Implication de la voie de signalisation de Fanconi
dans la régulation du cycle cellulaire via stathmine,
un nouveau partenaire de FANCC.
Thèse
Audrey MAGRON
Doctorat en biologie cellulaire et moléculaire
Philosophiae Doctor (Ph.D.)
Québec, Canada
© Audrey MAGRON, 2015
III
Résumé
L’anémie de Fanconi (FA) est une maladie génétique rare qui se caractérise par une insuffisance médullaire, des malformations congénitales et une prédisposition accrue au développement de cancers. Aujourd’hui, 16 protéines sont identifiées pour coopérer ensemble dans la voie métabolique de Fanconi, impliquées principalement dans la réparation de l’ADN. Pour mieux comprendre le rôle des protéines Fanconi dans d’autres mécanismes cellulaires, un criblage en double hybride a été réalisé au laboratoire afin d’identifier plusieurs nouveaux partenaires protéiques d’intérêt. La protéine associée aux microtubules, la stathmine (STMN) a ainsi été identifiée comme un nouveau partenaire potentiel de la protéine Fanconi C (FANCC).
L’étude de l’interaction a confirmé un lien direct entre FANCC et STMN et révèle qu’une mutation ponctuelle, ou une délétion de l’exon 1 de FANCC, cause une perte de cette interaction. Afin d’éclaircir la fonction du complexe FANCC-STMN, nous avons étudié la localisation des protéines durant le cycle cellulaire par immunofluorescence. Les résultats ont permis de mettre en évidence une localisation commune de FANCC et STMN dans les centrosomes de cellules en mitose. Une variation de l’état de phosphorylation de STMN dans les centrosomes a été observée dans plusieurs lignées cellulaires de patients. Ainsi, les protéines FANC semblent participer à la régulation de l’état de phosphorylation de STMN durant la progression du cycle cellulaire. Pour finir, les cellules de patients FA présentent de nombreuses malformations mitotiques, tels qu’un nombre important de centrosomes surnuméraire et un fuseau mitotique de petites tailles.
Notre étude a permis de montrer que la protéine FANCC participe à la régulation de l’état de phosphorylation de STMN durant la division cellulaire, via une interaction directe. Nos résultats suggèrent également que les protéines FANC semblent participer à la progression du cycle cellulaire, en régulant de manière indirecte l’activité de STMN. Puisque la dérégulation de STMN est connue pour être impliquée dans la cancérogenèse, une augmentation de l’activité de STMN dans les cellules FA semble expliquer la sensibilité des patients à développer différents types de cancer et représente une nouvelle cible prometteuse pour le traitement des cancers chez les patients FA.
V
Abstract
The Fanconi Anemia (FA) is a genetic recessive disease characterized by a bone marrow failure, various congenital malformations, and predispose to the development of cancers. Currently, 16 proteins are identified to cooperate together in Fanconi pathway, known mainly to play a role in DNA reparation. For better understanding the involvement of Fanconi proteins in other cellular mechanisms and to identify new partners of interest, a double hybrid screen had been realized at the laboratory. The microtubule-associated protein, the stathmin (STMN), had been identified as a new potential protein partner of the Fanconi protein C (FANCC).
Our study has confirmed a direct interaction between FANCC and STMN proteins, and identified that a punctually mutation or exon 1 deletion in FANCC induced a loss of this interaction. In order to clarify the function of the FANCC-STMN complex, we have study the localization of these proteins during the cell cycle by immunofluorescence experiments. These results have unveiled a co-localization of FANCC and STMN proteins in the centrosome of cells in mitosis. A decrease of STMN phosphorylation in the centrosome had been observed in fibroblast cells of FA patients. So, the FANC protein seems to be involved in the regulation of STMN phosphorylation during the cell cycle progression. Finally, the cells from FA patients display many mitotic spindle abnormities, such as an elevated number of supernumerary centrosomes and a decrease of mitotic spindle sizes.
Our study demonstrates that the FANCC protein is involved in the regulation of STMN phosphorylation during the cell division. Our results suggest also that the FANC protein seems participate in the cell cycle progression. Knowing that the STMN deregulation is involved in the carcinogenesis, an increase of STMN activity in the FA cells seems explain the sensibility of patients to develop various cancers. The STMN protein represents a possible therapeutic target to cancer treatment of FA patients.
VII
Table des matières
Résumé……….. ... III Abstract……… ... V Table des matières ... VII Liste des figures………… ... XI Liste des tableaux ... XIII Liste des abréviations ... XV Remerciement………….. ... XXIII Avant-propos…….. ... XXV
CHAPITRE 1 : INTRODUCTION ... 1
1.1 L’anémie de Fanconi ... 1
1.1.1 Origine et contexte actuel ... 1
1.1.2 Caractéristiques cliniques et cellulaires ... 3
1.1.2.1 Les malformations congénitales de l’anémie de Fanconi ... 3
1.1.2.2 Anomalies hématologiques de l’anémie de Fanconi ... 5
1.1.2.3 Développement de cancers chez les patients FA ... 7
1.1.2.4 Aspects cellulaires de l’anémie de Fanconi et diagnostics ... 8
1.1.3 Génétique moléculaire de l’anémie de Fanconi ... 10
1.1.3.1 Gènes associés à l’anémie de Fanconi et diagnostics moléculaires ... 10
1.1.3.2 Protéines associées à l’anémie de Fanconi ... 12
1.1.3.3 Voie de signalisation de l’anémie de Fanconi ... 20
1.1.4 Les fonctions exercées par les protéines Fanconi ... 23
1.1.4.1 Réplication et réparation de l’ADN ... 23
1.1.4.2 Régulation du cycle cellulaire ... 26
1.1.4.3 Rôle de la voie FA dans le processus de cancérogenèse ... 28
1.1.4.4 L’apoptose et la détoxification des radicaux oxygénés ... 30
1.1.4.5 Hématopoïèse dans l’anémie de Fanconi ... 32
VIII
1.2 Régulation du cycle cellulaire et mitose ... 35
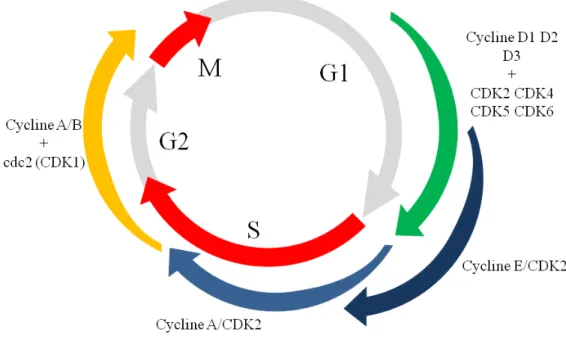
1.2.1 Le cycle cellulaire ... 35
1.2.2 Régulation du cycle cellulaire ... 39
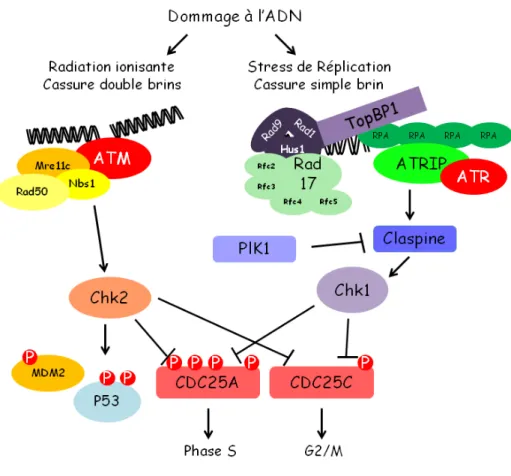
1.2.2.1 Réponse à un dommage à l’ADN par les kinases ATM et ATR ... 39
1.2.2.2 Régulation du cycle cellulaire par les CDKS ... 43
1.2.2.3 Régulation du cycle cellulaire par les protéines du centrosome ... 45
1.2.2.4 Régulation de la mitose ... 45
1.2.3 Dérégulation du cycle cellulaire et cancer ... 49
1.3 La protéine stathmine ... 52
1.3.1 Famille protéique de la stathmine ... 52
1.3.2 Régulation du cycle cellulaire par la stathmine ... 57
1.3.3 Implication de stathmine dans le développement de cancer ... 59
1.3.4 Rôle de stathmine dans l’hématopoïèse ... 60
CHAPITRE 2 : Contexte, hypothèses et objectifs de recherche ... 63
2.1 Caractérisation de la liaison entre FANCC et STMN1 et son implication dans la régulation de la phosphorylation de STMN1 ... 64
2.2. Implication des protéines FANC dans la régulation de l’activité de STMN1 ... 65
CHAPITRE 3 : La protéine C de l’anémie de Fanconi lie et régule la phosphorylation de stathmine ... 67 3.1 Avant propos... 68 3.2 Résumé ... 69 3.3 Abstract ... 70 3.4 Introduction ... 71 3.5 Results ... 74 3.6 Discussion ... 86
3.7 Materials and methods ... 89
3.8 Acknowledgments ... 93
3.9 Références ... 94
CHAPITRE 4 : Régulation de la phosphorylation de STMN1 dans un contexte d’anémie de Fanconi : conséquence sur la division cellulaire ... 99
IX
4.2 Introduction ... 101
4.3 Matériel et méthode ... 103
4.4 Résultats ... 105
4.5 Discussion ... 115
CHAPITRE 5 : Discussion et Perspectives ... 119
XI
Liste des figures
CHAPITRE 1 : INTRODUCTION
Figure 1. 1: Anomalies caractéristiques de l’anémie de Fanconi ... 4
Figure 1. 2 : Atteintes hématologiques chez les patients FA ... 6
Figure 1. 3 : Aspect cellulaire de l’anémie de Fanconi ... 9
