Global transcriptome analysis of the heat shock response of Bifidobacterium longum
10
0
0
Texte intégral
Figure
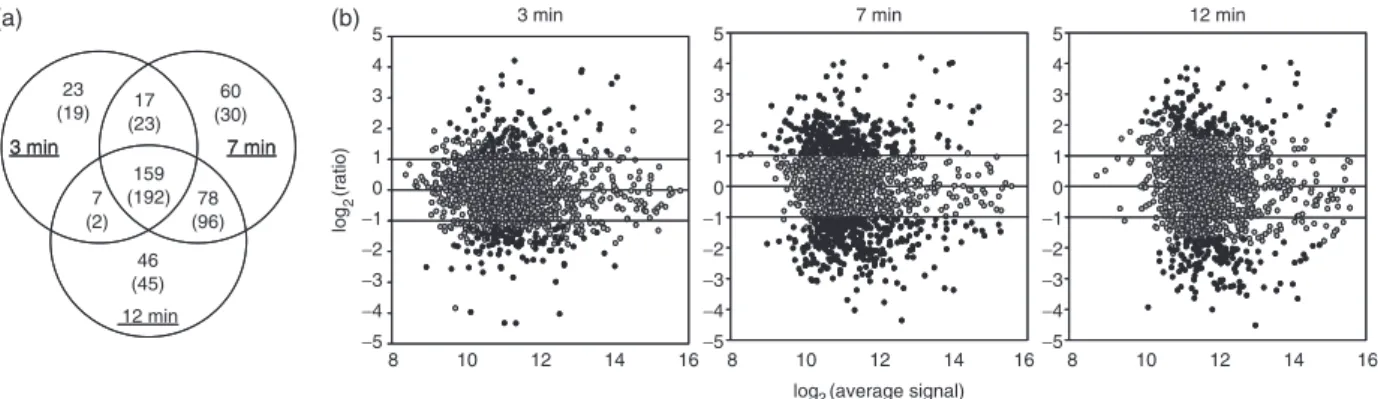
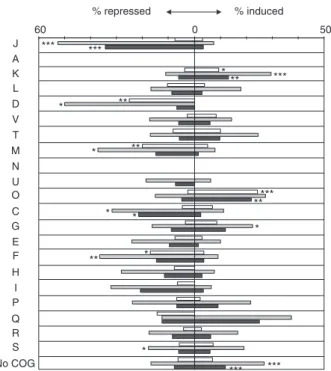
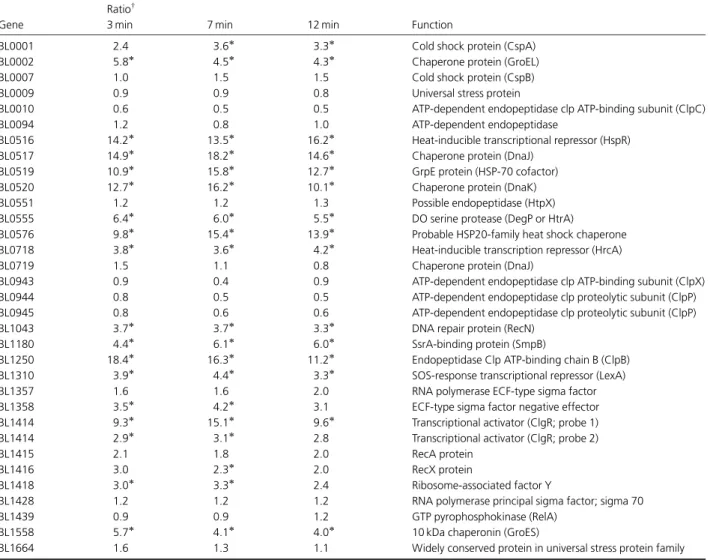
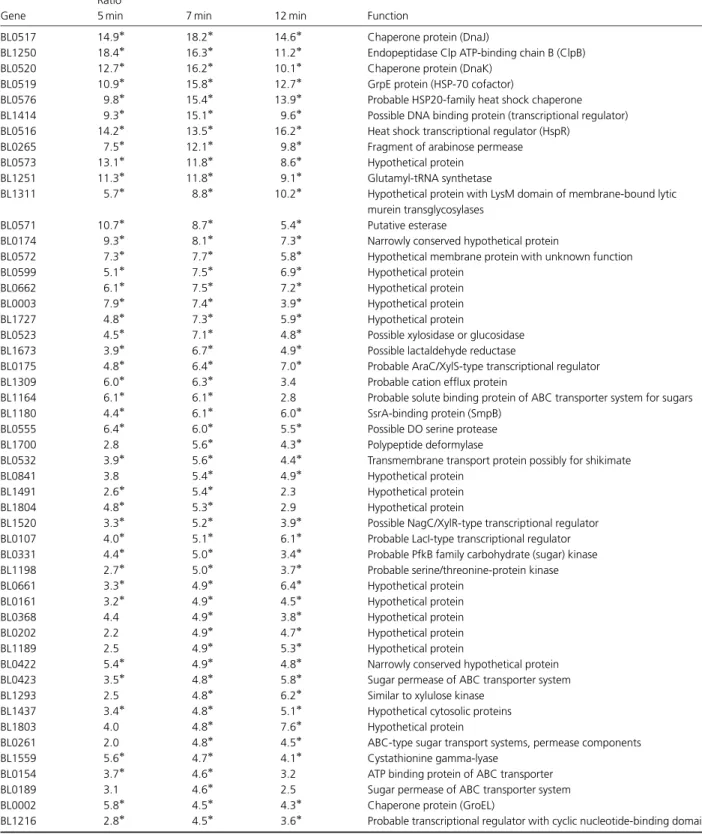
Documents relatifs