Réponse des macrophages pulmonaires au
surfactant pulmonaire oxydé
Mémoire
Mélanie Hamel-Auger
Maîtrise en Médecine expérimentale
Maître ès sciences (M. Sc.)
Québec, Canada
Réponse des macrophages pulmonaires au
surfactant pulmonaire oxydé
Mémoire
Mélanie Hamel-Auger
Sous la direction de :
Résumé
Problématique : Le surfactant pulmonaire est une structure vitale essentielle à
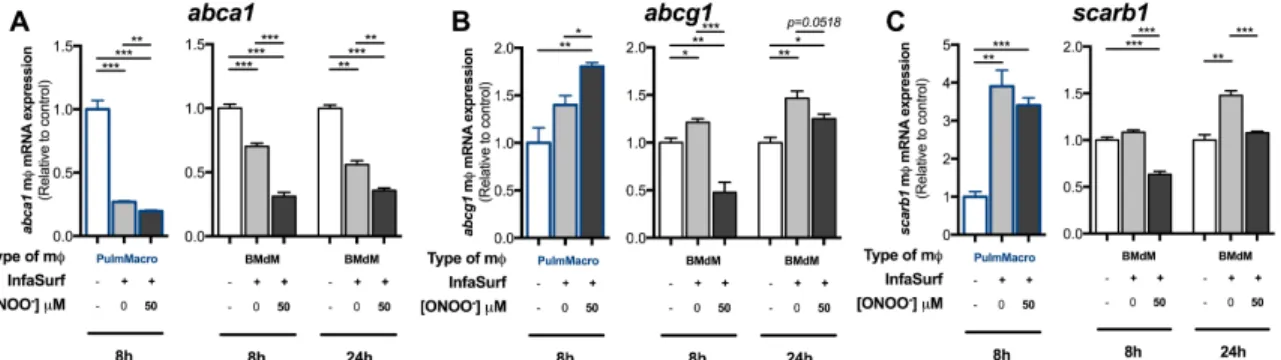
l’homéostasie pulmonaire. Étant composé majoritairement de phospholipides, il est particulièrement susceptible à l’oxydation puisqu’il est en constante interaction avec l’environnement extérieur. Par des mécanismes encore inconnus, les macrophages pulmonaires jouent un rôle majeur dans l’élimination du surfactant pulmonaire endommagé. Objectifs : 1) Caractériser la réponse transcriptionnelle et fonctionnelle des macrophages au surfactant pulmonaire sain et traité au peroxynitrite (ONOO-) et 2) investiguer le rôle de l’endocytose dans l’initiation de cette réponse. Méthodes : Des macrophages murins isolés par lavages bronchoalvéolaires ou différenciés de la moelle osseuse ont été exposés à du surfactant pulmonaire sain ou traité au ONOO-, avec ou sans Latrunculine A, un inhibiteur des mécanismes d’endocytose dépendant de l’actine. L’expression de gènes impliqués dans la capture (marco, msr1, cd36), l’accumulation intracellulaire (plin2) et l’export (abca1, abcg1, srb1) lipidique a été évaluée par qPCR, ainsi que les capacités fonctionnelles d’export lipidique. Résultats : L’addition de surfactant pulmonaire sain ou traité au ONOO- augmente l’expression de marco, msr1, cd36 et plin2 et diminue l’expression d’abca1, abcg1 et scarb1 chez les macrophages. Toutefois, certaines différences transcriptionnelles ont été observées entre les macrophages pulmonaires primaires et ceux dérivés de moelle osseuse. Également, qu’il soit sain ou oxydé, le surfactant engendre une diminution des capacités d’efflux de cholestérol des macrophages différenciés. De plus, ces impacts transcriptionnels et fonctionnels ne semblent pas affectés par un traitement à la Latrunculine A. Conclusion : En présence de surfactant, et ce, indépendamment son niveau d’oxydation, les macrophages pulmonaires semblent favoriser les mécanismes de capture et d’accumulation lipidique. Ainsi, aux dépens de ces mécanismes priorisés, les deux espèces présentent conjointement une diminution à la fois transcriptionnelle et fonctionnelle des mécanismes d’export lipidique.