Figure 1. 4: Représentation schématique des protéines FANC ... 13
Figure 1. 5: Voie canonique de l’anémie de Fanconi. ... 22
Figure 1. 6 : Réparation des ponts inter-brins durant la réplication de l’ADN ... 25
Figure 1. 7 : Les phases majeures du cycle de division cellulaire. ... 35
Figure 1. 8: La mitose. ... 38
Figure 1. 9 : Arrêt du cycle cellulaire suite à un dommage de l’ADN et l’activation de la voie ATM/ATR. ... 42
Figure 1. 10 : Contrôle du cycle cellulaire par les CDKs. ... 44
Figure 1. 11 : Régulation des différentes phases de la mitose. ... 48
Figure 1. 12 : La famille protéique stathmine. ... 52
Figure 1. 13 : Régulation de la phosphorylation de STMN1 ... 56
Figure 1. 14: Mode d’action de STMN1 sur la formation des microtubules. ... 58
CHAPITRE 3 : La protéine C de l’anémie de Fanconi lie régule la phosphorylation de stathmine Figure 3.1: STMN directly interacts with FANCC. ... 76
Figure 3.2 : FANCC colocalizes with STMN1 during the cell cycle. ... 78
Figure 3.3: FANCC is required for STMN1 phosphorylation during mitosis. ... 82
Figure 3.4 : Mutation in FANCC reduces STMN1 phosphorylation during mitosis. ... 83
Figure 3.5 : Reduced levels of phosphorylated STMN1 in FANCC mutant cells. ... 84
Figure 3.6: Mitotic spindles abnormalities in FANCC-deficient cells. ... 85
Figure 3.7 : Schematic interpretation of the role of FANCC in the control of mitotic events through its interaction with STMN1. ... 88
CHAPITRE 4 : Régulation de la phosphorylation de STMN1 dans un contexte d’anémie de Fanconi : conséquence sur la division cellulaire Figure 4.1: Une mutation de FANCA réduit la phosphorylation de STMN1 durant la mitose. ... 106
Figure 4.2: Une mutation de FANCD2 réduit la phosphorylation de STMN1 durant la mitose. ... 107
Figure 4.3 :L’absence de FANCA ou FANCD2 induit des défauts mitotiques. ... 110
Figure 4.4 : Niveau d’expression des kinases capables de phosphoryler STMN1 dans les cellules de patients FA. ... 112
XII
CHAPITRE 5 : Discussion et Perspectives
Figure 5.1 : Modèle proposé d’un mécanisme par lequel FANCC régule la phosphorylation de STMN1. ... 122 Figure 5.2: Modèle proposé d’un mécanisme par lequel les protéines FA régulent la
XIII
Liste des tableaux
XV
Liste des abréviations
ADN Akt APC ARM ATM ATP ATR ATRIP BRCA1/2 BACH1 BiP BRIP1 BRG1 BTB/POZ Bub3 BubR1 CaMK CD34 Cdc2 CDC14/20 CDC25C Cdh1 CDK Chk1/2 Cip/Kip CKI CRM1 CSH CTBP1 CYP2E1 DEB DKK1 DSB E2F-1 ECT2 EME1 ERCC1 ERCC4 FA FAAP100 FAAP24 FAN1 Acide DésoxyRibonucléique Thymoma viral proto-oncogene Anaphase-Promoting Complex Armadillo
Ataxia Telangiectasia and RAD3 related protein Adénosine TriPhosphate
Ataxia Telangiectasia Mutated protein kinase ART-interacting protein
BReast Cancer susceptibility protein 1/2 BRCA1-Associated C-terminal helicase 1 Binding Immunoglobulin Protein
BRCA1 interacting protein C-terminal helicase 1 Brahma-related gene 1
BR-C, Ttk and Bab / POx virus and Zinc finger Budding uninhibited by benzimidazole 3
Budding uninhibited by benzimidazole-Related 1
Ca2+/calmodulin-dependent protein kinase Cluster of Differentiation 34
Cell division control protein 2 Cell-Division Cycle protein 14/20 Cell division cycle 25 C
Cadherin 1
Cycline Dépendantes des Kinases Checkpoint kinase-1/2
CDK interacting protein/kinase inhibitory protein CDK Inhibitor
Chromosome Region Maintenance 1 Cellule Souche Hématopoïétique C-terminal binding protein-1 Cytochrome P450 2E1 DiEpoxyButane Dickkopf-1
Double Strand Break
E2 promoter-binding factor (E2F)-1 Epithelial Cell Transforming 2 Essential Meiotic Endonuclease 1
Excision Repair Cross-Complementing group 1 Excision Repair Cross-Complementing group 4 Fanconi Anemia – Anémie de Fanconi
FancA associated protein of 100kDa FancA associated protein of 24 kDa FA-associated nuclease 1
XVI FANC FAZF FOXM1 GM-CSF GTP HEF1 HEK293T Hela HES1 HL-60 Hsc70 Hsp70 Hus1 ICL IFAR IFN IL INK4 K562 KEAP1 KIS LMA Mad2 MAP MCC MDM2 MHF1/2 MUS81 MMC MRE11 MRN MT1-MMP Myt1 NADPH NBS1 Nek2 NER NES Nf-kB NHEJ NLS NRP1 Op18 p21cip/waf1
Fanconi Anemia protein – protéine de l’anémie de Fanconi (groupe de complémentation A à Q)
Fanconi Anemia Zinc Finger protein Forkhead box protein M1
Granulocyte / Macrophage Colony Stimulating Factors Guanosine-5'-triphosphate
Human Enhancer of Filamentation 1
Human Embryonic Kidney 293T cell – Cellules embryonnaires de rein humain 293T
Henrietta Lacks human epithelial carcinoma cell line Hairy and Enhancer of split 1
Human promyelocytic leukemia cells Heat Shock cognate 70
Heat shock protein 70
HUS1 checkpoint homolog (S. pombe) Interstand CrossLink
International Fanconi Anemia Registry Interféron
Interleukine
Inhibitors of CDK4
Human erythromyeloblastoid leukemia cell line Kelch-like ECH-associated protein 1
Kinase Interacting Stathmin Leucémie Myéloïde Aigue Mitotic Arrest Deficient 2 Microtubule Associeted Protein Mitotic Checkpoint Complex
Mouse Double Minute 2 homolog
FANCM-Associated Histone Fold protein ½ Methyl methansulfonate, UV Sensitive 81 MitoMycine C
Meiotic Recombination 11
Complexe MRE11, RAD50 et NSB1
Membrane Type 1-Matrix MetalloProteinase Myelin Transcription Factor 1
Nicotinamide Adenine Dinucleotide Phosphate reduced form Nuclear Export Signal – Signal d’export nucléaire
Nuclear factor kappa B
Never-in-mitosis-gene (NIMA)-related kinase 2 Nucléotide Excision R - excision de nucléotides Nuclear Export Signal – Signal d’export nucléaire Non-homologous end-joining
Nuclear Localization Signal – Signal de localization nucléaire
Neuropilin 1 Oncoprotéine 18
XVII p34cdc2 p33cdk2 PALB2 PCNA PIN1 PH PHD PHF9 PKA PKR PLK1 Polζ PP2A PRC1 PRDX3 PRL Rad17-RFC RAD50/51 Rb RB3 REV1 RING ROM RPA ROS SAC SAK/plk4 SCG10 SCLIP SLX1/4 STAT1 STMN SWI/SNF T2S TFDP TLE TNF TOME1 TopBP1 TPR TPX2 UAF1 UBE2T UBE2W UBF UBZ UNC5A
p34 associé à cdc2 (Cell Division Control protein 2 ) p33 associé à cdk2 (Cyclin Dependant Kinase 2) Partner And Localizer of BRCA2
Proliferating Cell Nuclear Antigen
Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 Potentiel Hydrogène
Plant Homeo Domain PHD finger 9
Protein Kinase
cAMP-dependent protein kinase Polo-Like Kinase 1
Polymerase zeta
Protein phosphatase 2A
Protein Regulator of cytokinesis 1
Thioredoxin-dependent peroxide reductase Protein tyrosine kinase
Rad17-Replication Factor C RADiation sensitive 50/51 Retinoblastoma protein
Rhodopirellula baltica SH 1
REVersionless 1
Really Interesting New Gene RNA I modulator protein Replicative Protein A Reactive Oxygen Species Spindle Assembly Checkpoint
Serine/threonine-protein kinase/polo-like kinase 4 Superior Cervical Ganglion Clone 10
SCG10-like protein
Synthetic Lethal of unknown (X) function 1/4
Signal Transduction and Activator of Transcription protein 1 Stathmine
Switch/Saccharose Non Fermentescibles Tubulin-2-stathmin
Transcription factor Dp-1 Transducing-like-enhancer Tumor Necrosis Factor Trigger of mitotic entry
DNA topoisomerase 2-binding protein 1
Several tetratricopeptide repeat Targeting protein for Xklp2 USP1-Associated Factor 1
UBiquitin-conjugating enzyme E2T Ubiquitin-conjugating enzyme E2W Ultrafine DNA bridges
Ubiquitin-binding domain Uncoordinated-5A
XVIII USP1 WD Wee1 Wnt XPF XRCC3 ou 9
Ubiquitine Specific Peptidase 1 Tryptophan-Aspartic acid dipeptide WEE1 G2 checkpoint kinase Wingless Int
Xeroderma Pigmentosum group F
XIX kDa ng µg mg µL mL nM µM mM α β γ ζ Étape G0 Étape G1 Étape S Étape G2 Étape M kilo Dalton nanogramme microgramme miligramme microlitre mililitre nanomolaire micromolaire milimolaire alpha beta gamma zeta Quiescence
Croissance et préparation à la réplication de l’ADN Réplication de l’ADN
Croissance et préparation à la mitose Mitose ou division cellulaire
Unité de mesure
Abréviation relative au cycle cellulaire
XXI Là où se trouve une volonté,
il existe un chemin.
