by Tim Wang B.S. Bioengineering University of California, Berkeley
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY JUNE 2017
Tim Wang. All rights reserved.
The author hereby grants to MIT permission to reproduce and to distribute publicly paper and electronic copies of this thesis in whole or in part in any medium now known or hereafter
created.
Signature of Author:
Signature redacted
Certified by:
Signature redacted
Tim Wang Department of Biology
David M. Sabatini Professor of Biology Member, Whitehead Institute
Certified
by:
Signature redacted
Accepted by:
'7
Eric S. Lander Professor of Biology Founding Director, Broad Institute
Signature redacted
OF TECHNOLOGYMAY 2
4
2017
LIBRARIES
Amy Keating Professor of Biology Co-Chair, Committee for Graduate StudentDevelopment and application of a general method for somatic cell qenetics by Tim Wang
Submitted to the Department of Biology on May 1st, 2017 in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy ABSTRACT
Genetic analysis, or genetic screening, is an efficient method for identifying the set of genes involved in a biological process. In microorganisms, powerful techniques enable systematic loss-of-function screens, which have yielded critical insights into the molecular basis of many fundamental cellular pathways. In human cells, however, prevailing screening methods fall short - leaving our understanding of human genes incomplete.
Here, I describe the development of a general method for genetic screening in mammalian cells using the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system. In this approach, libraries of knockout mutants can be generated in any cultured cell line using large-scale lentiviral single-guide RNA (sgRNA) pools and screened under both positive and negative selection. After the initial, proof-of-principle experiments, I constructed a genome-wide sgRNA library and screened for genes required for the proliferation and survival of the near-haploid chronic myeloid leukemia cell line, KBM7. The unusual karyotype of these cells also allowed an independent screening approach, involving gene-trap insertional mutagenesis, to find cell-essential genes. Together, these screens converged on a highly overlapping gene set that was enriched for genes that encode components of fundamental pathways, are expressed at high levels, and contain few inactivating polymorphisms in the human population.
To broadly survey patterns of human gene essentiality, I conducted CRISPR-based screens across many more cell lines and cancer types. Overall, these studies revealed a large set of common essential genes involved in housekeeping processes but, interestingly, also pinpointed differences specific to each cell line that reflected its developmental origin, oncogenic drivers,
paralogous gene expression pattern, and chromosomal structure. By mapping sets of genes displaying variable but correlated essentiality across lines, I uncovered functionally interacting gene networks. Using this dataset, I also identified genetic vulnerabilities associated with
oncogenic Ras which may be exploited for anti-cancer therapy.
Last, in collaboration with Bruce Walker and Nir Hacohen's labs, I have conducted a screen for host factors (i.e. human genes) that are required to support HIV infection in a physiologically relevant cell line model. Collectively, these results establish CRISPR-based screens as a powerful tool for systematic genetic analysis in mammalian cells.
Thesis co-supervisor: David M. Sabatini Title: Professor of Biology
Thesis co-supervisor: Eric S. Lander Title: Professor of Biology
Acknowledgements
First and foremost, this thesis is dedicated to my parents and my grandparents who have provided me with constant love, encouragement, and support throughout the years.
I also owe an enormous debt of gratitude to my thesis co-advisors, David Sabatini and Eric Lander, who have both generously shared with me their wisdom, their experience, and their passion for science and discovery. I truly could not have chosen a better pair of mentors from whom to learn the craft of scientific research.
Finally, I thank all of my friends, without whom, I would have probably graduated a few years sooner. Thank you for the many wonderful distractions along the way.
Table of Contents
Title page 1
Abstract 3
Acknowledgements 5
CHAPTER 1: Introduction 7
CHAPTER 2: Genetic screens in human cells using the CRISPR/Cas9 system 52 CHAPTER 3: Identification and characterization of essential genes in the human genome 96 CHAPTER 4: Gene essentiality profiling reveals gene networks and synthetic lethal 143 interactions with oncogenic Ras
CHAPTER 5: A genome-wide CRISPR screen identifies a restricted set of HIV host 207 dependency factors
Chapter 1
IntroductionParts of this chapter were first published as:
Wang, T., Lander, E.S., and Sabatini, D.M. Large-Scale Single Guide RNA Library Construction
and Use for CRISPR-Cas9-Based Genetic Screens. Cold Spring Harbor Protocols.
pdb.top086892. (2016).
SECTION I. Introduction
A central goal in the analysis of a complex system, such as the cell, is to identify its constitutive elements and to define their functions. Knowledge of the function of each element and the coordinated interactions between individual elements may in turn provide insights into the emergent properties of the system as a whole. For living systems, studies at the level of individual genes and their encoded proteins have been highly productive and have elucidated the mechanistic underpinnings of complex cellular pathways. The success of these studies stems, in part, from the availability of a wide array of powerful methods for gene and protein analysis.
Genetic analysis, in which mutants are generated in a random or systematic manner and screened for a phenotype, is an efficient method for identifying the set of genes involved in a biological process. In model organisms, powerful tools have enabled loss-of-function genetic screens that have yielded insights into many fundamental processes including the cell cycle, programmed cell death, and embryonic development (Ellis and Horvitz, 1986; Hartwell et al., 1970; Nusslein-Volhard and Wieschaus, 1980). The development of similar methods for the analysis of cultured mammalian cells has lagged behind - limiting our understanding of mammalian-specific biological processes and diseases.
In this thesis, I describe the development and application of a general method for loss-of-function genetic screening in cultured mammalian cells. In this chapter, to provide context for this work, I review fundamental genetic concepts, mammalian cell culture as it pertains to somatic cell genetics, screening designs and strategies, and methods for mutagenesis. I conclude this chapter with a summary of the specific contributions of this thesis.
SECTION II. Classes of mutations
Armed with a method of inducing mutations using X-ray radiation, Hermann J. Muller was able to generate and characterize a large collection of Drosophila mutants (within weeks of developing the method Muller had isolated about half of the Drosophila mutants ever discovered up to that point in time) (Muller, 1928). From detailed observations of these mutants, Muller proposed the first systematic classification of mutations in 1932 (Muller, 1932). This classification scheme: (1) assessed the strength or severity of the observed phenotype (as the chemical basis of genes and mutations was still to be elucidated) and (2) compared the phenotypes of the mutant allele crossed into strains carrying either additional copies or a deletion spanning the gene of interest. Though this latter process is infrequently performed today, and impractical to perform in somatic mammalian cells generally, this formalism is nonetheless helpful for thinking about mutations.
In total, Muller designated 5 classes of mutants that can be sub-divided into two groups: loss-of-function mutants, which include amorphs and hypomorphs, and gain-of-function mutants, which include hypermorphs, antimorphs, and neomorphs.
