Robustness of the Parsimonious Reconciliation Method in Cophylogeny
Texte intégral
Figure

Documents relatifs
When expressed in relation to body weight, pancreas, pancreatic protein weights and enzyme activities measured were higher in Roe deer than in calf.. The 1 st original feature is
Indeed since the mid- 1980s, lung cancer has overtaken breast cancer as the most common form of female cancer in the United States-the first country to show this
Last but not least, the next challenge will probably be to overcome the alternative between repression and control or deforestation, and to design sustainable and feasible
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
The cunning feature of the equation is that instead of calculating the number of ways in which a sample including a given taxon could be drawn from the population, which is
In section 4, we point out the sensibility of the controllability problem with respect to the numerical approximation, and then present an efficient and robust semi-discrete scheme
(The old Raja, who remained in Chumbi, died in 1863.) Eden was satisfied that “… under the advice of Cheeboo Lama, an entirely new state of things will now be inaugurated in
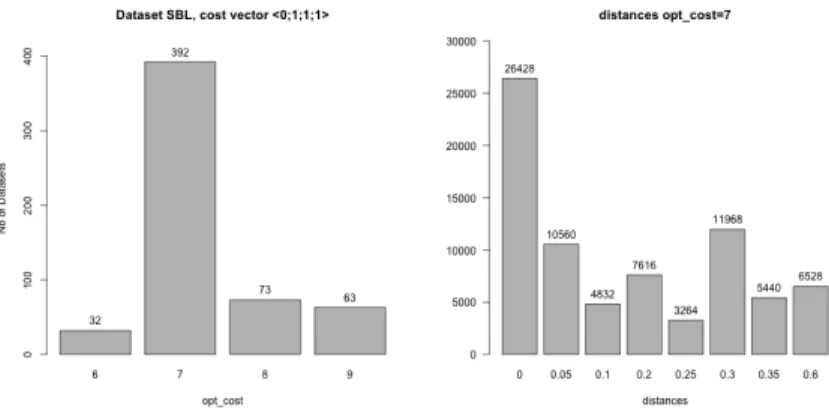
Barplots of optimum cost (left) and dissimilarity between pairs of reconciliations with the most frequent optimum cost (right) obtained on the datasets derived from the SBL x%