Winston Churchill
Le succès, c’est d’aller d’échec en échec sans perdre son enthousiasme.
XXIII
Remerciement
Pour commencer, j’aimerais remercier Madeleine Carreau de m’avoir offert la chance de réaliser mon doctorat au sein de son laboratoire. Je souhaiterais également remercier notre collaborateur Sabine Elowe pour son expertise dans le domaine de la division cellulaire. Je voudrais remercier l’ensemble des équipes du Dr Carreau et du Dr Lévesque pour leur soutien indéfectible durant ces 4 dernières années.
Merci à tous les étudiants présents et passés de m’avoir offert d’innombrables moments de rire et de légèreté durant ma thèse. Je souhaiterais remercier Kevin Goggin, pour son aide informatique, Fang Fei Huang, Carolina Koutras et Tagrid Kaddar pour leur aide technique durant mes débuts au laboratoire. Merci à vous, Caroline Tendron, Delphine Masi et Mélody Mazon d’avoir fait souffler un vent de folie au laboratoire depuis 1 an. Un grand merci aux assistantes de recherche Marie-Chantal Delisle et Chantal Godin pour leurs connaissances mais surtout pour leur soutien et leur amitié durant toutes ces années. J’aimerais remercier tout particulièrement Caroline Huard pour son écoute durant les moments difficiles et son aide technique. Merci Caro pour toutes les conversations scientifiques et non-scientifiques que nous avons partagé. Merci à vous tous de m’avoir permis d’arriver au bout de cette expérience professionnelle.
J’aimerais remercier l’ensemble de mes amis pour ces quatre merveilleuses années que je viens de passer. Je n’oublierais jamais les nombreuses soirées, sorties et voyages que nous avons faits. Merci à vous tous, petit Mathieu, grand Matthieu, Caro, Anne-claire, Max, Benoit, Nadège, Audrey, Yanis, Marie-Lucie, Romain et surtout aux bretons Aurélie, Nico et le petit Evan. Merci à mes amis restés en France et dont l’amitié est restée intact malgré la distance. Merci à vous, Aurélie Houche, Aurélie Mathieu et Aline Villaume, d’être des amies fidèles depuis toujours et de partager ma vie depuis aussi longtemps. Merci à ma petite Cécile d’avoir eu le courage de venir faire sa thèse sur ce continent. Tu me permets de retrouver mes racines à chaque fois que nous nous voyons.
XXIV
J’aimerais également remercier mes parents Dominique et Jean-luc, ma sœur Mélanie et mon frère Clément pour leur soutien tout au long de mes études. Vous m’avez toujours soutenu dans les choix que j’ai pu faire, même dans celui de tout quitter pour venir m’installer à l’autre bout du monde pour poursuivre mes rêves. Merci de m’avoir laissé voler de mes propres ailes! Merci également à la famille Poulin, d’être présente.
Pour terminer, j’aimerais te remercier Éric, mon amour, pour ton aide, ton soutien et ta gentillesse durant ces deux dernières années. Tu es rentré dans ma vie et dans mon cœur pour ne jamais plus en ressortir. Sans toi, je ne pense pas avoir eu la force d’aller jusqu’au bout de mon doctorat. Tu m’as permis de croire qu’avec beaucoup de volonté et d’envie tout est possible.
XXV
Avant-propos
Cette thèse présente l’ensemble des analyses et des résultats obtenus au cours de mon doctorat portant sur l’étude de l’implication de la voie de signalisation de Fanconi dans la régulation du cycle cellulaire, via la protéine stathmine. Ce projet de recherche a été développé suite à l’identification de la protéine neuronale SGC10 ou STMN2 (Superior Cervical Ganglion clone 10), appartenant à la famille de la stathmine, comme un nouveau partenaire de la protéine Fanconi C (FANCC) par un criblage en double hybride. Mes travaux ont ensuite permis d’établir un lien étroit entre la voie de signalisation de Fanconi et la progression du cycle cellulaire, à travers la régulation de l’activité de stathmine. L’ensemble des résultats obtenus permet d’éclaircir en partie la susceptibilité accrue des patients atteints de l’anémie de Fanconi à développer différents types de cancers. Cette thèse représentant l’aboutissement de plus de 4 ans de travail, et apporte de nouvelles connaissances permettant le développement d’outils thérapeutiques dans le traitement de cancer chez les patients Fanconi.
Dans l’ensemble, ce mémoire présente 5 chapitres répartis en : une introduction générale (chapitre 1), la description du contexte du projet et des hypothèses (chapitre 2), les résultats obtenus en rapport avec mes deux objectifs principaux (chapitres 3 et 4) et une discussion permettant de commenter les points soulevés dans mes résultats (chapitre 5).
Le chapitre 3 fait l’objet d’une soumission dans le journal Biology Open. Pour ce chapitre, j’ai été responsable de la mise en place et de l’élaboration de l’ensemble des expériences présentées dans cet article. J’ai interprété les résultats, rédigé le manuscrit et mis en forme les figures. Dr Madeleine Carreau a dirigé les recherches, analysé les résultats et édité le manuscrit à l’aide des co-auteures. Dr Sabine Elowe a contribué à l’écriture du manuscrit et a fourni un soutient logistique.
Le chapitre 4 ne fait pas encore l’objet d’une soumission pour publication. Il correspond à l’ensemble des résultats obtenus suite à l’étude décrite dans le chapitre 3. J’ai participé à l’élaboration et à l’analyse de l’ensemble des expériences présentées dans ce chapitre. J’ai ensuite mis en formes les figures, interprété les résultats et rédigé le texte. Dr
XXVI
Madeleine Carreau a dirigé les recherches et analysé les résultats. Dr Sabine Elowe a contribué à l’écriture du manuscrit et a fourni un soutient logistique.
1
CHAPITRE 1 : INTRODUCTION
1.1 L’anémie de Fanconi
1.1.1 Origine et contexte actuelL’anémie de Fanconi (FA) a été décrite pour la première fois en 1927, par le pédiatre suisse Guido Fanconi, suite à la caractérisation d’une combinaison d’anomalies somatiques retrouvée dans une fratrie de trois enfants provenant d’une vallée alpine [1]. Ces trois frères présentaient une anémie aplasique progressive et diverses malformations congénitales, telles qu’une petite stature, un hypogonadisme et une hyperpigmentation de la peau. Depuis la découverte de la maladie en 1927, plus de 2000 nouveaux cas ont été rapportés dans la littérature [2-4].