A. Loss-of-function 1. Amorphic
Amorphs or nullomorphs are mutants characterized by a complete absence of gene function and are indistinguishable from a deletion of the entire gene. Such null alleles or 'knockout' alleles may be produced by (1) frameshifting insertions or deletions, (2) nonsense
mutations occurring early in the coding sequence of a gene, (3) missense mutations in a
critical codon in a gene, (4) deletions spanning an entire gene or large portion thereof, (5)
mutations in the promoter or distal enhancer elements of a gene that result in a complete loss
of transcriptional activity, or (6) the insertion of a transposable element that disrupts a gene
entirely.
Mutant Deletion Mutant Deletion
2. Hypomorphic
Hypomorphic mutations are characterized by reduced wild-type gene function. The
defining feature of this class of mutants is the retention of some level of gene function such
that the homozygous mutant phenotype is distinguishable and less severe than that of the
homozygous deletion mutant. Hypomorphic, or partial loss-of-function, alleles may be the
result of mutations that reduce the transcriptional activity or stability of the mRNA or missense
mutations that reduce the activity or stability of the encoded protein. Because of the remaining
gene activity, it can prove difficult to determine the wild-type function of a gene solely from the
examination of hypomorphs. Nonetheless, hypomorphs, which can be readily generated using
RNA interference (RNAi)-based reagents (see Section VI.B.1), can be very useful, particularly
for studying essential cellular processes where the complete loss of gene function is
incompatible with life.
Deletion Mutant Wild-type Deletion Mutant Wild-type
B. Gain-of-function 1. Hypermorphic
Hypermorphs possess increased wild-type gene activity. Hypermorphic mutations can
be distinguished from loss-of-function mutations as they display a more severe phenotype
when situated in trans to a wild-type allele than a deletion of the gene. Hypermorphic
mutations can be caused by gene duplication events, mutations that increase gene
transcription, and insertions of transposons or viruses containing DNA elements that drive transcription of the gene. Experimentally, hypermorphs can also be readily generated by the overexpression of a cDNA.
Mutant Mutant Mutant
Mutant Wild-type Deletion
2. Antimorphic
Antimorphic mutations, or dominant negative mutations, produce changes in the encoded gene product that counteract the function of the wild-type gene product. Such mutants may appear phenotypically similar to hypomorphs or amorphs but can be
distinguished (1) on the basis of their inheritance pattern (dominant rather than recessive) and (2) by the fact that inactivation of the mutant allele results in a reversion of the phenotype (or alternatively that the heterozygous dominant negative mutant displays a more severe
phenotype than a heterozygous null mutant). Dominant negative mutations are typically missense or truncating mutations and occur predominantly in genes encoding proteins which form homo-oligomeric complexes or polymers such that the incorporation of one mutant copy of the gene product can 'poison' the entire complex. In contrast to the mutations described previously, it is not possible to systematically generate dominant negative alleles for a given gene.
Mutant Mutant Deletion Wild-type
Deletion Wild-type Wild-type Wild-type
3. Neomorphic
A neomorphic mutation endows a gene with a novel function. On an organismal level, a neomorphic mutation may be caused by (1) a mutation in the transcriptional control unit of a gene or (2) a translocation of the coding sequence of a gene into a novel genomic context that results in ectopic expression of the gene at a different anatomical location, in a different cell
type, or at a different stage during development. Alternatively, neomorphic mutations can also
be the result of missense mutations that alter the binding partners of the encoded protein or,
in the case of enzymes, change their substrate specificity. Neomorphic mutations do not yield
predictable phenotype and, like antimorphic mutations, they cannot be generated in a
systematic manner.
C. Loss-of-function versus gain-of-function approaches
Although examination of the null phenotype may provide the most direct means to infer
the wild-type function of a gene, other classes of mutations can be highly informative as well.
In some cases, these other types of mutations may represent the only means to study a gene.
As discussed above, genes involved in essential cellular processes fall into this category as
null mutations are inviable and thus do not provide much insight into the wild-type function of
the gene. The availability of conditional mutants, such as temperature sensitive mutants, can
be helpful but are rarely used in mammalian cells, which can only be cultured under a narrow
range of temperatures.
It is particularly difficult to perform loss-of-function studies in cultured human cells for
three major reasons. First, cultured somatic cells exist, for the most part, in the diploid (or
hyperdiploid) state and, unlike model organisms, cannot be induced to undergo meiosis or
'mated' with other cells to generate mutant homozygotes. Second, the vast majority of genes
are likely haplosufficient - that is, cells retaining a single wild-type copy of a given gene will
not display a visible phenotype (Kondrashov and Koonin, 2004). On a molecular level, this is
because the majority of genes are not dosage-limited so that a 50% reduction in the levels of
a protein will have minimal impact on a given cellular process. (Some notable exceptions
include rate-limiting enzymes, structural proteins, and highly abundant cellular components
such as the ribosome). Third, many genes in the mammalian genome have undergone
duplication events (Li et al., 2001). After duplication, each paralog in a gene family may retain
partially overlapping functions with other family members; thus, the loss of any particular family member may not elicit a detectable phenotype. For these reasons, loss-of-function
manipulations of cultured mammalian cells have been primarily limited to ones which confer a dominant phenotype. Such manipulations include the expression of RNAi-based reagents or, when possible, a dominant-negative cDNA.
SECTION III. Genetic interactions
For over 100 years, it has been recognized that genes do not act as independent units responsible for determining the individual traits or characteristics of an organism. Rather, through crossing experiments, a more complex picture emerged, namely that alleles of one gene can often influence (and, in some cases, completely mask) the expression of a trait conferred by an allele of a second gene. This phenomenon of 'interacting alleles', termed epistasis by William Bateson in 1909, was originally used described dihybrid crosses (AaBb X AaBb) that did not produce offspring at the expected phenotypic ratios (9:3:3:1) (Bateson,
1909). Later, the definition of epistasis was expanded to encompass interactions between pairs of genes in which the phenotype of the double mutant deviated from the additive combination of effects of the two individual mutants. (Fisher, 1918).
More recently, with the availability of high-throughput screening methods, epistasis analysis in budding yeast has been systematized to enable the quantitative analysis of double
knockouts of all viable, single-gene knockout strains (Costanzo et al., 2010; Costanzo et al., 2016). Through this unbiased approach, it has become possible to construct large-scale genetic networks that define the functional gene groups within the eukaryotic cell.
