Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development
Texte intégral
Figure
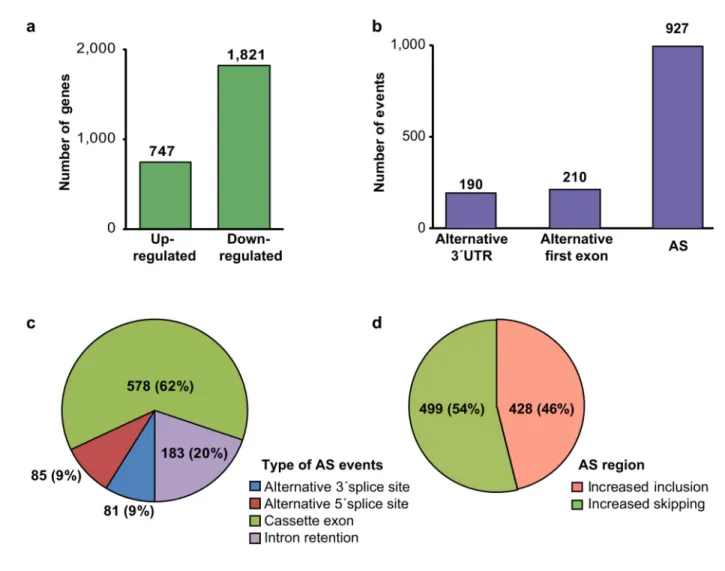
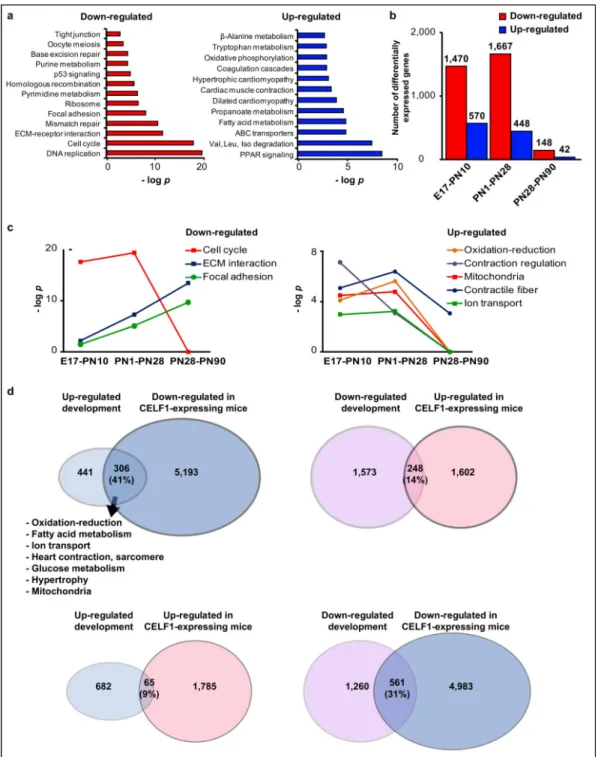
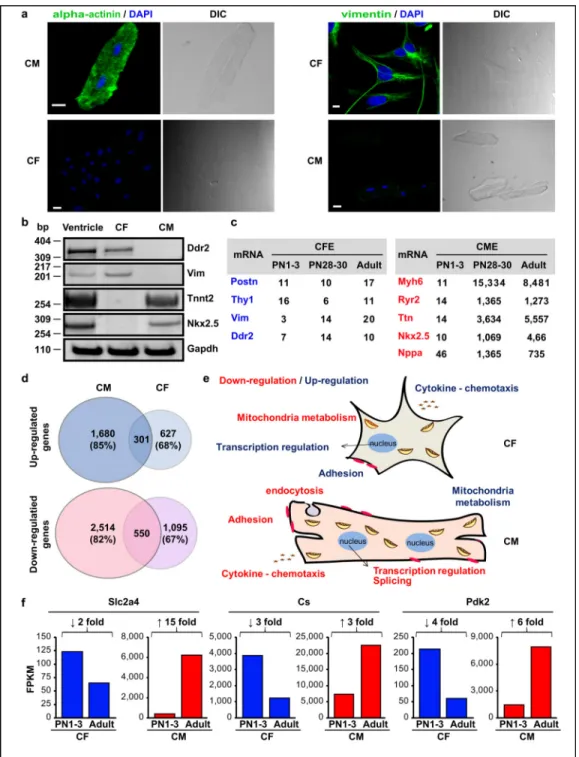
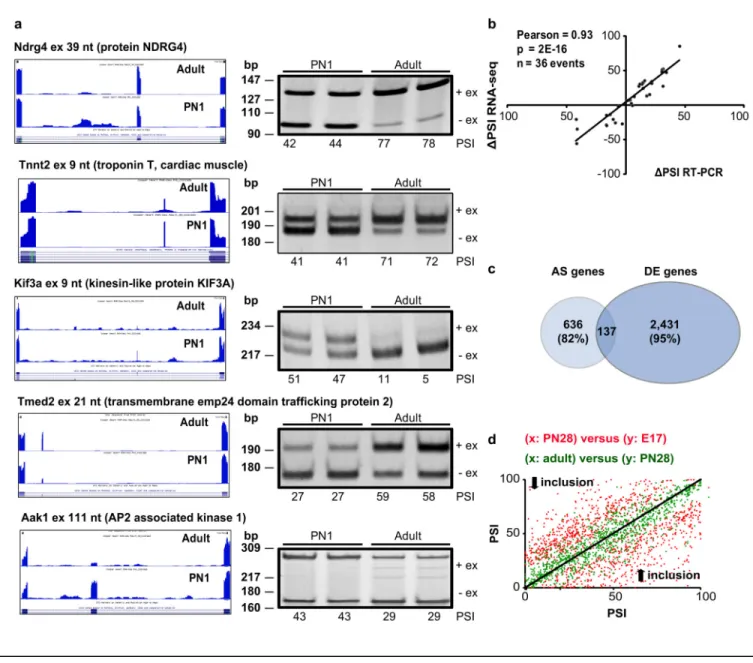
Documents relatifs
The goal of the present study was to provide quantita- tive information about the postnatal cellular maturation of the rat amygdala. Our major findings are as follows: 1)
Note that the total number of neurons (mature + immature) remains constant throughout postnatal development. Figure 5: Numbers of oligodendrocytes in the six main nuclei of the monkey
Apelin and apelin receptor (APJ) gene expression in myocardial tissue mRNA extracts from young (24 days) postnatally normal-fed (NF) or overfed (OF) mice.. The mRNA expression of
In contrast, specific maximal force (relative maximal force per unit of muscle mass was decreased in all 6-month old male and female KO mice, except in 6-month old female KO Grobet
Summary ― Changes in the concentrations of cholecystokinin, gastric inhibitory peptide, gastrin, motilin, pancreatic polypeptide, secretin, somatostatin,
Coupez et collez le prochain image qui devrait apparaitre dans la suite1.
No changes in excitatory Purkinje cell synapses in Lgals3 knockout mice. (C and D) Immunostaining for the Purkinje cell marker CABP (green) and the
While the role of Lgi1 in epilepsy remains to be further defined, three main hypotheses underlying Lgi1 func- tion have emerged: 1/Lgi1 may potentiate excitatory synaptic