La FA est une maladie génétique rare à transmission autosomique récessive, appartenant à un groupe de pathologies appelées maladies cassantes. Ces pathologies se caractérisent par l’observation de cassures chromosomiques spontanées ou induites par des agents susceptibles d’endommager l’ADN. L’anémie de Fanconi apparait dans toutes les régions du globe et touche toutes les ethnies avec une incidence d’environ 3 naissances par millions chaque année [2]. La fréquence des hétérozygotes est d’environ 1 personne sur 300 en Europe et aux États-Unis [5, 6]. De manière inexpliquée, la maladie semble être légèrement plus fréquente chez l’homme que chez la femme avec un ratio d’environ 1,2 :1 [4]. Comme toutes maladies récessives rares, sa fréquence augmente dans un contexte de consanguinité. Un effet fondateur est ainsi retrouvé chez la population blanche Afrikaner d’origine Boër d’Afrique du sud [7, 8], des juifs de descendance Ashkenazi [9, 10] ainsi que chez les gitans espagnols [11]. Entre 1982 et 2008, 1075 cases d’anémie de Fanconi sont reporté dans le nord du continent américain par IFAR (International Fanconi Anemia Registry) [2]. L’âge moyen lors du diagnostic de la maladie est de 6,5 ans. Toutefois l’identification de la pathologie peut se faire aussi bien dès la naissance, qu’à l’âge adulte. Le premier signe de l’anémie de Fanconi est l’observation d’une pancytopénie lors d’un hémogramme, associée à une augmentation du volume des globules rouges et de l’hémoglobine fœtale [12, 13]. La pancytopénie observée est due à une diminution du renouvèlement de
2
l’ensemble des lignées cellulaires hématopoïétiques et à la perte des cellules souches hématopoïétiques (CSH) de la moelle osseuse [12]. L’insuffisance médullaire chez les patients FA apparait durant les 10 ou 20 premières années de vie de la personne [14]. En plus de défauts hématologiques 70% des patients présentent une combinaison de malformations congénitales de sévérités variables dont les plus fréquentes sont des anomalies squelettiques pour 2/3 d’entre eux. L’observation de l’ensemble de ces premiers symptômes suggère un diagnostic d’anémie de Fanconi. Ce diagnostic posé par le médecin est crucial pour le développement d’une thérapie adéquate contre l’aplasie médullaire, car les patients FA présentent une hypersensibilité aux agents de chimiothérapie utilisés durant le processus de transplantation conventionnel des CSH [15].
L’identification d’anomalies cytogénétiques spécifiques des syndromes myélodysplasiques est recherchée annuellement par un myélogramme chez les patients FA. Lors de l’identification d’une anémie aplasique, un traitement par transfusions sanguines et par antibiothérapies est dans un premier temps mis en place. Toutefois, ce traitement n’est efficace que pour une courte durée. Lorsque l’insuffisance médullaire des patients FA s’aggrave, ou lors de l’apparition d’une anomalie clonale (leucémie), les médecins préconisent une transplantation allogénique en utilisant soit le sang du cordon ou les cellules souches hématopoïétiques [12]. Chez les patients FA, l’utilisation d’un traitement immunosuppresseur moins toxique, le fludarabine, est recommandée avant la transplantation. Le moment optimal de la greffe est également un facteur important pour la réussite du traitement. Les données actuelles suggèrent que le moment optimal d’une greffe est avant l’âge de 10 ans et avant 20 transfusions sanguines en prè-greffe [16]. Néanmoins, la transplantation des CSH corrige seulement les manifestations hématologiques de la maladie, puisque les autres anomalies dans les organes persistent et nécessitent un suivi pluridisciplinaire. Sachant que le développement de tumeurs solides est augmenté après une transplantation en particulier chez les patients FA qui développent une grave réaction de greffon contre l’hôte, la mise en place d’un dépistage précoce des tumeurs et une prise en charge des lésions précancéreuses est aussi nécessaire [17].
3 1.1.2 Caractéristiques cliniques et cellulaires
Les principales caractéristiques cliniques des patient FA peuvent se diviser en trois catégories : les malformations congénitales, les anomalies hématologiques et une susceptibilité aux développements des cancers.
1.1.2.1 Les malformations congénitales de l’anémie de Fanconi
L’expression clinique de FA est marquée par une grande hétérogénéité phénotypique, y compris dans la même famille, ce qui reflète l’hétérogénéité génétique de la maladie. Toutefois une majorité des patients, environ 70% présentent des malformations congénitales de sévérité variable [4]. Parmi ces anomalies congénitales, on retrouve fréquemment un retard statural débutant dès la vie intra-utérine par un retard de croissance, sans rattrapage post-natal pour la majorité (Figure 1.1A) [18]. Les causes de ce retard sont multifactorielles et dépendent des contextes génétiques, endocriniens, nutritionnels et digestifs. Plusieurs malformations squelettiques sont observables telles qu’une hypoplasie radiale caractérisée par l’absence du radius et/ou du pouce, des scolioses et des malformations vertébrales (Figure 1.B-C) [14]. Des anomalies cutanées sont présentes chez 40% des patients FA, de type hyper-pigmentation de la peau appelée tâche « café au lait » ou de types hypo-pigmentation (Figure 1.1D). Environ un quart des patients présente une dysmorphie faciale caractérisée par un visage triangulaire et une microcéphalie se traduisant par un retard mentale et une microphtalmie (Figure 1.1E).
4
Figure 1. 1:Anomalies caractéristiques de l’anémie de Fanconi
Les patients atteints de l’anémie de Fanconi peuvent présenter une petite stature (A), une absence du pouce ou du radius (B), une duplication du pouce (C), des tâches de type « café au lait » (D) et une microcéphalie (E). Photos tirées de [19-22]
Des malformations des reins et de l’appareil urinaire sont retrouvées chez environ 20% des patients [16]. Des anomalies oto-rhino-laryngologiques (ORL) telles que des malformations de l’oreille externe, une surdité, ou des malformations des osselets de l’oreille moyenne touchent environ 10% des patients [16]. Les anomalies associées au tube digestif concernent 7% des patients et sont associées à une anorexie, des nausées, des diarrhées et des douleurs abdominales [16]. Suite à une malformation du système génital, les patients des deux sexes présentent une diminution de leur fertilité. Chez la femme, l’infertilité est causée par des cycles menstruels irréguliers et anovulatoires durant lesquels aucun ovocyte n’est libéré par les ovaires. Chez l’homme, le problème de fertilité est dû à un défaut de l’appareil reproducteur, à un hypogonadisme et à une spermatogenèse anormale [23, 24]. La FA est généralement associée à un retard de croissance qui est lié à une déficience en hormone de croissance ou à de l’hypothyroïdisme [25]. Le retard de croissance peut entrainer un retard du développement mental [23]. D’autres anomalies endocriniennes sont reportées chez les patients FA, tel qu’un déséquilibre du métabolisme du glucose et de l’insuline (diabète), une anomalie du taux de gras dans le sang (dyslipidémie) et des syndromes métaboliques. (Tableau 1)
5 La gravité des défauts de développement chez les patients FA corrèle avec une hématopoïèse embryonnaire compromise [26]. L’atteinte hématologique est le plus souvent absente à la naissance et n’apparait que plus tard dans la vie du patient.
1.1.2.2 Anomalies hématologiques de l’anémie de Fanconi
Les caractéristiques cliniques les plus importantes de l’anémie de Fanconi sont hématologiques et constituent la cause principale de la mortalité de la maladie. Les patients FA, à leur naissance, ne présentent aucun défaut dans la numération des cellules sanguines. Le premier signe détectable est une macrocytose correspondant à une augmentation de la taille des globules rouges. Les patients développent ensuite progressivement une thrombopénie due à une diminution des plaquettes circulantes, puis une neutropénie due à une chute des neutrophiles dans le sang et pour finir une anémie aplasique.
Le principal symptôme développé chez les patients FA est une anémie aplasique ou pancytopénie causée par un défaut dans le renouvèlement des cellules souches hématopoïétiques (CSH) de la moelle osseuse [14]. Une coloration à l’hématéine et à l’éosine d’une coupe d’os d’une personne développant une anémie aplasique montre une perte du renouvellement des CSH de la moelle osseuse (Figure 1.2 A) en comparaison d’un tissu sain (Figure 1.2 B). La pancytopénie apparait chez les patients FA vers l’âge de 5 à 10 ans avec un âge médian à 7 ans, bien que cet âge puisse varier énormément [5, 27, 28]. Les patients atteints de la maladie FA présentent par conséquence une fatigue chronique, due à une perte des globules rouges, des problèmes d’hémorragies, dues à un déficit en plaquette, et des difficultés à combattre les infections dus à une diminution des leucocytes circulants.
6
Figure 1. 2 :Atteintes hématologiques chez les patients FA
Coupe d’os d’une personne développant une anémie aplasique (A) caractérisée par une hypocellularité de la moelle osseuse, en comparaison d’une personne saine (B). Les coupes de tissus ont été colorées à l’hématéine et à l’éosine.