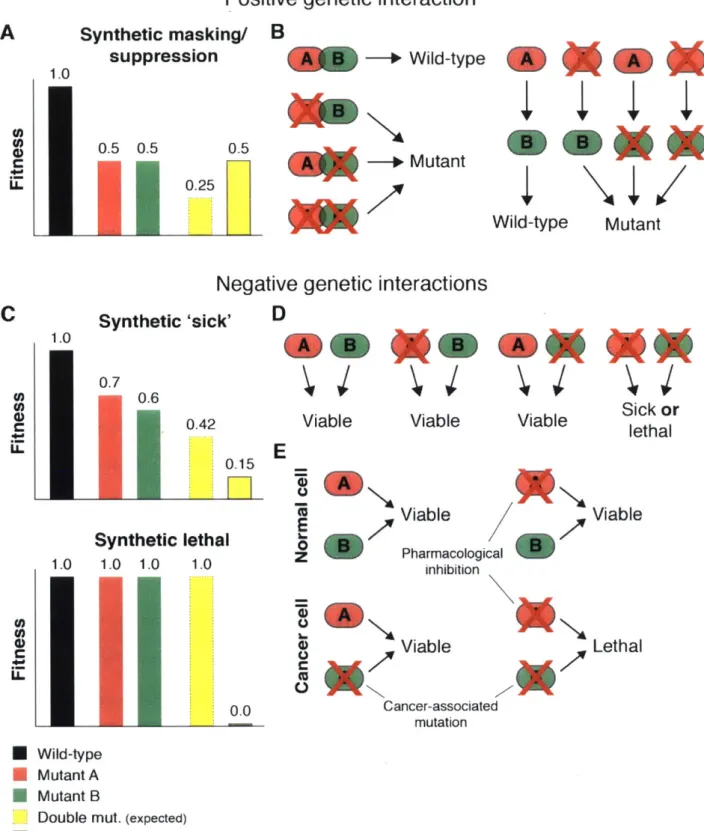
Below, I describe positive and negative genetic interactions between knockout mutants and outline the potential relevance of the latter class for the development of anti-cancer therapies (Fig. 1).
A. Positive genetic interactions
Two genes are involved in a positive genetic interaction if the observed phenotype of
the double knockout is less severe than would be expected given the phenotypes of the single
knockouts. Such interactions can arise between genes encoding non-redundant members of a
protein complex, in which loss of either component completely destabilizes the complex. Thus,
cells deficient for a single member of the complex are unaffected by the loss of additional
complex members. Genes that function as enzymes in a linear metabolic or biochemical
pathway may also form positive genetic interactions. For instance, in the simple case where
cell fitness is determined only by the levels of the end product of a linear metabolic pathway,
the loss of any intermediate enzyme would be equally detrimental. Furthermore, the presence
of a single mutation in the pathway would prevent the generation of the final product and
therefore mask the effects of any additional mutations in the pathway.
B. Negative genetic interactions
If the knockout of two genes exhibits a phenotype that is more severe than would be predicted on the basis of the single-gene knockout phenotypes, the gene pair forms a
negative genetic interaction. Gene pairs for which the individual knockouts are viable but the
combined knockout is inviable are termed 'synthetic lethal' [first described as complementary
lethal in (Sturtevant, 1956)]. If the combined knockout shows an exacerbated proliferation
defect but remains viable, the pair is said to be 'synthetic sick'. In the vast majority of cases,
genes forming negative genetic interactions either (1) perform highly similar (or even identical)
molecular functions or (2) are involved in highly related or redundant cellular pathways.
Many genes that are involved in essential cellular processes (as well as those
participating in non-essential cellular processes) have been duplicated either by transposition
or genomic amplification. For genes within these families, knockout of an individual family
member will often produce no observable phenotype because of the presence of one or more
'back-up' paralogs; deletion or chemical inhibition of the entire family, however, can result in
lethality (Malumbres et al., 2004). Genetic redundancy of this variety is relatively
commonplace in mammalian genomes, which contain hundreds paralogous gene pairs and gene families (Li et al., 2001). However, the degree of redundancy between paralogs is often poorly defined as members of the same gene family may show only partial or no functional overlap. To further complicate matters, proteins that perform identical molecular functions may
not necessarily show sequence conservation; as such, the identification of functional paralogs presents an even greater challenge.
Gene pairs involved in negative genetic interactions may also have distinct molecular functions or encode components of distinct proteins complexes. In such cases, the two
pathways are often functionally overlapping, such that the activity provided by either one of the pathways can partially or fully compensate for loss of the other; the simultaneous
inactivation of both pathways, however, results in a substantial loss of viability. Whereas functional paralogs tend to show interactions with only each other, interactions between genes participating in parallel cellular processes can be distinguished by the extensive interactions observed between all members of the two pathways. Systematic studies of double knockout strains in budding yeast have largely confirmed these predicted interaction patterns (Costanzo et al., 2016; Schuldiner et al., 2005).
C. Exploiting synthetic lethality for targeted anti-cancer therapies
Recent cancer genome sequencing studies have provided a detailed survey of the mutational events that occur during tumor development (Lawrence et al., 2014). This has enabled the systematic identification of frequently mutated 'driver' genes that play a casual role in carcinogenesis. Broadly, these genes can be categorized into two classes (proto-oncogenes and tumor suppressor genes (TSG)) with distinct mutational patterns and molecular functions. As mutations in the former class of genes (i.e. oncogenic mutations) typically result in hyperactive protein function, they have been the focus of intensive
Positive genetic interaction
A
Synthetic masking/ B suppression 1.0 0 0.5 0.5 C_ 0.5 0.25 -0 Wild-type Mutant Wild-typeNegative genetic interactions
Synthetic 'sick' 0.7 0.6 0.42
I0.15
Synthetic 1.0 1.0 1.0 U) U) 0 CU-D
Viable Viable Viable ck or
E
E 0z
lethal 1.0 0.0A
Viable Viable Pharmacological inhibition ViabletoA0
Lethal Cancer-associated mutation * Wild-type * Mutant A * Mutant BDouble mut. (expected)
L Double mut. (observed)
Figure 1. Genetic interactions.
(A) Epistasis analysis of genes involved in positive genetic interactions. Double mutants show a less severe phenotype than predicted by the single mutant phenotypes.
Chapter 1- Introduction Mutant
C
1.0 LL Mao**(B) Positive genetic interactions may occur between genes encoding members of a protein complex or genes acting in a linear pathway.
(C) Epistasis analysis of genes involved in negative genetic interactions. Double mutants show a more severe phenotype than predicted by the single mutant phenotypes.
(D) Negative genetic interactions may occur between genes involved in a distinct, but functionally overlapping pathways.
(E) Somatic mutations present in cancer, but not normal, cells may confer a genetic liability that can be exploited to selectively target cancers.
drug development efforts aimed at pharmacological inhibition of the mutant gene product. In contrast to conventional chemotherapy, these targeted approaches have the potential to be highly cancer-specific and thus spare normal tissues. The use of imatinib in chronic myeloid leukemia to target the BCR-ABL fusion protein serves as a prime example of how activating mutations in cancer cells can be successfully exploited (Druker et al., 1996).
