters. 437
The electrostatic multipole parameters in AMOEBA were derived using the DMA 438
procedure. They can be further optimized to the electrostatic potentials of chosen ab 439
initio theory and basis sets. The AMOEBA valence parameters were derived from ab
440
initio data such as molecular geometries and vibrational frequencies of the gas-phase
441
monomer. The vdW parameters are estimated from gas-phase cluster calculations, and 442
subsequently refined in liquid simulations using experimental data (e.g., densities and 443
heats of vaporization). The torsional parameters the last obtained during the 444
parameterization scheme are derived by fitting to ab initio QM conformational energy 445
profiles. An automated protocol (PolType) that can generate AMOEBA parameters for 446
small molecules is under development.200 Because force field parameterization is a 447
tedious process, such an automated tool is convenient and reduces the likelihood of 448
human error. 449
The CHARMM-Drude force field developers devoted much of their efforts on organic 450
compounds. Their parameterization scheme starts from an initial guess of charge (based 451
22
on the CHARMM22 force field), and invokes changes at some lone pair sites. Those 452
parameters are then fit to a series of “perturbed” ESP maps. The vdW parameters are then 453
optimized to match neat liquid properties as is done many other force fields.115 Overall, a 454
systematic improvement over the CHARMM22 additive force field has been observed for 455
both gas-phase and condensed-phase properties. These studies on small molecules lay the 456
groundwork for developing a Drude-based polarizable force field for proteins and nucleic 457
acids. 458
3.4. Proteins
459
One of the goals for polarizable force fields is to model accurately protein structures, 460
dynamics, and interactions. Proteins are a ubiquitous class of biopolymers whose 461
functionalities depend on the details of their 3D structures, which, in turn, are largely 462
determined by their amino acid sequences. Fixed-charge force fields for proteins, like 463
AMBER, CHARMM, and OPLS-AA, have been developed and for years subjected to 464
various tests and validations. The development of polarizable protein force fields is still 465
in its infancy. Although the importance of including polarization effects was recognized 466
long ago, polarizable protein force fields emerged only in the past decade.9, 21, 28, 29, 37, 138, 467
201-205 468
The use of polarizable electrostatics in protein simulations dates back to 1976,1 when 469
Warshel and Levitt simulated lysozyme via single point calculations. Kaminski et al. 470
reported in 2002 an ab initio polarizable protein force field (PFF) based on inducible 471
dipoles and point charges.16,137 Simulations on bovine pancreatic trypsin inhibitor using 472
PDFF showed a satisfactory root mean square displacement (RMSD) compared to the 473
23
experimental crystal structure and polarization was found to affect the solvation 474
dynamics.138 The fluctuating-charge based ABEEM/MM force field was used to examine 475
protein systems like trypsin inhibitors206 and the heme prosthetic group.207 The SIBFA 476
force field has been used to study the interaction between focal adhesion kinase (FAK) 477
and five pyrrolopyrimidine inhibitors.208 The energy balances accounting for the 478
solvation/desolvation effects calculated by SIBFA agree with experimental ordering. 479
Water networks in the binding pocket were shown to be critical in terms of binding 480
affinity. Moreover, the polarization contribution was considered as an indispensable 481
component during the molecular recognition. In comparison, the continuum reaction field 482
procedure fails to reproduce these properties. In addition kinases, the SIBFA protein 483
force field has been used to study a variety of metalloproteins encompassing cations such 484
as Cu+, Zn++, Ca++ or Mg++, as well as enabling inhibition studies.91, 209-211 Future 485
molecular dynamics simulations should extend the applicability of SIBFA to protein-486
ligand binding. 487
Ren and coworkers have been systematically developing the AMOEBA protein force 488
field, and using it to study to several protein systems to understand protein-ligand 489
binding.57-59, 61 More recently an X-Pol force field for proteins has been developed and 490
demonstrated in a simulation of solvated trypsin.32 491
The first attempt to compute the protein-ligand binding free energy using a polarizable 492
force field was made on the trypsin-benzamidine systems using AMOEBA.57, 61, 62 The 493
absolute binding free energy of benzamidine to trypsin, and the relative binding free 494
energies for a series of benzamidine analogs, were computed using a rigorous alchemical 495
transformation. AMOEBA was successful in evaluating the binding free energies 496
24