Comparaison de frottis sanguins colorés au giemsa d’une personne développant un syndrome myélodysplasique (C) en comparaison d’une personne saine (D). [29-31]
Plus de 85% des patients présentent une aplasie médullaire avant l’âge de 20 ans [32]. Il est estimé que la mortalité chez 12% des patients FA serait due à des complications associées à une défaillance de la moelle osseuse et peut augmenter jusqu’à 38% à l’âge de 45 ans [28, 33]. Une greffe de moelle osseuse est aujourd’hui le seul traitement curatif préconisé contre les manifestations hématologiques liées à la défaillance médullaire des patients FA. Les patients qui survivent à l’âge adulte présentent un fort risque de développer un syndrome myélodysplasique, une leucémie aigüe ou des tumeurs solides [34-37]. Une coloration au giemsa du frottis sanguin d’une personne développant un syndrome myélodysplasique montre une prolifération aberrante des cellules myéloïdes dans le sang de patients atteints d’une leucémie myéloïde (Figure 1.2 C) en comparaison d’une personne saine (Figure 1.2 D).
7 1.1.2.3 Développement de cancers chez les patients FA
Les patients FA présentent un risque accru au développement de leucémies myéloïdes aiguës [36] d’environ 30% avant l’âge de 40 ans [5, 27, 33, 38] et peuvent développer d’autres syndromes myélodysplasiques avec un risque de 5%, tout âge confondu [39]. Les syndromes myélodysplasiques se caractérisent par une prolifération clonale et maligne d’une ou plusieurs lignées cellulaires myéloïdes, bloquées à un stade précoce de la différenciation cellulaire et incapable de maturation terminale [13]. La présence de 10% au minimum de cellules dysplasiques dans la moelle osseuse suffit pour déposer le diagnostic de syndrome myélodysplasique. La population cellulaire clonale du syndrome myélodysplasique envahit ensuite la moelle osseuse puis le système sanguin, et perturbe le fonctionnement des progéniteurs normaux en entrainant une insuffisance médullaire et le développement d’une leucémie myéloïde aiguë (LMA). Le risque de développer une LMA pour les personnes atteintes de FA est 500 fois supérieur à la population générale [40].
Les patients présentent également une susceptibilité accrue au développement de différents types de cancers solides [34, 41]. Il est estimé que les patients FA présentent une probabilité 50 fois plus élevé de développer un cancer que dans la population générale [33]. Plus particulièrement, le risque de développer un cancer avant l’âge de 45 ans chez les patients FA est de 76% [40]. Pour les patients FA, l’âge moyen de développement d’une tumeur solide est de 26 ans, alors que l’âge moyen d’apparitions des leucémies est de 11 ans [17]. Les tumeurs solides les plus développées sont des cancers des régions de la tête et du cou, du pancréas, des voies aériennes et digestives supérieures ainsi que celles des voies uro-gynécologiques [33, 38, 39, 42]. De plus, le risque de développer un cancer de la tête et du cou chez les patients FA est augmenté suite à une infection par le papillomavirus [43, 44]. Toutefois, d’autres études montrent que le papillomavirus ne semble pourtant pas jouer un rôle majeur dans le développement chez les patients FA des carcinomes de la tête et du cou [42]. Les patients FA ayant subit une transplantation de la moelle osseuse présentent un accroissement du risque de développer des tumeurs solides pouvant aller jusqu’à 42% à l’âge de 12 ans [39]. L’augmentation du risque serait reliée aux différents traitements d’irradiation ou à l’exposition d’agents cytotoxiques administrés en préparation de la
8
transplantation, ainsi qu’aux complications associées à cette transplantation, telles que les réactions du greffons contre l’hôte [40]. Toutefois l’impact de la transplantation sur le développement de cancer n’est pas encore bien défini.
L’apparition des anomalies hématologiques et cancéreuses au cours de la vie des patients FA rendent le diagnostic difficile à partir des simples observations cliniques. Le diagnostic définitif de la maladie est basé sur l’observation de cassures chromosomiques suite au traitement à des agents pontant l’ADN lors de la culture de lymphocytes de patients.
1.1.2.4 Aspects cellulaires de l’anémie de Fanconi et diagnostics
Le développement de cancers chez les patients FA serait due à une incapacité des cellules à maintenir l’intégrité génomique [45]. En effet, les lymphocytes provenant de patients FA présentent une instabilité chromosomique et une hypersensibilité aux agents pontant l’ADN comme le cisplatine, la mitomycine C (MMC) ou le diépoxybutane (DEB) [46]. Plus précisément, le traitement avec ces agents alkylants induit la formation de liens covalents entre les deux brins d’ADN d’une même hélice, et entraine la formation de cassures chromosomiques aléatoires à une fréquence anormalement élevée dans les cellules FA comparativement aux cellules saines. Au cours de la métaphase, les chromosomes s’apparient de manière imparfaite, formant ainsi des chromosomes quadrilatéraux, caractéristiques des cellules FA [47-49]. La formation de chromosomes quadrilatéraux est causée par une fusion de deux chromosomes suite à des erreurs de réparation de l’ADN double-brins.
Aujourd’hui, l’observation des dommages chromosomiques suite à un traitement à la MMC sur une culture de cellules lymphocytaires est couramment utilisée comme outil de diagnostic pour l’anémie de Fanconi [50]. Plus précisément, ce test consiste à cultiver les lymphocytes provenant du sang périphérique du patient avec un agent alkylant, comme la MMC, puis d’observer grâce à un caryotype l’apparition de chromosomes altérés qui peuvent être visualisés et quantifiés en microscopie au cours de la métaphase (Figure 1.3 A). D’autres tests sanguins incluant une analyse du cycle cellulaire et une évaluation de la mono-ubiquitination de FANCD2 pour certaines mutations, peuvent être réalisés comme
9 complément pour confirmer ou infirmer le diagnostic [51, 52]. Cependant des résultats faux négatifs peuvent être observés chez certains patients qui possèdent une mosaïque de cellules d’apparence normale et de cellules FA [53, 54]. Plus particulièrement, certains patients FA possédant des mutations différentes sur chaque allèles du gène peuvent, par un mécanisme de réversion génique, permettre la correction du gène muté dans certaines cellules souches [54, 55].
La formation des aberrations chromosomiques observése chez les patients FA entraîne un arrêt spontané du cycle cellulaire au cours de la phase S [56, 57] ou G2/M [58], observable en cytométrie en flux (Figure 1.3 B). Toutefois, la gravité des aberrations chromosomiques et l’arrêt en phase G2/M est variable d’un patient à l’autre. La grande hétérogénéité des anomalies phénotypiques observées chez les patients FA ainsi que les différentes caractéristiques des cellules FA traduisent l’implication de plusieurs protéines Fanconi dans le développement de la maladie.
Figure 1. 3 : Aspect cellulaire de l’anémie de Fanconi
Les lymphocytes des patients FA traités au DEB présentent des cassures chromosomiques ou des chromosomes quadrilatéraux au cours de la métaphase (A).
Les cellules de patients (HSC356N), présentant une mutation pour le gène FANCC, sont traitées à la MMC puis colorées au iodure de propidium et leur cycle cellulaire est analysé par cytométrie en flux. Les cellules de patients FA présentent un blocage du cycle cellulaire en G2/M suite à un traitement à la MMC en comparaison de cellules de patient complémentées avec un gène FANCC fonctionnel (HSC536N (+FAC)) (B). Les illustrations sont extraites de [59-61].
10
1.1.3 Génétique moléculaire de l’anémie de Fanconi
1.1.3.1 Gènes associés à l’anémie de Fanconi et diagnostics moléculaires
Jusqu’à présent, les chercheurs ont mis en évidence 16 gènes mutés responsables du développement de la maladie. Les gènes FANC identifiés jusqu’à présent se répartissent sur l’ensemble du génome humain et sont nommés : FANCA, FANCB, FANCC,
FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI,
FANCJ/BRIP1/BACH1, FANCL/PHF9, FANCM/HEF, FANCN/PALB2, FANCO/Rad51, FANCP/SLX4, et FANCQ/ERCC4 [62-83](Tableau 1). Ces gènes codent pour 16 protéines
connues pour coopérer ensemble dans la voie de signalisation de l’anémie de Fanconi. Les gènes FANC ne présentent aucune homologie de séquence entre eux et codent pour des protéines ne possédant aucun motif semblable qui pourrait les relier à une fonction commune. La majorité des gènes FANC est localisés sur des chromosomes homologues ou autosomes. Les chromosomes homologues sont des chromosomes de même taille et de même forme, possédant le même contenu génique. En opposition aux chromosomes homologue, les chromosomes hétérologues ou sexuels ne sont pas identiques. Un seul gène
FANC a été retrouvé sur le chromosome sexuel X, et il a été nommé FANCB.
Suite à un diagnostic d’anémie de Fanconi, une étude génétique des personnes est ensuite indispensable. Les mutations sont recherchées dans un premier temps sur les gènes
FANC les plus fréquemment mutés. Environs 70% des patients présentent une mutation
dans le gène FANCA, 10% dans le gène FANCC et 10% dans le gène FANCG, les 13 autres gènes mutés sont plus rares [49]. Plus de 100 mutations différentes sont reportées aujourd’hui pour le gène FANCA, 10 pour FANCC et 18 pour FANCG [84-87]. Pour les trois gènes les plus courants, une complémentation des cellules par un système de lentivirus ou par hétérocaryon, cellules à deux noyaux résultant d’une fusion cellulaire de deux patients distincts, peut être utilisée pour poser le diagnostic moléculaire. Cette dernière technique permet la fusion de deux cellules de patients FA induisant un phénotype normal, permettant ainsi d’identifier les patients comme appartenant à deux groupes de complémentation différents, alors que la fusion de deux cellules de patients appartenant au
11 même groupe de complémentation conduit à l’obtention d’un phénotype caractéristique des cellules FA.