However, these targeted therapies are only effective against gain-of-function mutations in proto-oncogenes encoding proteins are considered to be 'druggable' (primarily enzymes such as protein kinases). Activating mutations in so-called 'undruggable' proto-oncogenes and
mutational events that result in the loss of gene function (i.e. TSG inactivation) cannot be directly targeted. It has been proposed that cancer cells bearing this class of lesions could be exposed to genetic vulnerabilities that are not shared by their wild-type counterparts, thereby
providing a therapeutic window (Kaelin, 2005). This concept of synthetic lethality in cancer is perhaps best illustrated by the negative genetic interaction observed between BRCA1/2, DNA double-strand break repair genes frequently mutated in sporadic and familial breast and ovarian cancers, and Poly(ADP-ribose) polymerase (PARP), another class of DNA repair enzymes (Farmer et al., 2005). PARP inhibitors selectively kill BRCA-mutant tumor cell, but not BRCA-wild-type normal cells. While this example demonstrates the power of the general therapeutic strategy, it remains unclear whether such synthetic lethal interactions are
commonplace and how they can be systematically identified. Genetic screens offer a promising strategy for identifying such vulnerabilities and, in combination with increasingly
sophisticated chemical approaches for targeting non-enzymatic proteins, will greatly expand the repertoire of somatic cancer mutations that can be selectively targeted.
SECTION IV. Mammalian cell culture
A. Rationale for culturing mammalian cells
Mammalian cell culture provides a convenient method for examining cellular processes with a degree of experimental control not available in the analysis of an intact organism. Additionally, mammalian cell culture provides several advantages as compared to similar studies in microorganisms, such as budding yeast. First, many genes and gene families are unique to mammals or may have evolved specialized functions in mammalian cells. Second, some biological processes cannot be appropriately modeled in microorganisms. For example, as single-celled organisms, yeast do not undergo apoptosis and do not require hormonal input (i.e growth factor signals) in order to divide. Last, many disease processes, such as HIV infection, are most appropriately modeled in human cells.
In addition their relevance for biomedical research, cultured mammalian cells also serve as the starting material for many therapies. For instance, cultured cells serve as a source for vaccines (Cox and Schneider, 1976; Ehrlich et al., 2008; Monath et al., 2011). Moreover, some recombinant protein therapeutics, such as monoclonal antibodies, can only
be produced reliably in mammalian cells, which possess the necessary post-translational modification machinery (Hossler et al., 2009). Finally, recent advances in cell culture and gene therapy have enabled cell-based therapies in which cells from a patient, or an immuno-compatible donor, are genetically modified (if necessary), expanded in culture, and
transplanted into the patient (Dimmeler et al., 2008; Yee et al., 2002). B. Characteristics of cultured mammalian cells
A diverse array of cell types can be grown in culture. These can be broadly categorized into two groups: primary cells and continuous cell lines.
1. Primary cells
Primary cells are those derived from tissues excised from an organism. These cells
can retain some specialized characteristics of the native tissue from which they were derived (e.g. electrical activity in neurons) and are therefore be suitable for examining many
physiological processes that are not well-modeled in cell lines that have been continuously
cultured. However, primary cells are typically used for short-term experiments only as they
undergo senescence after serial passaging. Thus, primary cells are not well-suited for
carrying out genetic screening experiments.
2. Continuous cell lines
Continuous cell lines, on the other hand, are capable of proliferating indefinitely. The
vast majority of continuous human cell lines are derived from cancers that, during the course
of tumorigenesis, have acquired the mutations necessary to sustain continuous proliferation.
Recent large-scale efforts have begun to genomically annotate collections of cancer cell lines, providing mutational, DNA copy number, mRNA expression, and drug sensitivity information.
This information will greatly aid future in vitro studies (Barretina et al., 2012; Garnett et al., 2012). Continuous cells lines may also be derived from primary cells that have undergone
immortalization. This process can occur spontaneously in rodent (but rarely in human) cells or
induced experimentally. Immortalization can be achieved by (1) the forced expression of
telomerase (Meyerson et al., 1997), pro-tumorigenic factors (Hahn et al., 1999), or
reprogramming factors (Takahashi and Yamanaka, 2006), (2) transduction with tumor-causing
viruses, such as Epstein-Barr Virus for lymphocytes or Simian virus 40 for a wide range of
other cell types (Miller et al., 1974; Todaro et al., 1963), or (3) cell-fusion with immortalized
cells to form hybridomas (Williams et al., 1977). Finally, embryonic stem (ES) cells derived
from the inner cell mass of an embryo are a unique cell type that can proliferate indefinitely if
cultured under the appropriate conditions (Martin, 1981).
C. Specialized cell lines for genetic analysis
Though it is possible, at least in principle, to isolate mutants and perform screens in any cell line, most mammalian cell lines are not well-suited for the analysis of recessive phenotypes. First, cultured mammalian cells are asexual - it is not possible to 'mate' cell lines and induce meiosis to generate homozygous mutants (though complementation and
dominance tests can sometimes be performed via the generation of cell hybrids). Second, the vast majority of cultured cells are diploid (or polyploidy) making it difficult to inactivate all copies of a gene in a single cell. Whereas the former limitation has yet to be overcome, a number of unique cell lines have been identified over the years, that are naturally or
engineered to be haploid or functionally haploid, which allow for recessive genetic screening. 1. Chinese hamster ovary (CHO) cells
In 1958, Theodore Puck and colleagues generated a cell line using cells from the ovary of an inbred strain of the Chinese hamster (Chinese hamster ovary; CHO) that would become a workhorse for somatic cell genetics (Tjio and Puck, 1958). Unlike other lines generated in the same study (human fibroblasts and the testis cells of the American
opossum), CHO cells displayed an unusually elevated degree of karyotypic instability (whole genome duplications and single chromosome gains/losses were the most commonly found aberrations). Through further experimentation, Puck and colleagues were able to isolate a
panel of phenotypically stable clones with various nutrient auxotrophies using the BrdU technique (see Section V.B.2) (Kao and Puck, 1968). Careful karyotypic analysis revealed that, while the CHO sub-clone used in the genetic studies contained roughly 97% of the total
DNA content of wild-type CHO cells, the majority of the chromosomes had undergone
extensive rearrangement - an observation consistent with the idea that single-gene mutations were produced as a result of 'loss of part of the chromosomal complement and mutation of the
critical gene on the homologous chromosome' (Deaven and Petersen, 1973; Tjio and Puck,
1958). Furthermore, the high rates at which recessive phenotypes could be recovered and the fact that autosomal and X-linked recessive mutations appeared to occur at similar frequencies led to the proposal that CHO cells were 'functionally hemizygous' (Siminovitch, 1976). Later, based on the revertability of some mutant clones upon low-dose treatment with the DNA methylation inhibitor 5-azacytidine, it was suggested that the functional hemizygosity of CHO cells could also be attributed, at least in part, to silencing of one allele by DNA methylation (Holliday and Ho, 1990). Whatever the root cause, the highly unusual, functional pseudo-haploidy of CHO cells turned out to be greatly enabling for recessive genetic screening. Throughout the years, CHO cells have been employed for many screens - perhaps most notable are a series of highly productive experiments dissecting the cholesterol biosynthetic pathway conducted by the Brown and Goldstein lab (Chin et al., 1982; Hua et al., 1996; Krieger et al., 1981; Mosley et al., 1983). Interestingly, how these cells are able to tolerate such dramatic changes in gene dosage and why they are chromosomal unstable remain mysterious.