accurately with an average error well within 1.0 kcal/mol. A similar study on trypsin, 497
thrombin and urokinase was reported using another ab initio QM-based polarizable force 498
field.212 A thermodynamic integration scheme was used to compute the relative binding 499
free energies, which were in excellent agreement with experimental data (root mean 500
squre error (RMSE)=1.0 kcal/mol). 501
AMOEBA was later used to examine an “entropic paradox” associated with ligand 502
preorganization discovered in a previous study of conformationally constrained 503
phosphorylated-peptide analogs that bind to the SH2 domain of the growth receptor 504
binding protein 2 (Grb2).59 The paradox refers to the unusual trend in which the binding 505
of unconstrained peptides (assumed to lose more entropy upon binding) is actually more 506
favorable entropically than are the constrained counterparts. AMOEBA correctly 507
reproduced the experimental trend and at the same time repeated a mechanism in which 508
the unconstrained peptide ligands were “locked” by intramolecular nonbonded 509
interactions. The simulations uncovered a crucial caveat that had not been previously 510
acknowledged regarding the general design principle of ligand preorganization, which is 511
presumed by many to have a favorable effect on binding entropy. 512
More recently, Zhang et al. demonstrated the ability of AMOEBA in dealing with 513
systems with a metal ion.58 Those authors studied the Zinc-containing matrix 514
metalloproteinases (MMPs) in a complex with an inhibitor where the coordination of 515
Zn++ waswith organic compounds and protein side chains. Polarization was found to play 516
a key role in Zn++ coordination geometry in MMP. In addition, the relative binding free 517
energies of selected inhibitors binding with MMP13 were found to be in excellent 518
agreement with experimental results. As with the previous trypsin study, it was found that 519
25
binding affinities are likely to be overestimated when the polarization between ligands 520
and environments is ignored. 521
Having a more rigorous physical model for treating polarization, the ability to model 522
protein-ligand interactions has been improved significantly. Systems involving highly 523
charged species, like metal ions, can now be treated with confidence. This in turn, 524
provides tremendous opportunities for investifating important proteins for drug discovery 525
and for protein engineering. 526
3.5. Lipids
527
With the rapid development of computational resources, simulations of large systems like 528
lipid bilayers with membrane proteins is feasible.126, 213 Patel and coworkers have been 529
developing a polarizable force field for biomembranes to study the structure and 530
dynamics of ion channel systems.40, 113, 128, 214 Simulations of solvated DMPC 531
(dimyristoyl phosphatidylcholine) and dipalmitoylphosphatidylcholine (DPPC) bilayers 532
were reported.113, 214 The distribution of the membrane components along the lipid bilayer 533
is similar to that from a fixed charge model. The water dipole moment was found to 534
increase from about 1.9 Debye in the middle of the membrane plane to the average bulk 535
value of 2.5~2.6 Debye. The lipid surface computed with the polarizable force field was 536
not improved from those of non-polarizable ones however. In addition, ion permeation in 537
a gramicidin A channel embedded in a DMPC bilayer was investigated.113 Davis and 538
Patel concluded that including the electronic polarization lowered the ion permeation free 539
energy barrier significantly, from 12 kcal/mol to 6 kcal/mol. 540
3.6. Continuum Solvents for Polarizable Biomolecular Solutes
26
A continuum solvent replaces explicit atomic details with a bulk, mean-field response. It 542
is possible to demonstrate from statistical mechanics that an implicit solvent potential of 543
mean force (PMF) exists, wihch preserves exactly the solute thermodynamic properties 544
obtained from explicit solvent.215 It is possible to formulate a perfect implicit solvent in 545
principle, but in practice approximations are necessary to achieve efficiency. This 546
remains an active area of research.216 An implicit solvent PMF can be formulated via a 547
thermodynamic cycle that discharges the solute in vapor, grows the uncharged (apolar) 548
solute into a solvent and finally recharges the solute within a continuum 549
dielectric 550
[12] 551
The continuum electrostatic energy, including mobile electrolytes, can be described by 552
either the nonlinear Poisson-Boltzmann Equation (NPBE) or the simplified linearized 553
Poisson-Boltzmann Equation (LPBE) 554
[13] 555
where the coefficients are a function of position r, is the potential, is the pemittivity, 556
is the modified Debye-Hückel screening factor, and is the solute charge density.217, 557
218
Implementations of a Poisson-Boltzmann continuum for many-body quantum 558