Les protéines FANC sont connues pours coopérer ensemble à travers 3 complexes protéiques, conduisant à l’activation de la voie Fanconi. Une des étapes intermédiaire mais essentielle de cette voie est la mono-ubiquitination de la protéine FANCD2. Afin d’orienter les recherches vers le gène muté pouvant coder pour une protéine du complexe en amont ou en aval de la voie FA, un test de mono-ubiquitination de FANCD2 dans les fibroblastes de patients peut être réalisé [13]. Le gène en cause de la maladie peut être identifié également par la technique d’hybridation in situ en fluorescence par l’utilisation d’une sonde fluorescente complémentaire ou par séquençage [16, 88]. Certains patients ayant reçu un diagnostic d’anémie de Fanconi ne présentent aucune mutation dans les gènes Fanconi connus, suggérant la possibilité qu’il existe encore des gènes inconnus impliqués dans cette pathologie [89].
L’identification du gène muté est important pour la prise en charge du patient. Les personnes portant une mutation sur les gènes FANCD2 et FANCD1/BRCA2 présentent un phénotype plus sévère avec une progression plus rapide de la maladie et le développement de tumeurs [90-92]. Toutefois peu de preuves existent aujourd’hui pour associer le phénotype des patients FA au gène muté.
12
Tableau 1 : Gènes impliqués dans l’anémie de Fanconi
Les gènes responsables de la maladie sont décrits dans la 1ère colonne et codent pour des protéines possédant
des fonctions moléculaires très différentes (3e colonne). La localisation chromosomique (locus) de chacun des
gènes est identifiée dans la 2e colonne. Données provenant de l’IFAR (International Fanconi Anemia Registry).
1.1.3.2 Protéines associées à l’anémie de Fanconi
Les gènes FANC sont constitués de 1 à 44 exons et codent pour des protéines de différentes tailles, qui ont révélé très peu d’homologie entre elles. De plus, plusieurs protéines FANC possèdent certaines séquences non-répertoriées ne permettant pas de les relier à une fonction commune. Toutefois, la présence de domaines conservés suggère une fonction potentielle à ces protéines (Figure 1.4). Le rôle des protéines FANC dans la réparation de l’ADN commence à être bien connu (chapitre 1, 1.1.4.1). Depuis quelques années, la littérature montre que les protéines FANC jouent également un rôle dans d’autres mécanismes cellulaires tels que la régulation du cycle cellulaire (chapitre 1, 1.1.4.2), la cancérogenèse (chapitre 1, 1.1.4.3), l’apoptose et la détoxifications des radicaux oxygénés (chapitre 1, 1.1.4.4), le processus de l’hématopoïèse (chapitre 1, 1.1.4.5) et la régulation transcriptionnelle (chapitre 1, 1.1.4.6) [93]. Néanmoins, l’ensemble des mécanismes moléculaires cités précédemment restent encore peu connus jusqu’ à ce jour. Les protéines FANC répertoriées si dessous participent ensembles à la voie de signalisation FA qui est discutée dans le chapitre 1 partie 1.1.3.3.
13
Figure 1. 4: Représentation schématique des protéines FANC
Les protéines FANC sont classées en trois groupes. Le groupe I, constitué de FANCA, FANCG, FANCB, FANCL, FANCE, FANCC, FANCF, FANCM et des protéines associées FAAP100 et FAAP24 possède un rôle dans la monoubiquitination du complexe II, composé de FANCD2 et FANCI. Les protéines FANC possèdent différents domaines dont les domaines d’interaction protéine-protéine, comme les motifs TPR (Several tetradicopeptide repeat) ou BRC (motif d’interaction entre BRCA2 et Rad51), des répétitions WD40 (Tryptophan-Aspartic acid dipeptide) et ARM (Armadillo), des domaine BTB/POZ (BR-C Thk and Bad/Pox virus and Znc finger) et PDH/ring finger (Plant Homeo Domain), mais aussi un domaine d’attachement à l’ADN comme OB-fold (oligonucleotide/oligosaccharide-binding fold) ou des motifs coiled-coil impliqué dans la régulation de la transcription. De plus les protéines FANCM et FANCJ possèdent une activité hélicase et FANCO possède une activité recombinase. Tirée et modifiée de [94].
14
FANCA est une protéine majoritairement nucléaire, qui possède une séquence de
localisation nucléaire (NLS) et cinq domaines d’export nucléaire (NES) nécessaires à la translocation de la protéine au cytoplasme, dépendamment du CRM1 (Chromosome Region Maintenance 1) [81, 95]. Le gène FANCA est le premier gène Fanconi à avoir été cloné. C’est le consortium Fanconi qui est à l’ origine de cette découverte [80]. La protéine FANCA possèdent également une séquence « leucine zipper », caractéristique de certains facteurs de transcription [81]. Elle est retrouvée mutée dans deux cas sur trois des patients FA et représente le groupe de complémentation le plus important. Le complexe protéique formé par FANCA et FANCG est nécessaire au maintien du niveau des deux protéines et à l’accumulation nucléaire du complexe FA [96]. De plus, les cellules renfermant une mutation de FANCA présentent une diminution du niveau d’expression de FANCG [97].
FANCB se localise dans les compartiments nucléaire et cytoplasmique des cellules.
Elle possède toutefois une séquence, suggérant un rôle de FANCB dans le compartiment nucléaire. Le gène codant pour FANCB est le seule gène FANC se trouvant sur le chromosome X, expliquant ainsi que les patients de ce groupe sont exclusivement masculin [64].
FANCC ne possède aucun domaine de localisation cellulaire, et se retrouve
majoritairement dans le cytoplasme [98-101]. Toutefois, la translocation de la protéine FANCC dans le compartiment nucléaire est une étape essentielle à son activité. Une mutation ponctuelle dans le gène FANCC entrainant la substitution de la proline 554 en leucine empêche la translocation de la protéine FANCC au noyau et inhibe son activité fonctionnelle [102, 103]. FANCC est une protéine multifonctionnelle qui contrôle l’apoptose induite par le TNF-α (Tumor Necrosis Factor) et IFN-γ (Interféron) en se liant à la protéine Hsp70 [104] et protège les CSH des dommages oxydatifs [105, 106]. FANCC semble également jouer un rôle dans l’immunité inné [107] et l’inflammation [108]. Suite à un dommage à l’ADN, FANCC est nécessaire au maintien du point de contrôle G2 durant la réparation des dommages à l’ADN [109] et semble donc participer au contrôle du cycle cellulaire. Pour finir, FANCC est clivée lors de l’apoptose par les caspases 3 et 8 grâce à plusieurs sites putatifs de clivages [110].
15
FANCD1 communément appelée BRCA2, est une protéine majoritairement
nucléaire, dont la mutation biallelic du gène a été identifié en 2002 [62]. La protéine BRCA2 présente une capacité de liaison à l’ADN et un rôle dans la réparation des cassures double-brins de l’ADN par réparation homologue [111, 112]. L’étape clé de la réparation homologue est le recrutement de la recombinase Rad51 au site de cassure. Pour ce faire, BRCA2 possède plusieurs domaines BRC permettant une interaction avec Rad51. L’interaction BRCA2/Rad51 conduit à une séquestration de Rad51 aux foci nucléaires [113]. De plus, la protéine BRCA2 participe à la régulation du cycle cellulaire via une liaison avec les kinases Plk1 et CDK connues pour être impliquées dans la progression du cycle cellulaire [114]. Plus précisément, la kinase CDK (Cyclin Dependent kinase) phosphoryle BRCA2, favorisant ainsi son interaction avec Rad51. En parallèle, la kinase Plk1 phopshoryle Rad51 en association avec BRCA2, puis permet le recrutement de Rad51 aux foci de stress de réplication [114]. La protéine BRCA2 semble donc participer à la régulation du cycle cellulaire mais aussi de la mitose. Une étude montre que BRCA2 se localise au centrosome durant la mitose et participe à la duplication et à la séparation des centrosomes [115]. La phosphorylation de BRCA2 par Plk1 contribue aussi à la régulation de la cytokinèse durant la division cellulaire [116]. Pour finir, le gène BRCA2 est identifié comme responsable des formes familiales du cancer du sein [92] et des ovaires [117].