2. Bim-deficient cells
BLM encodes an ATP-dependent DNA helicase involved in DNA replication and repair
and mutations in this gene cause Bloom's syndrome, an autosomal, recessive disorder characterized by short stature and a predisposition to cancer (Ellis et al., 1995). Interestingly, B/m-deficient cells display an elevated rate of mitotic recombination and loss of heterozygosity (LOH) (in murine ES cells the rate of LOH increases 18-fold from 2.3x10~5 to 4.2x104
events/cell/generation) (Luo et al., 2000). Because of this unique property, mutations
generated in these cells can be homozygosed due to mitotic recombination or hemizygosed due to LOH, thereby allowing for the study of recessive phenotypes (Guo et al., 2004).
To perform genome-wide loss-of-function screens in B/m-deficient murine ES cells, Allan Bradley and colleagues performed chemical mutagenesis using N-Ethyl-N-nitrosourea
(ENU) and insertional mutagenesis using either gene-trap retroviruses or the piggyBac (PB) transposon (see Section VI) (Guo et al., 2004; Wang et al., 2009; Yusa et al., 2004). After mutagenesis, a stringent selection was applied. In the surviving clones, the causal genes were mapped by cDNA complementation or inverse PCR of the viral or transposon integration sites.
3. Hypodiploid cancer cell lines
Another method of studying recessive phenotypes relies on the use of chromosomally stable, hypodiploid cell lines. In such lines, like CHO cells, mutations in a gene can unmask
recessive phenotypes due to the absence of a homologous chromosome. Whereas the vast majority of aneuploid cancer cells have more than 2N DNA content, karyotyping studies have identified a few cancer subtypes that are characterized by relatively frequent hypodiploidy (Harrison et al., 2004). In the majority of these cancers, the modal chromosome number lies just slightly below 46, making them of limited utility for genetic analysis. In 1987, a myeloid
leukemia cell line with a highly unusual karyotype was derived from a patient presenting with blast phase chronic myeloid leukemia (CML) (Andersson et al., 1987). This line, named KBM7, is haploid for all chromosomes except chromosome 8 (25, XY, +8, Ph(+)) (Ph: the Philadelphia chromosome is found in 95% of CML cases and is a translocation of
chromosomes 9 and 22 that generates the BCR-ABL oncogene). By serial sub-culture, a KBM7 sub-clone that stably maintained the near-haploid state was derived (Kotecki et al., 1999).
The unique karyotype of KBM7 enabled genome-wide loss-of-function screens using insertional mutagenesis (Carette et al., 2009). Initially, individual colonies were picked and the insertion site mapped via inverse PCR. The insert mapping process was then adapted for high-throughput sequencing to enable rapid mapping of causal genes in a mutant population (Carette et al., 2011). Screens using this cell line have identified host factors that are essential
for the pathogenicity of microbial factors and transporters of toxic molecules (Birsoy et al.,
2013; Chen et al., 2014). To expand the repertoire of cellular processes that could be interrogated in these screens, KBM7 cells were transduced with the reprogramming factors
SOX2, KLF4, OCT4, and MYC to generate an adherent, fully haploid induced pluripotency
stem (iPS)-like cell line, termed HAP1 (Carette et al., 2010).
4. Haploid embryonic stem cell lines
Haploid ES cell lines have also been experimentally generated (Leeb and Wutz, 2011;
Li et al., 2012). Parthenogenetic haploid ES cells have been generated from activated,
unfertilized oocytes. Androgenetic haploid ES cells have been generated by sperm injection
into enucleated oocytes followed by SrCI2-induced activation. To date, only a handful of
proof-of-principle genetic screens have been performed in these cells using insertional
mutagenesis.
SECTION V. Large-scale genetic analysis in cultured mammalian cells A. Screen format
1. Pooled screens
Screens in cultured cells can be performed in a pooled or in an arrayed format. In
pooled screens, the mutagenesis is carried out in the cell population en masse. Because each
cell is perturbed an unknown manner, the mutants of interest must be isolated and the causal
mutations mapped. With the availability of genomic tools, the entire mutant population may be
surveyed altogether using microarray- or high-throughput sequencing-based approaches.
2. Arrayed screens
Aided by advances in automation, collections of reagents targeting (or expressing)
single genes can be systematically generated and tested in individual wells in an arrayed
fashion. In this approach, the perturbation induced in each well is defined and, importantly,
each well can be monitored using imaging methods to assay for high-content phenotypes
(See Section V.C.3). Arrayed screens can also be used to study cell non-autonomous phenotypes. However, arrayed screens are not without their drawbacks. First, there are immense upfront costs for generating and maintaining the reagent collection. Second, arrayed screens can suffer from severe artifacts due to well-to-well and plate-to-plate variability. Finally, large-scale arrayed studies require expertise in automation to carry out the screens and sophisticated methods for analyzing them.
B. Screen selection
1. Positive selection screens
Pooled screens can be further divided into two classes, positive selection and negative selection, which can often reveal complementary biological information (Fig. 2). In positive selection screens, the desired class of mutants should possess a selective advantage
compared to their wild-type counterparts or mutants with unrelated phenotypes. In this setting, the mutants of interest should rise to high frequency in the population. As a result, gene candidates can often be readily identified.
Due to the relative ease of mutant isolation and gene identification, it is generally desirable to conduct screens via positive selection. Some biological processes, such as drug resistance or anchorage-independent growth, are ideally suited for positive selection
screening as they are intrinsically linked to cellular proliferation and survival. For studying other pathways, additional selection strategies can be devised. For example, cells can be engineered to express a selectable marker in a pathway activity-dependent manner or
isolated in screens using fluorescence-activated cell sorting (FACS). Together, these approaches can greatly broaden the diversity of phenotypes amenable for screening. 2. Negative selection screens
Negative selection screens seek to identify mutants that are at a selective
disadvantage. As this class of mutants should decrease in frequency over the course of a
screen, they can be difficult to detect. In microorganisms, negative selection experiments can be performed by replica plating whereby mutants are plated under permissive conditions and then transferred onto secondary plates under restrictive conditions (e.g. by shifting the
temperature or removal of a non-essential nutrient). Because the relative spatial orientation of all the colonies is preserved, a 'missing' mutant can be easily recognized against the
background of wild-type cells. Although possible (Stamato and Waldren, 1977), replica plating is impractical for most mammalian cell lines - most adherent cell lines do not grow as colonies (and instead migrate across the plate) and suspension cells are dispersed throughout the culture. Interestingly, negative selection screens can also be 'converted' into positive selection screens. Cells can be placed into a deficient media, in which the desired mutant should fail to proliferate, and treated with a chemical agent, such as BrdU, that will selectively kill dividing cells (Puck and Kao, 1967). However, this approach can be not be used to recover mutants that are actively killed or which do not arrest entirely but proliferate at a reduced rate.