mechanical potentials have been applied to small molecules for decades. Examples 559
include the Polarizable Continuum Model (PCM) 219, 220, COSMO 221 and the Solvent 560
Model series (SMx).222 In contrast, applications of biomolecular continuum electrostatics 561
have been limited mainly to fixed partial charge solute descriptions for reasons of 562
computing efficiency force field availability. However, as a result of increasing 563
27
computational power and the completion of the polarizable force fields for biomolecules 564
described above, the coupling of classical many-body potentials to continuum 565
electrostatics is now possible. 566
An important initial demonstration of polarizable biomolecules within a Poisson-567
Boltzmann continuum used the Polarizable Force Field (PFF) of Maple et al. to model 568
protein-ligand interactions.223 A second demonstration used the Electronic Polarization 569
from Internal Continuum (EPIC), which accounts for intramolecular polarization using a 570
continuum dielectric.224, 225 Finally, the polarizable multipole Poisson-Boltzmann (PMPB) 571
model based on the AMOEBA force field demonstrated that the self-consistent reaction 572
field (SCRF) of proteins within a continuum solvent is consonant with the ensemble 573
average response of explicit solvent.71 Contrarily, end-state calculations of protein-ligand 574
binding affinity using the PMPB model were shown to not recapitulate explicit solvent 575
alchemical free energies to chemical accuracy.61 This motivates development of analytic 576
continuum electrostatics (discussed next), which are fast enough to allow binding 577
affinities to be computed using alchemical sampling, rather than merely relying on end-578
states. A key advantage of EPIC is that the biomolecular self-consistent field (SCF) is 579
determined by a single numerical finite-difference (FD) solution of the PBE, unlike the 580
aforementioned atom-centered PFF and PMPB models that require a new solution for 581
each SCF iteration. However, a tradeoff of EPIC’s efficiency gain is a reduction in model 582
flexibility because electrostatic masking rules cannot be incorporated into the FD solver 583
(i.e., the permanent field due to 1-2 or 1-3 interactions cannot be neglected). Although 584
masking of short-range bonded interactions is the standard approach used by essentially 585
all biomolecular force fields, this is not possible for an EPIC style energy model. 586
28
The first example of an analytic continuum electrostatic model for polarizable 587
biomolecules is the generalized Kirkwood (GK) model for the AMOEBA force field.73 588
The AMOEBA/GK approach has been combined with alchemical sampling to predict 589
trypsin-ligand binding affinity with a correlation coefficient of 0.93. This is a significant 590
improvement over the PMPB end-state approach.226 A second example, based on the 591
ABEEMσπ fluctuating charge force field combined with a generalized Born (GB)
592
continuum electrostatic model, showed promising results for the computation of solvation 593
free energies for small organic molecules and peptide fragments.227 594
3.7. Macromolecular X-ray Crystallography Refinement
595
X-ray crystallography is the dominant experimental method for determining the 3-596
dimensional coordinates of macromolecules. Collected diffraction data is the Fourier 597
transform of the ensemble average electron density of the macromolecular crystal. While 598
reciprocal space amplitudes of Bragg diffraction peaks are measured, their phases are not. 599
Instead, phase information is derived from the Fourier transform of a model structure that 600
is sufficiently close to the actual experimental ensemble. This is known as molecular 601
replacement (MR). After an initial model has been built into the electron density, further 602
refinement is based optimizating a target function of the form 603
[14] 604
where evaluates the agreement between measured and calculated diffraction 605
amplitudes, restrains the model using prior knowledge of intra- and 606
intermolecular chemical forces and weights the relative strength of the two terms.77, 607
29 228
We now focus on the evolution of the prior chemical knowledge used during the X-608
ray refinement process, and we culminate in ongoing work using polarizable force fields 609
in combination with PME electrostatics algorithms to obtain the most accurate, 610
informative biomolecular models possible. 611
The first application of molecular mechanics to macromolecular X-ray crystallography 612
refinement (based on fixed partial charge electrostatics evaluated using a spherical cutoff) 613
was on influenza-virus hemagglutinin by Weis et al. in 1990.229 This work demonstrated 614
that electrostatics maintained chemically reasonable hydrogen-bonding, although charged 615
surface residues were sometimes observed to form incorrect salt bridges.229 The latter 616