FANCD2 présente de nombreuses modifications post-traductionnelles. Le gène
codant pour FANCD2 a été cloné pour la premier fois en 2001 [74]. Lors d’un dommage à l’ADN, FANCD2 est monoubiquitinilée et se localise avec FANCD1/BRCA2, BRCA1 et Rad51 aux foci nucléaires [118]. Le site de monoubiquitination de FANCD2 a été identifié chez l’homme sur la lysine 561 et chez le poulet sur la lysine 563 [119]. La monoubiquitination de FANCD2 grâce à son rôle de plateforme de recrutement, permet le recrutement de protéines impliquées dans la réparation à l’ADN au site de dommage à l’ADN. La protéine FANCD2 joue donc un rôle important dans la réparation des cassures doubles-brins de l’ADN [118, 120, 121]. A la sortie de la phase S, FANCD2 est déubiquitinilée par USP1 (Ubiquitine Specific Peptidase 1) [122]. Cette inactivation de la protéine permet une libération de FANCD2 aux sites de réparation de l’ADN et le recyclage de la protéine. FANCD2 peut également être phosphorylée par deux kinases ATM (Ataxia Telangiectasia Mutated protein kinase) et ATR (Ataxia Telangiectasia and
16
RAD3 related protein) en présence de radiation ionisante en plus d’être monoubiquitinilé [123].
FANCE renferme deux motifs NLS, suggérant une localisation majoritairement
nucléaire de la protéine [78]. En 2000, une étude met en évidence le gène FANCE contenant 10 exons et codant pour une protéine de 536 acides aminsé [78]. L’accumulation nucléaire de FANCE n’est pas seulement dépendante des motifs de localisation au noyau, mais aussi de FANCC [124]. FANCE présente aussi la capacité de se lier à FANCC et à FANCD2, jouant ainsi un rôle de pont moléculaire entre le complexe I et FANCD2 [125, 126]. Suite à un dommage à l’ADN, FANCE est phosphorylée par Chk1 sur deux sites conservés, permettant l’activation de la voie de Fanconi [127]. Durant la progression normale de la phase S, FANCE colocalise avec FANCD2 aux foci de réparation de l’ADN [124]. FANCE renferme également des motifs « FANC repeat » appartenant à la famille des « two- and three-helical motifs » que l’on peut retrouver sur FANCD2 et FANCI [128].
FANCF est une protéine majoritairement nucléaire qui joue un rôle de pont entre
les sous-complexes FANCC/FANCE et FANCA/FANCG [129]. Elle a un rôle essentiel à l’interaction des membres protéiques du complexe I et permet la bonne monoubiquitination de FANCD2 [130]. FANCF possède également une région homologue à la protéine ROM (RNA I modulator) capable de lier les acides désoxyribonucléiques, suggérant ainsi une liaison potentielle à l’ADN de la protéine FANCF [67].
FANCG/XRCC9 (X-ray Repair Cross Complementing) est une protéine
majoritairement nucléaire qui présente la capacité d’interagir avec FANCA sur sa région NLS, permettant ainsi son accumulation dans le noyau et le prolongement de la demi-vie du complexe I [79, 95-97, 131]. L’interaction entre FANCGG et FANCA est nécessaire à la résistance à la MMC [132]. FANCG est également capable de recruter plusieurs protéines FANC via une répétition de 7 motifs TRP (Several tetratricopeptide repeat) permettant des interactions protéine-protéine nécessaires à la formation du complexe I [133]. La protéine FANCG phosphorylée sur la sérine 7 est requise pour la formation d’un complexe protéique contenant BRCA2, FANCD2 et XRCC3, suggérant un rôle de FANCG dans la réparation de l’ADN par recombinaison homologue indépendant de la voie FA [134].
17 FANCG constitue un des groupes de complémentation les plus courants chez les patients FA.
FANCI possède une similarité de séquence avec la protéine FANCD2. Tout comme
FANCD2, FANCI est monoubiquitinilée lors de dommage à l’ADN et peut être phosphorylée par les kinases ATM et ATR [135, 136]. Les deux protéines FANCI et FANCD2 jouent un rôle important dans leur monoubiquitination mutuelle et dans leur maintenance réciproque [137, 138]. Les protéines FANCI et FANCD2 colocalisent ensemble sur la chromatine en réponse à un dommage à l’ADN et possèdent des motifs de répétition Armadillo (ARM), impliqués dans les interactions protéine-protéine [139].
FANCJ/BRIP1 (BRCA1 interacting protein C-terminal helicase 1) /BACH1
(BRCA1-Associated C-terminal helicase 1) est une protéine nucléaire possédant une séquence NLS et un domaine hélicase qui permet de résoudre les structures secondaire de l’ADN au cours de la réplication [82, 83, 140-142]. Elle est impliquée dans le redémarrage de la réplication de l’ADN durant la réparation des ICL (Interstand Crosslink) et des cassures doubles brins. FANCJ s’associe à la protéine RPA (Replicative Protein A), connue pour lier l’ADN simple brin dans les foci nucléaires lors de la réparation ou de la réplication de l’ADN [142]. Cette liaison favorise l’activité hélicase de FANCJ en déroulant les intermédiaires de l’ADN afin de maintenir la stabilité génomique, ce qui prouve un rôle direct de FANCJ dans la réparation à l’ADN. La protéine FANCJ peut inhiber Rad51 par son activité ATPase et ainsi participe à régulation de la réparation homologue [143]. La protéine FANCJ est également connue pour être impliquée dans le développement du cancer du sein [144].
FANCL/PHF9 (PHD finger 9) renferme un motif en doigt de zinc de type PHD
(Plant Homeo Domain) responsable de l’activité E3 ubiquitine ligase de la protéine, suggérant que FANCL est responsable de la monoubiquitination des protéines FANCD2 et FANCI lors d’un dommage à l’ADN [70]. FANCL joue ainsi un rôle crucial dans l’activation de la voie métabolique de Fanconi [71]. L’activité de monoubiquitination de FANCL est régulée par une liaison avec l’enzyme conjuguante de l’ubiquitine UBE2W (UBiquitine-conjugating enzyme E2W) [145]. La fonction de FANCL peut être stabilisé
18
par la voie PI3K/Akt1 impliquée dans la survie des CSH, suggérant ainsi un rôle de FANCL dans le maintien des cellules souches [146]. De plus, la protéine FANCL participe au processus d’hématopoïèse en favorisant l’augmentation de l’activité et l’expression de la β-catenine, un facteur clé de la voie de Wnt [147].
FANCM/HEF1 (Human Enhancer of Filamentation 1) appartient à la superfamille des
hélicases 2, dont l’activité est nécessaire à la résolution de structures secondaires observables au cours de la réplication de l’ADN [148]. FANCM présente également une la capacité de se lier à l’ADN au niveau de structure spécifique tel que les fourche de réplication ou les D-loop [148]. La protéine FANCM possède également une activité ATPasique permettant sa liaison à l’ADN et une activité de translocation permettant à FANCM de se déplacer le long du brin d’ADN [148]. De plus, la protéine FANCM permet la résolution des jonctions de Holliday, les D-loop et les fourches de réplication mobiles [148]. Durant la phase S, FANCM est modérément phosphorylée, tandis qu’elle est hyper phosphorylée durant la mitose [149]. L’augmentation de la phosphorylation de FANCM durant le cycle cellulaire est inversement proportionnelle avec la monoubiquitination de FANCD2 et l’association de FANCG à la chromatine, suggèrant ainsi un rôle de régulation de la voie FA durant la mitose par FANCM [121, 150]. La phosphorylation de FANCM est réalisée par la kinase Plk1 qui conduit ensuite à une dégradation de FANCM durant la mitose [151]. De plus, une mutation de FANCM conduit à une diminution du niveau des protéines FANCA, FANCG et moindrement de FANCL dans le noyau, laissant supposer l’importance de FANCM dans l’intégrité du complexe I dans le noyau [71].
FANCN/PALB2 (Partner And Localizer of BRCA2) est une protéine majoritairement
nucléaire jouant un rôle déterminant dans l’interaction de BRCA1 et BRCA2 [152]. La protéine FANCN possède un domaine WD40 nécessaire à sa liaison avec BRCA2 et Rad51c [153]. Sa présence est essentielle au processus de recombinaison homologue. FANCN participe également à la régulation du stress oxydatif en se liant à KEAP1 (Kelch-like ECH-associated protein1), un senseur du stress oxydatif. En se liant à KEAP1, FANCN permet l’accumulation du facteur de transcription anitioxydant NRF2 dans le noyau et l’activation de ses gènes cibles. L’interaction KEAP1-FANCM compétitionne avec KEAP1-NRF2, permettant ainsi l’augmentation de l’activité de NRF1 au noyau. En se
19 liant à KEAP1, FANCM diminue l’export de NRF2 vers le cytoplasme et ainsi sa dégradation [154]. PALB2 est mutée dans les cancers du sein [155] et les cancers pédiatriques [156].