Abstract
Problematic: Pulmonary surfactant is a vital structure essential to reduce the surface tension at the liquid-air interface. It is mainly composed of phospholipids and is susceptible to oxidation due to its close interaction with the external environment. Pulmonary macrophages play a major role in degrading damaged surfactant. However, the mechanisms used by pulmonary macrophages to detect and initiate its degradation are still unknown. Objectives: 1) To characterize the transcriptional and functional response of macrophages to native and peroxynitrite
(ONOO-)-treated pulmonary surfactant and 2) to investigate the role of endocytosis
in the initiation of this response. Methods: Primary mouse pulmonary
macrophages isolated from bronchoalveolar lavages or differentiated from the
bone marrow were exposed to native or ONOO--treated pulmonary surfactant with
or without endocytosis inhibitors (Latrunculin A). Expression of key genes implicated in lipid capture (msr1, marco, cd36), intracellular lipid accumulation (plin2), and lipid export (abca1, abcg1, scarb1) was assessed by quantitative PCR, as well as functional lipid export capacities. Results: The addition of native and
ONOO--treated pulmonary surfactant increased marco, msr1, cd36 and plin2
expression and decreased abca1, abcg1 and scarb1 expression in macrophages. However, some transcriptional differences were observed between primary and
bone marrow macrophages. Also, both native and ONOO--treated pulmonary
surfactant generated a decrease in differentiated macrophages cholesterol efflux capacities. Moreover, those transcriptional and functional impacts do not seem to be affected by Latrunculin A treatment. Conclusion: In the presence of pulmonary surfactant and this, independently its oxidation level, lung macrophages seem to promote lipid capture and storage mechanisms. Thus, depending on these prioritized mechanisms, the two species jointly showed a transcriptional and functional decrease in lipid export mechanisms.
Table des matières
Résumé ... iii
Abstract ... iv
Liste des figures ... vii
Liste des abréviations et des sigles ... viii
Remerciements ... xi
Avant-propos ... xii
Chapitre I : Introduction générale et problématique ... 1
1.1 Le surfactant pulmonaire ... 3
1.1.1 Les rôles du surfactant pulmonaire ... 3
1.1.2 La formation du surfactant pulmonaire ... 4
1.1.3 Les rôles et les proportions des composantes du surfactant pulmonaire . 7 1.2 L’oxydation du surfactant pulmonaire ... 11
1.2.1 Les causes de l’oxydation du surfactant pulmonaire ... 11
1.2.2 Les différentes sources d’espèces oxydatives ... 11
1.2.3 L’oxydation lipidique ... 14
1.2.4 L’impact fonctionnel de l’oxydation du surfactant pulmonaire ... 16
1.3 La biologie lipidique des macrophages pulmonaires ... 18
1.3.1 Le transport lipidique par les macrophages pulmonaires ... 19
1.3.1.1 La capture lipidique ... 21
1.3.1.2 L’accumulation intracellulaire lipidique ... 24
1.3.1.3 L’export lipidique ... 26
1.3.1.4 Les mécanismes d’endocytose des macrophages ... 30
1.4 Hypothèses et objectifs de recherche ... 33
1.4.1 Hypothèses de travail ... 33
1.4.2 Objectifs spécifiques (manipulations in vitro) ... 33
Chapitre II : Impact of pulmonary surfactant on cholesterol efflux and expression of genes associated with lipid transport in macrophages ... 34
2.1 Avant-propos ... 35
2.1.1 Contributions des auteurs ... 35
2.2 Résumé ... 36
2.3 Abstract ... 38
2.4 Introduction ... 39
2.5 Methods ... 41
2.5.1 Culture of mouse pulmonary and bone marrow-derived macrophages .. 41
2.5.1.2 Isolation and culture of bone marrow-derived macrophages (BMdM)
... 41
2.5.2 Macrophage in vitro treatments and processing ... 42
2.5.2.1 Treatment with native and ONOO--treated pulmonary surfactant .... 42
2.5.2.2 Bone marrow-derived macrophages treatment with endocytosis inhibitor Latrunculin A ... 43
2.5.2.3 Cholesterol efflux assay with bone marrow-derived macrophage. ... 43
2.5.3 Quantitative PCR ... 43
2.5.4 Statistical analysis ... 45
2.6 Results ... 46
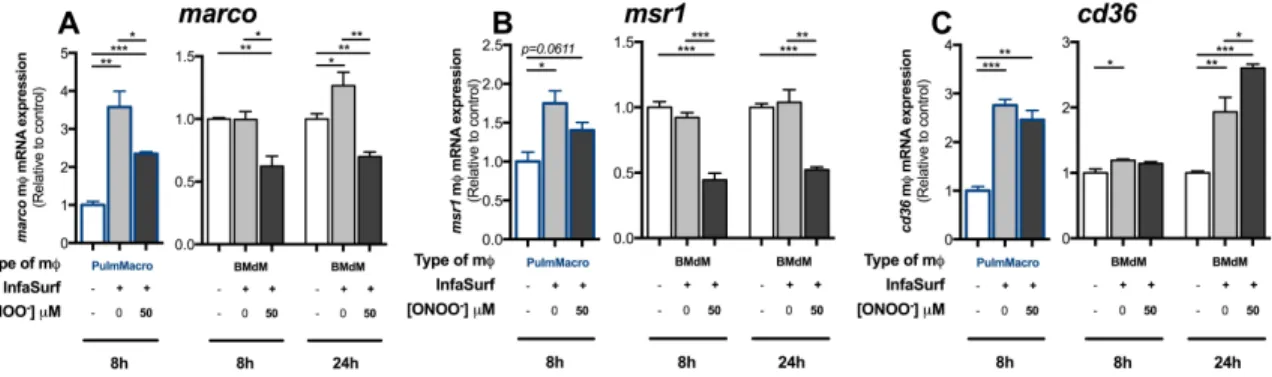
2.6.1 Impact of native and ONOO--treated pulmonary surfactant on key genes involved in reverse lipid export ... 46