observation highlights the importance of accounting for dielectric screening arising from 617
the heterogeneous distribution of solvent within a macromolecular crystal, by using one 618
of the above described continuum electrostatics models. For example, the generalized 619
Born (GB) model for fixed charge electrostatics has been described, albeit with a 620
spherical cutoff approximation.230 Comparing refinements with and without GB 621
screening showed that roughly 10% of the amino acid side-chain conformations were 622
altered, with 75% of these side-chain differences due to residues at the macromolecular 623
surface.230 Although these first applications of fixed charge force field electrostatics were 624
encouraging, the use of spherical cutoffs to approximate crystal lattice sums is now 625
known to be only conditionally convergent and therefore prone to a variety of artifacts.231 626
In 1921, Ewald introduced an absolutely convergent solution to the problem of evaluating 627
electrostatic lattice summations in crystals. He did this by separating the problem into a 628
short-ranged real space sum and a periodic, smoothly varying, long-range sum that can be 629
evaluated efficiently in reciprocal space.232 This approach, now known as Ewald 630
30
summation, has been described for both fixed partial charges and atomic multipoles.233, 631
234
More recently, the efficiency of Ewald summation was improved via the particle-mesh 632
Ewald (PME) algorithm, wherein the reciprocal space summation leverages the fast 633
Fourier transforms (FFT)235 via b-Spline interpolation236 for both fixed partial charge and 634
atomic multipole descriptions.237 635
The speed of the PME algorithm has been further improved for crystals by incorporating 636
explicit support for space group symmetry and by parallelization for heterogeneous 637
computer architectures.63 Combining the polarizable AMOEBA force field with 638
electrostatics evaluated using PME has been shown to improve macromolecular models 639
from X-ray crystallography refinement in a variety of contexts.64, 77, 238-240 At high 640
resolution (~1 Å or lower), the information contained within a polarizable atomic 641
multipole force field can be used to formulate the electron density of the scattering model 642
( ), in addition to contributing chemical restraints ( ).64, 238 The importance 643
of the prior chemical information contained in a polarizable force field is most significant 644
when positioning parts of the model that are not discernable from the experimental 645
electron density, as in the orientation of water hydrogen atoms239 or secondary structure 646
elements for mid-to-low resolution data sets (~3-4 Å).63 647
Let use consider an example, the AMOEBA-assisted biomolecular X-ray refinement with 648
electrostatics evaluated via PME in the program Force Field X. This program was used to 649
re-refine nine mouse and human DNA methyltransferase 1 (Dnmt1) data sets deposited in 650
the Protein databank (PDB). Significant improvements in model quality (presented in 651
Table 1) were achieved as assayed by the MolProbity 241 structure validation tool. The 652
MolProbity score is calibrated to reflect the expected resolution of the X-ray data. After 653
31
re-refinement, the average MolProbity score was reduced to 2.14, indicating a level of 654
model improvement consistent with collecting data to 0.67 Å higher resolution. For 655
example, the pose of S-adenosyl-L-homocysteine (SAH) from mouse (3PT6) and human 656
(3PTA) structures differed by an RMSD of 1.6 Å before re-refinement, but only 0.9 Å 657
afterwards. 658
Table 1. DNA Methyltransferase 1 (Dnmt1) Models Before and After Polarizable X-Ray 659
Refinement with the Program Force Field X. 660
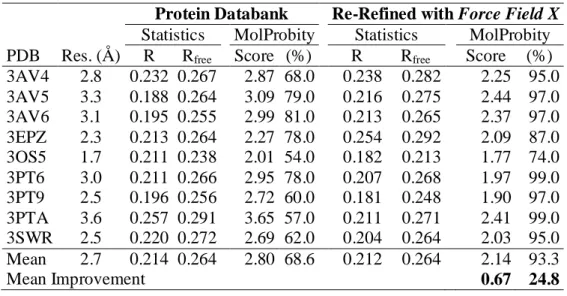
Protein Databank Re-Refined with Force Field X Statistics MolProbity Statistics MolProbity PDB Res. (Å) R Rfree Score (%) R Rfree Score (%) 3AV4 2.8 0.232 0.267 2.87 68.0 0.238 0.282 2.25 95.0 3AV5 3.3 0.188 0.264 3.09 79.0 0.216 0.275 2.44 97.0 3AV6 3.1 0.195 0.255 2.99 81.0 0.213 0.265 2.37 97.0 3EPZ 2.3 0.213 0.264 2.27 78.0 0.254 0.292 2.09 87.0 3OS5 1.7 0.211 0.238 2.01 54.0 0.182 0.213 1.77 74.0 3PT6 3.0 0.211 0.266 2.95 78.0 0.207 0.268 1.97 99.0 3PT9 2.5 0.196 0.256 2.72 60.0 0.181 0.248 1.90 97.0 3PTA 3.6 0.257 0.291 3.65 57.0 0.211 0.271 2.41 99.0 3SWR 2.5 0.220 0.272 2.69 62.0 0.204 0.264 2.03 95.0 Mean 2.7 0.214 0.264 2.80 68.6 0.212 0.264 2.14 93.3 Mean Improvement 0.67 24.8 661
Figure 1. Polarizable biomolecular X-ray refinement on two Dnmt1 data sets. The left 662
panel shows the deposited pose of SAH from data sets 3PT6 (mouse, grey) and 3PTA 663
(human, cyan) do not agree (coord. RMSD 1.6 Å). In the right panel, the poses of SAH 664
from mouse and human structures are more consistent (coord. RMSD 0.9 Å) after Force 665
32
Field X refinement.