FANCO/ Rad51 (RADiation sensitive 51)a été récemment identifiée comme une protéine
FANC [157]. Dans le génome humain, on retrouve 5 gènes paralogues de Rad51. La protéine Rad51 est connue comme une recombinase, jouant un rôle clé dans la recombinaison homologue des cassures double-brin [157, 158]. Suite à un traitement à l’hydroxyuré ou à une irradiation aux ultra-violets, Rad51, BRCA1 et BRCA2 forment un complexe localisé aux fourches de réplications de l’ADN [159].Durant la phase S et G2 du cycle cellulaire, Rad51 se localise aux foci de réparations avec les protéines RPA, BRCA2 et PALB2 [159].
FANCP/SLX4 (Synthetic Lethal of unknown (X) function 4) est une des protéines Fanconi
contenant le plus de domaines d’interactions protéiques suggérant que FANCP agit comme une plateforme de recrutement de plusieurs partenaires protéiques impliqués dans la réparation à l’ADN et la formation d’un complexe fonctionnel [160]. La protéine FANCP possède deux domaines de liaison à l’ubiquitine de type UBZ, nécessaire à son recrutement aux ICL (Interstand CrossLink) ou à la résolution des jonctions de Holliday [161]. FANCP a pour fonction le maintien de la stabilité du génome en orchestrant les réparations ICL, la résolution des jonctions d’Holliday, la restauration des fourches de réplication et le maintien des télomères [162].
FANCQ/XPF (Xeroderma Pigmentosum group F)/ERCC4 (Excision Repair
Cross-Complementing group 4) est impliquée dans la voie de réparation de l’ADN par son activité d’excision de nucléotides (NER) [72]. Récemment, ERCC4 a été associée à un nouveau groupe de complémentation de l’anémie de Fanconi (FANCQ). Une mutation sur le gène FANCQ/ERCC4 est également retrouvée dans les cancers du sein héréditaire [163].
Pour conclure, les protéines FANC possèdent des caractéristiques structurales et fonctionnelles différents suggèrent l’implication des protéines dans plusieurs mécanismes
20
de régulation cellulaire (Figure 1.4). L’absence de fonction commune entre les différentes protéines FANC suggère qu’elles agissent comme intermédiaires dans une voie de canonique sous forme de plusieurs complexes multi-protéiques.
1.1.3.3 Voie de signalisation de l’anémie de Fanconi
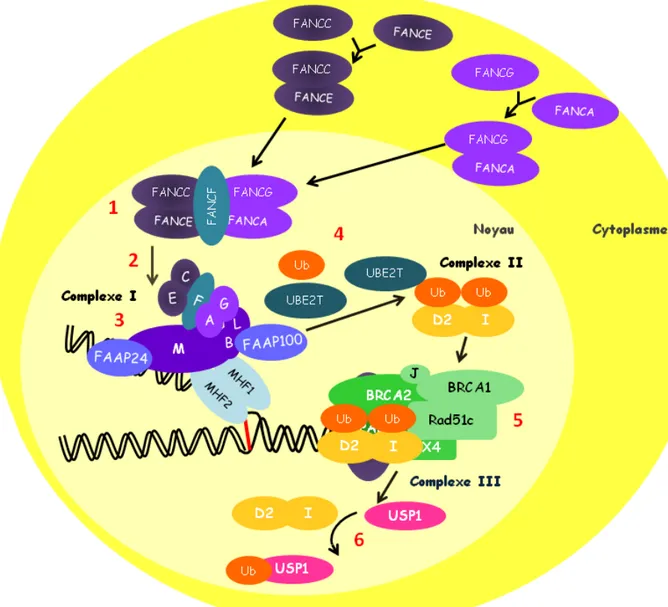
La majorité des protéines FANC ont la particularité de s’accumuler dans le compartiment nucléaire pour former trois complexes protéiques, conduisant à l’activation de la voie de Fanconi, impliqués dans différents mécanismes cellulaires dont la réparation à l’ADN (Figure 1.5).
Suite à un dommage à l’ADN, les protéines FANCA et FANCG d’une part puis FANCC et FANCE d’autre part, s’associent dans le cytoplasme et relocalisent ensuite dans le compartiment nucléaire. Les deux sous-complexes ainsi formés interagissent ensemble par l’intermédiaire de la protéine FANCF [164]. Les protéines FANCB, FANCM, FANCL viennent s’associer ensuite pour former un complexe d’environs 650 kDa, appelé complexe I [165]. D’autres protéines ont été identifiées comme des partenaires essentiels du complexe I, dont FAAP100 (FancA associated protein of 100kDa) [166], FAAP24 (FancA associated protein of 24 kDa) [167], MHF1/MHF2 (FANCM-Associated histone fold protein 1 et 2) [168] et Hes1 (Hairy and Enhancer of split 1) [169]. Toutefois aucune mutation sur ces protéines chaperons n’a été identifiée comme induisant l’anémie Fanconi. Le complexe I est donc une association de protéines chaperons et de protéines FANC, qui interagissent ensemble grâce à plusieurs domaines protéiques nécessaires à la stabilité du complexe. En réponse à un dommage à l’ADN, FANCM associée aux protéines FANC du complexe I, reconnait les fourches de réplication bloquées et participe à leur résolution grâce à son activité hélicase et translocase [170]. Pour finir, FANCM recrute à la chromatine le complexe I via une interaction impliquant une hétérodimérisation de FANCM avec FAAP24 [167, 171].
La fonction principale du complexe I ainsi formé est de modifier par mono-ubiquitination FANCD2 et FANCI sur leurs résidus lysine 561et 523 respectivement [73, 137, 172]. Les protéines FANCD2 et FANCI sont les deux seules cibles connues du complexe I. Elles interagissent ensemble pour former le complexe II ou complexe ID. Les
21 protéines FANCD2 et FANCI ont besoin de s’associer avant d’être mono-ubiquitinilées par le complexe I [73, 137]. FANCL, grâce à son domaine PHD caractéristique des ligases ubiquitines de type RING, est la protéine du complexe I responsable de la mono-ubiquitination de FANCD2 et FANCI [173]. Les protéines FANCE et FANCD2 possèdent la capacité d’interagir ensemble, suggérant un rôle de FANCE dans l’identification d’un substrat spécifique du complexe I [174]. L’ubiquitination d’une protéine nécessite la participation de plusieurs molécules, dont une enzyme activatrice (E1), un conjugateur (E2) et une ligase (E3), chacune responsable d’une activité précise [175, 176]. La protéine UBE2T (Ubiquitine-conjugating enzyme E2T), un conjugateur de l’ubiquitine E2, interagit directement avec la ligase FANCL grâce à son motif PHD. La protéine UBE2T transfère ensuite la molécule d’ubiquitine au complexe I activé, ce qui permet la mono-ubiquitination du complexe II [177, 178]. L’activateur d’ubiquitine E1 du complexe I reste encore aujourd’hui inconnu.
Une fois activé, le complexe II est recruté à la chromatine et s’associe avec les protéines FANC impliquées dans la réparation à l’ADN. Plus précisément, FANCD2-FANCI se lient aux protéines FANCD1, FANCJ, FANCN, FANCP et Rad51c, formant le complexe III et agissant en aval de la voie Fanconi [75, 179, 180]. Durant la transition G2/M et suite à la réplication ou à la réparation de l’ADN, FANCD2 et FANCI sont dé-ubiquitinées par le complexe enzymatique UAF1 (USP1-Associated Factor 1)//USP1 (Ubiquitine Specific Peptidase 1), permettant ensuite le relargage des deux protéines et le retour à un niveau basal de la voie FA [122, 181].
Pour conclure, la voie canonique de l’anémie de Fanconi est constituée d’une cascade signalétique comprenant la formation du complexe I, la mono-ubiquitination des protéines du complexe II, et le recrutement à la chromatine des protéines du complexe III (Figure 1.4). La voie FA est impliquée dans divers mécanismes cellulaires tels que la réparation à l’ADN [182], l’apoptose [183], la détoxification des radicaux oxygénés [184], l’hématopoïèse [185] et la régulation du cycle cellulaire [45].
22
Figure 1. 5: Voie canonique de l’anémie de Fanconi.
1- Suite à un dommage à l’ADN, les hétérodimères FANCA/FANCG et FANCC/FANCE sont stabilisés par une interaction avec la protéine FANCF. 2- Le sous-complexe FANCA/FANCG/FANCC/FANCE/FANCF s’associe ensuite avec les protéines FANCB et FANCL. 3- Le complexe I est chargé à la chromatine suite à sa liaison avec la protéine FANCM. 4- L’ubiquitination de complexe II est ensuite réalisé par le complexe I via le conjugateur de l’ubiquitine UBE2T et l’ubiquitine FANCL. 5- Les protéines FANCD2 et FANCI du ubiquitinilées, s’associent avec les constituants du complexe III sur la chromatine. 6- Pour finir, le complexe II est ensuite reconnu par la peptidase d’ubiquitine USP1, qui retire les molécules d’ubiquitine et le recyclage du complexe II, le rendant de nouveau disponible pour être activé par le complexe I.