In order to reliably detect the depletion of mutants in mammalian cell screens, mutants must either be (1) arrayed in individual wells under permissive conditions and screened by sub-plating into restrictive conditions or (2) tracked as a population using DNA-based barcodes that can be deconvoluted using high-throughput approaches. For insertional mutagenesis-based screens, integration sites can be mapped using inverse PCR (or related insertion mapping techniques) and subjected to massively parallel sequencing. For screens with lentiviral libraries, the viral cassette is stably integrated into the genome and can therefore serve as a barcode to quantify the relative abundance of each mutant by high-throughput sequencing or microarray analysis (Berns et al., 2004a; Smith et al., 2010). In order to ensure that a mutant 'drop-out' can be reliably distinguished from random changes in abundance resulting from sampling fluctuations, each mutant (or each barcode) must be well-represented in the initial cell population. For this reason, it may be impractical or impossible to
conduct negative selection screens in settings where the starting number cell number is limiting.
A
.*..
Positive
selection
.0*.g4
Negative
selection
.000
000
0
00
Positive
selection
Initial
population
Negative
selection
Mutant frequency
Figure 2. Positive and negative selection screens.
(A) The mutants of interest (outlined in red) rise to high frequency in positive selection screens but are underrepresented in negative selection screens, and consequently require more precise methods for their detection.
(B) Relative mutant frequency in positive and negative selection screen
Chapter 1- Introduction
C. Screen readouts
Screens can be conducted for arbitrary phenotypes of interest that can be scored with various levels of precision. In general, a trade-off exists between the cost, speed, and ease with which a phenotype can be scored and the number of mutants that can be examined in a screen.
Below, I discuss several common readouts for screens in cultured cells in increasing order of phenotypic resolution.
1. Proliferation
Proliferation-based screens are the most common type of screen in cultured cells. Cell number is a very simple phenotype to score and is amenable to pooling provided that the abundance of each mutant in the population can be surveyed simaltaneously. At a first glance, it may appear that such screens are limited to phenotypes that are directly linked to cellular
proliferation and survival, such as drug resistance. However, mutant isolation strategies can be devised to probe a wide array of biological processes. For example, to isolate mutants involved in a particular pathway of interest, cells can be selectively sensitized to perturbations in the pathway if it is partially inhibited (for example, by treatment with a chemical inhibitor). In this setting, genes involved in the pathway may display enhanced essentiality thus score differentially between the compound-treated and -untreated conditions. Furthermore, any pathway that produces a specific transcriptional response can be screened in a proliferation-based format by using a selectable marker that is expressed under the control of a pathway activity-dependent gene promoter (Lee et al., 2013).
Because many pathways impact cellular proliferation, systematic screens for essential genes are also of great utility. In budding yeast, comparisons of gene essentiality across panels of engineered strains have been used to identify sets of genes that function in common cellular processes (Costanzo et al., 2010). Such 'guilt-by-association' approaches have been
successful in mapping functional gene networks in budding yeast, but the lack of a similar large-scale gene essentiality dataset has precluded such an analysis in human cells. 2. Fluorescence-activated cell sorting (FACS)
Although resistance marker-coupled reporters greatly increase the breadth of
phenotypes that can be screened in a proliferation-based manner, many phenotypes are still beyond the reach of this approach for several reasons. First, many pathways of interest do not produce a well-defined (or unique) transcriptional output. Second, for applications in which the endogenous expression pattern of a gene must be precisely mimicked, knock-in reporters must first be generated through a tedious process. Last, it may be relatively difficult to isolate mutants that have altered activity (either, increased or decreased) using reporters carrying antibiotic selection markers where low levels of expression can often confer resistance.
Screens employing fluorescence-activated cell sorting (FACS) to isolate the desired classes of mutants may help to address each of these issues. FACS can be used to isolate mutants expressing altered levels of a fluorescent reporter or changes in the expression of
endogenous proteins (Duncan et al., 2012). Notably, the set of suitable antigens need not be restricted to cell-surface antigens as cells may be fixed, stained, and sorted before being processed for PCR and sequencing analysis (Parnas et al., 2015).
3. Imaging screens
Although FACS-based readouts greatly increase the range of phenotypes that can be screened, some phenotypes still cannot be assessed via FACS for several reasons: (1)
alterations in many pathways do not necessarily result in a change in the abundance of a protein (2) the abundance of the target protein may be too low or too variable to reliably quantify by FACS or (3) the proteins which change in abundance not may be known a priori. Arrayed, imaging-based screens can provide richer details into the cellular state and may allow for open-ended studies into phenotypes that are not pre-specified or anticipated at the
outset of the screen (Carpenter et al., 2006). By applying machine learning algorithms, it is possible to define cells of interest based on their shape, nuclear staining pattern, or the staining (and co-staining) patterns of any number of arbitrary markers (Jones et al., 2009). Additionally, image-based analysis can be refined to only examine cells at a particular stage of the cell cycle, abnormalities related to a particular organelle within the cell, changes in the localization of protein (rather than its abundance) or even protein trafficking dynamics using live-cell imaging.
4. Expression screening
Profiling the abundance and/or localization of one or a handful of markers via imaging-or FACS-based screens can be highly infimaging-ormative; transcriptomic profiling offers an even higher-resolution and unbiased view of the cellular state. Gene expression profiling may also be able to tease apart seemingly similar phenotypes (e.g. loss of any essential gene will affect proliferation but may elicit different transcriptional responses before cell death occurs). With the advent of microarray technology, it became feasible to perform such studies in an arrayed fashion - the gene expression changes resulting from defined perturbations can be compiled as a reference data set and used to compare with the transcriptional changes elicited by drugs with a poorly defined mechanism of action (Lamb et al., 2006). Recent advances in single-cell RNA sequencing methods have enabled such screens to be performed in a pooled format, greatly enhancing the throughput and reducing the cost of such studies (Adamson et al., 2016; Dixit et al., 2016). Continuing improvements in single-cell profiling technology may enable other '-omics' readouts of the cellular state for pooled screens.
SECTION VI. Loss-of-function genetics in cultured mammalian cells A. Random mutagenesis
1. Spontaneously arising
Spontaneously arising mutations were the first class to be analyzed in the lab.
Historically, they have been extremely valuable for the genetic studies of mice and continue to
play a major role in human genetics today. The field of mouse genetics was greatly aided by
hobbyists who collected inbred strains of spontaneously arising 'fancy' mice that displayed
interesting morphological (e.g. dwarf), behavioral (e.g. twirler, waltzer), and coat color
phenotypes (Rader, 2004). In humans, spontaneously arising mutations that cause disease
may come to clinical attention and thus provide insight into the wild-type function of the
disease-related gene. In cases for which the disease manifests a phenotype in a cell type that
can be readily obtained, immortalized, and cultured (e.g. skin fibroblast or lymphocytes),
patient-derived cells can be used to study gene function (Brown and Goldstein, 1974).