666
3.8. Prediction of Organic Crystal Structure, Thermodynamics and
667
Solubility
668
It was emphasized in 1998 that predicting crystal structures from chemical composition 669
remained a major unsolved challenge.242 Significant progress has been made since then to 670
address this challenge, as evidenced by successes of the 4th and 5th blind tests of crystal 671
structure prediction (CSP) organized by the Cambridge Crystallographic Data Center 672
(CCDC).243, 244 Prediction of crystal structures is important in the pharmaceutical industry, 673
where extensive experimental screens are necessary to explore the range of stable 674
polymorphs a molecule may form. The unique three-dimensional molecular packing of 675
each polymorph determines its physical properties such as stability and bioavailability. 676
For this reason, both FDA approval and patent protection are awarded to a specific 677
crystal polymorph, rather than to the molecule itself. To illustrate this point, eight 678
companies have filed eleven patents on five possible crystal forms of the molecule 679
cefdinir.245 680
Prediction of thermodynamically stable crystal structures from chemical composition 681
requires a potential energy function capable of distinguishing between large numbers of 682
structures that are closely spaced in thermodynamic stability.246, 247 In this section, we 683
restrict our focus to energy models that explicitly account for electronic polarization 684
classically65, 248, 249 and neglect the more expensive electronic structure methods 685
sometimes used to (re)score favorable structures.250 686
The vast majority of CSP has been limited to using intermolecular potentials that lack 687
explicit inclusion of polarization,249, 251 although its importance has become a topic of 688
33
interest35, 252-254. Non-polarizable force fields, based on fixed partial charges or fixed 689
atomic multipoles, must implicitly account for the 20% to 40% of the lattice energy 690
attributable induction.249 On the other hand, polarizable models such as the AMOEBA 691
force field for organic molecules 54, 255 based on the Thole damping scheme45 and the 692
Williams-Stone-Misquitta (WSM) method256, 257 for obtaining distributed polarizabilities 693
allow one to include polarization during CSP explicitly. 694
Beyond polarization, modeling the conformational flexibility and corresponding 695
intermolecular energetics of organic molecules via sampling methods such as molecular 696
dynamics is essential to predicting the thermodynamic properties of crystals.258 For 697
example, the structure, stability and solubility of n-alkylamide crystals, from acetamide 698
through octanamide, can be predicted by an alchemical sampling method to compute the 699
sublimation/deposition phase transition free energy.65 700
4. Summary
701
Significant progress has been made in the past decade in developing general-purpose 702
polarizable force fields. Polarizable force fields have exhibited success in disparate 703
research areas including ion solvation, protein-ligand interactions, ion channels and lipids, 704
macromolecular structural refinement and so on. There remain plenty of challenges ahead. 705
The importance of polarization still needs to be established systematically for a wide 706
range of biological systems. While polarizable force fields in principle have better 707
transferability than do non-polarizable force fields, they are also expected to also perform 708
better in a broader range of systems, making parameterization a more elaborate process. 709
In addition to polarization, treatment of other physical effects, including high-order 710
34
permanent charge distributions interactions, short-range electrostatic penetration and 711
charge-transfer effects need further improvement to advance the overall quality of 712
classical electrostatic models. Because computational efficiency (including the need for 713
parallelization) has been a major barrier to the adoption of polarizable force fields, better 714
and more efficient algorithms are also required to advance the application of polarizable 715
force fields. A future area for advancement is to combine the polarizable force fields with 716
fixed-charge force fields in a multiscale fashion, as is done with QM/MM. Technically 717
this can be achieved straightforwardly but caution is needed to ensure the interactions 718
across the two resolutions are balanced. 719
Acknowledgement.
The authors are grateful to the support provided by Robert A. Welch 720Foundation (F-1691). 721
35 REFERENCES
723
1. A. Warshel and M. Levitt, Theoretical Studies of Enzymic Reactions - Dielectric, 724
Electrostatic and Steric Stabilization of Carbonium-Ion in Reaction of Lysozyme. Journal
725
of Molecular Biology, 1976. 103(2): p. 227-249. 726
2. J.A. Mccammon, B.R. Gelin, and M. Karplus, Dynamics of Folded Proteins. Nature, 1977. 727
267(5612): p. 585-590. 728
3. M.A. Spackman, The Use of the Promolecular Charge Density to Approximate the 729
Penetration Contribution to Intermolecular Electrostatic Energies. Chemical Physics
730
Letters, 2006. 418(1-3): p. 158-162. 731
4. D. Nachtigallova, P. Hobza, and V. Spirko, Assigning the Nh Stretches of the Guanine 732
Tautomers Using Adiabatic Separation: CCSD(T) Benchmark Calculations. Journal of
733
Physical Chemistry A, 2008. 112(9): p. 1854-1856. 734
5. W.D. Cornell, P. Cieplak, C.I. Bayly, I.R. Gould, K.M. Merz, D.M. Ferguson, D.C. 735
Spellmeyer, T. Fox, J.W. Caldwell, and P.A. Kollman, A 2nd Generation Force-Field for the 736
Simulation of Proteins, Nucleic-Acids, and Organic-Molecules. Journal of the American
737
Chemical Society, 1995. 117(19): p. 5179-5197. 738
6. A.D. MacKerell, D. Bashford, M. Bellott, R.L. Dunbrack, J.D. Evanseck, M.J. Field, S. 739
Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F.T.K. Lau, C. 740
Mattos, S. Michnick, T. Ngo, D.T. Nguyen, B. Prodhom, W.E. Reiher, B. Roux, M. 741
Schlenkrich, J.C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, 742
and M. Karplus, All-Atom Empirical Potential for Molecular Modeling and Dynamics 743
Studies of Proteins. Journal of Physical Chemistry B, 1998. 102(18): p. 3586-3616.
744
7. H. Valdes, K. Pluhackova, M. Pitonak, J. Rezac, and P. Hobza, Benchmark Database on 745
Isolated Small Peptides Containing an Aromatic Side Chain: Comparison between Wave
36
Function and Density Functional Theory Methods and Empirical Force Field. Physical
747
Chemistry Chemical Physics, 2008. 10(19): p. 2747-2757. 748
8. W.L. Jorgensen, D.S. Maxwell, and J. Tirado-Rives, Development and Testing of the OPLS 749
All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids.
750
Journal of the American Chemical Society, 1996. 118(45): p. 11225-11236. 751
9. J.W. Ponder and D.A. Case, Force Fields for Protein Simulations. Advances in Protein 752
Chemistry, 2003. 66: p. 27-85. 753
10. J. Rezac, P. Jurecka, K.E. Riley, J. Cerny, H. Valdes, K. Pluhackova, K. Berka, T. Rezac, M. 754
Pitonak, J. Vondrasek, and P. Hobza, Quantum Chemical Benchmark Energy and 755
Geometry Database for Molecular Clusters and Complex Molecular Systems
756
(Www.Begdb.Com): A Users Manual and Examples. Collection of Czechoslovak Chemical
757
Communications, 2008. 73(10): p. 1261-1270. 758
11. J. Rezac, K.E. Riley, and P. Hobza, S66: A Well-Balanced Database of Benchmark 759
Interaction Energies Relevant to Biomolecular Structures. Journal of Chemical Theory
760
and Computation, 2011. 7(8): p. 2427-2438. 761
12. K. Berka, R. Laskowski, K.E. Riley, P. Hobza, and J.i. Vondrášek, Representative Amino 762
Acid Side Chain Interactions in Proteins. A Comparison of Highly Accurate Correlated Ab
763
Initio Quantum Chemical and Empirical Potential Procedures. Journal of Chemical Theory
764
and Computation, 2009. 5(4): p. 982-992. 765
13. J.A. Barker, Statistical Mechanics of Interacting Dipoles. Proceedings of the Royal Society 766
of London. Series A, Mathematical and Physical Sciences, 1953. 219(1138): p. 367-372. 767
14. J.W. Caldwell and P.A. Kollman, Structure and Properties of Neat Liquids Using 768
Nonadditive Molecular Dynamics: Water, Methanol, and N-Methylacetamide. Journal of
769
Physical Chemistry, 1995. 99: p. 6208-6219. 770