However, because of the array of DNA repair pathways that are in place to allow for the
faithful transmission of genetic information, such mutations (especially phenotypically
interesting ones) are very rare. With the arsenal of mutagens available today, experimental
geneticists no longer need to rely on spontaneously arising mutations.
2. Ionizing radiation
In 1928, Hermann Muller described a method to induce heritable phenotypic changes
in Drosophila using X-ray radiation, thus marking the inception of the field of experimental
genetic analysis (Muller, 1928). Armed with this technique, Muller and colleagues were able to
collect mutants displaying either visible or 'latent' (e.g. recessive, sex-linked lethality scored
by the absence of male progeny in a test cross) phenotypes at an unprecedented pace. These studies lead to a greater understanding of the physical nature of the gene, mutational
processes, as well as the potential dangers of radioactivity (Muller, 1930; Muller, 1954).
Ionizing radiation, such as X-rays and higher energy gamma rays, tends to induce
DNA damage by creating double-strand breaks (DSBs) that can ultimately result in either small deletions or large-scale chromosomal rearrangements. Due to the unpredictable nature
of the resulting mutations (which may disrupt multiple genes at once), and the fact that X-ray mutagenesis is a rather inefficient process, ionizing radiation is rarely used to mutagenize mammalian cells.
3. Chemical mutagenesis
It was later recognized that chemical mutagens could also be used to alter DNA (Auerbach, 1949). Mutagenic compounds can act through a variety of mechanisms including: (1) the inhibition of DNA repair enzymes or the replication machinery, (2) the incorporation of non-natural bases into the DNA sequence, (3) the generation of inter-strand crosslinks, and (4) the modification the DNA bases themselves.
The alkylating agent N-ethyl-N-nitrosourea (as known as ENU) is highly efficient mutagen that preferentially induces single base pair A->T transversions and AT->GC transitions (Cordes, 2005). As a result, it tends to generate local mutations - typically
missense, nonsense, or splice site mutations - and is the mutagen of choice for mammalian cell screening.
4. Insertional mutagenesis
Mobile DNA elements can be randomly inserted into the genome to alter gene function. For this purpose, transposable elements (transposons) or integrating viruses
(primarily, retroviruses) can both be used (Ivics et al., 1997; Schnieke et al., 1983). Gain-of-function screens can be performed by including a strong promoter into the insert element to
drive the transcription of genes near the insertion site. Loss-of-function insertional
mutagenesis screens can also be performed by using a mobile element that contains a strong splice acceptor (a so-called 'gene-trap') so that inserts landing in an intronic region will disrupt the production of the full-length mRNA (Friedrich and Soriano, 1991).
The major advantage of insertional mutagenesis is the ease of mapping mutants. Because the relevant mutations induced by chemical and X-ray mutagenesis are not 'marked',
such mutations can only be mapped through laborious methods such as cloning by
complementation. For insertional mutagenesis, however, the site of the insertion (and thus causal gene) is marked and can be readily mapped using techniques such as inverse PCR (Ochman et al., 1990). More recently, insertion tracking can be coupled to high-throughput sequencing to greatly improve the mapping throughput in pooled populations (Schmidt et al., 2007).
B. Targeted approaches 1. Anti-sense RNA
Because it is difficult to inactivate both copies of a gene at the DNA level, several methods have been developed for perturbing genes in mammalian cells by targeting them at the RNA level. As these methods do not induce permanent changes in DNA, they are not genetic perturbations per se; nonetheless, mRNA targeting techniques are powerful tools for conducting loss-of-function studies. Prior to the discovery of the phenomenon of RNA
interference (RNAi; discussed below), scattered reports had suggested that naturally
occurring or exogenously introduced anti-sense RNA molecules can antagonize the function of their complementary counterparts (Eckhardt and LOhrmann, 1979; Stephenson and Zamecnik, 1978; Tomizawa et al., 1981). The idea to use anti-sense RNA as a general
method to perturb mammalian genes was first proposed by Harold Weintraub. In a pair of studies published in 1984-5, Weintraub and colleagues demonstrated efficient targeting of an exogenous reporter plasmid and the endogenous actin gene by using cDNAs cloned (and thus transcribed) in the reverse orientation (Izant and Weintraub, 1984, 1985).
Following the discovery of the post-transcriptional gene silencing phenomenon known as RNAi, it was recognized that synthetic microRNA- and small interfering RNA (siRNA)-based reagents could be introduced into mammalian cells to mediate targeted gene
repression (Brummelkamp et al., 2002; Elbashir et al., 2001; Harborth et al., 2001; Paddison
et al., 2002; Sui et al., 2002). Because target specificity is dictated by base pair
complementarity, it is easy to rapidly generate RNAi reagents for many genes at once. Thus, genome-wide shRNA and siRNA libraries were quickly developed and used to perform the first systematic loss-of-function studies in mammalian cells (Berns et al., 2004b; Paddison et
al., 2004). Later studies mapped the set of genes required for normal and cancer cell proliferation providing scores of promising gene targets for anti-cancer therapies (Luo et al., 2008; Luo et al., 2009; Schlabach et al., 2008; Scholl et al., 2009; Silva et al., 2008).
However, it is now clear that RNAi-based screens suffer from major drawbacks (Kaelin, 2012). First, many reagents only partially suppress their target mRNA; the remaining transcripts may be sufficient to carry out the wild-type gene function thus leading to potential false negatives. Perhaps more problematic is the fact that RNAi reagents can act on (hundreds of) unintended transcripts; these 'off-target' effects can lead to false positive results (Echeverri et al., 2006;
Jackson et al., 2003; Schultz et al., 2011). It should also be noted that the issue of off-target effects is greatly magnified when performing proliferation-based screens because of the
increased size of the potential 'off-target space'. For these reasons, RNAi-based screens have faced major challenges in reproducibility (Begley and Ellis, 2012).
2. Homologous recombination
Genomic sequences in eukaryotic cells can be manipulated by exploiting the endogenous homology-directed DNA repair (HDR) machinery (Dunham et al., 1989). If a double-strand break occurs in the late S or G2 phase of the cell cycle, the lesion can be repaired by HDR via the homologous recombination pathway (or alternatively by the error-prone non-homologous end joining pathway is which active in all phases of the cell cycle) (West, 2003). In the native setting, the sister chromatid (rather than the homologous chromosome) serves as the 'donor' template; in an experimental setting, linearized DNA molecules containing homologous 'arms' flanking the lesion site can serve as the template
and thus be incorporated into the genome. Importantly, mutations, ranging from a single base pair substitution to a large insertion encoding a fluorescent protein, can be introduced into the donor template thus allowing for arbitrary modification of the genome.
After the development of murine embryonic stem cell (mESC) culture, Oliver Smithies and Mario Capecchi adopted methods that had been developed for performing homologous recombination in budding yeast to edit the genomes of mESCs (Doetschman et al., 1987;
Martin, 1981; Thomas and Capecchi, 1987). Through the use of clever positive-negative selection strategies to ensure on-target (rather than random, non-homologous) integration of targeting cassettes, mESCs and mice bearing the desired mutations could be readily
generated.
3. Programmable nucleases - Zinc finger nucleases (ZFN) and Transcription activator-like effector nucleases (TALEN)
Despite the power and flexibility of homology recombination, the efficiency of this process is very low. DSBs can locally stimulate homologous recombination - in fact, the efficiency of genome editing can be increased up to 1000-fold if a DSB is present at the desired locus (Jasin, 1996; West, 2003). Additionally, reagents that can induce lesions in targeted loci could be simply used to inactivate genes - in the absence of a donor template,
DSB repair would simply occur via the error-prone NHEJ pathway, producing loss-of-function mutations.
The ability to cleave a specific genomic locus in mammalian cells began with the development of engineered zinc finger-nuclease fusion proteins (Kim et al., 1996). These
chimeric proteins are a fusion of a DNA-binding domain consisting of 3-6 zinc fingers repeats (with each unit recognizing three base pairs, thus providing a total of 9-18 base pairs of specificity) with a DNA cleavage domain comprised of the non-specific Fokl nuclease.
Because the Fokl is only active as a homodimer, pairs of ZFNs must be introduced in order to
induce cleavage, allowing for much greater targeting specificity. In principle, this
programmable design allows for targeting of arbitrary genomic sites with high specificity. In practice, the large, repetitive DNA-binding domains are difficult to clone and, more
importantly, the cleavage efficiency of each ZFN pair is highly variable and unpredictable because the targeting specificity of each zinc finger repeat is partially dependent on its neighbors (Sander et al., 2011).
The next generation of programmable nucleases was identified from plant pathogenic bacteria of the Xanthomonas genus. Remarkably, these bacteria produce TAL (transcription activator-like) effector proteins that direct bind to the DNA of their hosts and modulate host gene expression (Kay and Bonas, 2009). Furthermore, the DNA-binding domain of these effector proteins consists of individual modular repeats allowing them to be readily
programmed (Moscou and Bogdanove, 2009). Each 34-amino acid TALE repeat targets a single nucleotide with 2 amino acids within the repeat conferring specificity for the individual base pairs. Like ZFNs, the TALE repeats are fused with the Fokl nuclease to generate a TALE nuclease, or TALEN. Pairs of TALENs can be introduced into mammalian cells to induce site-specific DNA cleavage in a manner highly analogous to ZFN-mediated genomic targeting (Hockemeyer et al., 2011). Because the nucleotide binding specificity of each TALE repeat is independent of its neighbors, the activity of pairs of TALENs is much more predictable than ZFNs. However, the highly repetitive DNA binding domain prevents packaging into
lentiviruses. Furthermore, TALENs are relatively large and difficult to clone and are thus difficult to synthesize on a large scale. For these reasons, TALENs are not ideally suited for conducting systematic genetic screens.
4. CRISPR
CRISPRs were discovered in 1992 by Francisco Mojica and Francisco Rodriguez-Valera, a pair of researchers studying the salt regulation of the halophilic extremophile,
Haloferax mediterranei (Mojica et al., 1993). They focused in on a region of the H. mediterranei genome that appeared to be modified by the salinity of the microbial culture
medium. Sequencing of this stretch of DNA revealed an unexpected pattern: 30 nucleotide-long repeats tiled at regular intervals of 36 nucleotides. Though similar repetitive DNA sequences had been documented previously in E. coli and were later found in many more prokaryotic species, the functional significance of the CRISPR system remained unclear for the next decade (Ishino et al., 1987). Eventually, it was recognized that CRISPR served as a prokaryotic 'adaptive immune system' to protect bacteria from invading phage (Barrangou et al., 2007; Mojica et al., 2005; Pourcel et al., 2005). Subsequent biochemical studies
demonstrated that the CRISPR system functions by cleaving the genome of the invading viruses. The cleavage reaction is carried out by an effector nuclease that is directed to its target by a guide RNA (gRNA) molecule, which bears sequence complementarity to the target, that is in complex with a trans-activating CRISPR RNA (tracrRNA) (Deltcheva et al., 2011; Jinek et al., 2012).
Following the biochemical studies characterizing the class I CRISPR system, two reports demonstrated that the heterologous expression of two components of the system, the Cas9 nuclease and a single guide RNA (a chimeric RNA that fuses the gRNA and tracrRNA; sgRNA), was sufficient to induce DSBs at specific regions of the human genome (Cong et al., 2013; Mali et al., 2013). Because the targeting specificity of CRISPR/Cas9 is dictated by the 20-base pair sequence at the 5'-end of the sgRNA, reagents for each gene can be rapidly generated - providing a major advantage over previous genome editing methods. By default, the DSBs are resolved by the error-prone non-homologous end joining pathway which
typically generates a deleterious mutation at the target site; in the presence of a homologous DNA template, the DSB can be repaired by homologous recombination. Follow-up studies demonstrated the rapid generation of a menagerie of transgenic mammals, including mice,
pigs, rats, monkeys, and dogs, via co-injection of Cas9 and a sgRNA into fertilized zygotes (Hai et al., 2014; Li et al., 2013; Niu et al., 2014; Wang et al., 2013; Zou et al., 2015).
Since the initial reports, numerous modifications and improvements of the CRISPR genome editing system have been implemented. For instance, several strategies have been developed to increase targeting specificity: (1) fusion of catalytically inactive Cas9 with the
Fokl nuclease with paired guide RNAs (Guilinger et al., 2014; Tsai et al., 2014) (2) a Cas9 variant with only nickase activity with paired guide RNAs (Ran et al., 2013), (3) truncated sgRNAs (Fu et al., 2014) and, (4) structure-guided mutation of Cas9 for enhanced fidelity
(Kleinstiver et al., 2016; Slaymaker et al., 2016). Additionally, several strategies have been developed to increase the rate of homology-directed repair to allow for efficient 'knock-in' of desired mutations and reporter constructs (Chu et al., 2015; Maruyama et al., 2015; Paquet et
al., 2016).
Several Cas9 fusion proteins have also been engineered. Whereas wild-type Cas9 produces permanent changes in the genome, catalytically inactive variants of Cas9 (dCas9) have been generated which allow for inducible modulation of gene expression. When paired with an sgRNA targeting a gene promoter, dCas9 can be fused to transcriptional repressors,
such as the Kruppel associated box (KRAB) domain, to downregulate gene expression (a system termed CRISPR interference or CRISPRi) (Qi et al., 2013) or to transcriptional activators, such as the VP64 transactivation domain, to increase gene expression (a system termed CRISPR activation or CRISPRa) (Gilbert et al., 2014; Maeder et al., 2013). dCas9 can also be fused to GFP to enable live imaging of genomic loci and to cytidine deaminase to generate point mutations (predominantly C->T transitions) (Knight et al., 2015; Komor et al., 2016).
Last, many novel effector proteins from the class 1I (i.e. Cas9-related) and other CRISPR systems have been recently discovered as well. Cas9 variants that bind different