2.6.2 Impact of native and ONOO--treated pulmonary surfactant on cholesterol efflux capacity ... 46
2.6.3 Impact of native and ONOO--treated pulmonary surfactant on key genes implicated in lipid capture and intracellular lipid accumulation ... 47
2.6.4 Impact of endocytosis inhibition on the response to native and ONOO- -treated pulmonary surfactant ... 47
2.7 Discussion ... 49
2.8 References ... 52
2.9 Figure legends ... 55
2.10 Figures ... 57
Chapitre III : Discussion générale ... 60
3.1 Portait comparatif de la caractérisation transcriptionnelle des macrophages pulmonaires et des BMdM stimulés au surfactant pulmonaire sain ou traité au ONOO- ... 61
3.2 Caractérisation fonctionnelle de la réponse au surfactant pulmonaire sain ou traité au ONOO- chez les BMdM ... 63
3.3 Investigation du rôle de l’endocytose lipidique dans la réponse des BMdM au surfactant pulmonaire sain ou traité au ONOO- ... 64
3.3.1 Mécanismes d’action des macrophages proposés en réponse au surfactant pulmonaire sain ou traité au ONOO- ... 66
3.4 Forces et limitations de l’étude ... 69
3.5 Perspectives ... 73
3.6 Conclusion générale ... 73
Liste des figures
Chapitre I
Figure 1.1 Le métabolisme du surfactant pulmonaire. ... 6
Figure 1.2 Structure et composition du surfactant pulmonaire. ... 10
Figure 1.3 Le cycle lipidique dans les macrophages pulmonaires. ... 20
Figure 1.4 Les mécanismes d’endocytose chez les macrophages. ... 32
Chapitre II
Figure 1. Native and ONOO--treated pulmonary surfactant affect the expression of key genes involved in reverse lipid transport in both pulmonary macrophages and bone marrow-derived macrophages. ... 57Figure 2. In vitro treatments with native and OONO--treated pulmonary surfactant reduces macrophage cholesterol export capacity. ... 57
Figure 3. Native and OONO--treated pulmonary surfactant modulate the expression of key genes involved in lipid capture in both pulmonary macrophages and bone marrow-derived macrophages. ... 58
Figure 4. Native and OONO--treated pulmonary surfactant modulate the expression of key genes involved in lipid accumulation in both pulmonary macrophages and bone marrow-derived macrophages. ... 58
Figure 5. Endocytosis inhibition by Latrunculin A does not impact the transcriptional changes induced by native and OONO--treated pulmonary surfactant. ... 59
Figure 6. Endocytosis inhibition by Latrunculin A does not impact the reverse lipid transport functional change induced by native and OONO--treated pulmonary surfactant. ... 59
Liste des abréviations et des sigles
3H-cholesterol : Tritium cholesterolABCA1 : ABC-binding cassette subfamily A member 1 ABCG1 : ABC-binding cassette subfamily G member 1 APOA1 : Apolipoprotein A-1
APOE : Apolipoprotein E BAL : Bronchoalveolar lavage BALF : Bronchoalveolar lavage fluid
BMdM : Bone marrow derived macrophages BSA : Bovine serum albumin
CD36 : Cluster of differenciation 36
cDNA : Complementary deoxyribonucleic acid CE : Cholesterol ester
Ci : Curie
Cq : Quantitative cycle
CSF2RA : colony stimulating factor 2 receptor alpha subunit DMSO : Dimethyl sulfoxide
DPPC : Dipalmitoylphosphatidylcholine DSPC : Disaturated phosphatidylcholine FBS : Fetal bovine serum
Fwr : Forward
Gapdh : Glyceraldehyde 3-phosphate dehydrogenase
GM-CSF : Granulocyte macrophage colony stimulating factor HDL : High density lipoprotein
HEPES : 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
IU : International unit
LBA : Lavage bronchoalvéolaire LDL : Low density lipoprotein LXR : Liver X receptor
M-CSF : Macrophage colony stimulating factor
MARCO : Macrophage receptor with collagenous structure mRNA : Messenger ribonucleic acid
MSR1 : Macrophage scavenger receptor 1 nCEH : Neutral cholesterol ester hydrolase ONOO- : Peroxynitrite PBS : Phosphate-buffered saline PC : Phosphatidylcholine PE : Phosphatidyléthanolamine PG : Phosphatidylglycérol PI : Phosphatidylinositol PLIN2 : Perilipin 2
PPAR : Peroxisome proliferator-activated receptor PulmMacro : Pulmonary macrophages
qPCR : Quantitative polymerase chain reaction RE : Réticulum endoplasmique
Rv : Reverse
RMPI : Roswell Park Memorial Institute medium
RNA : Ribonucleic acid
sn-3 : Stereospecific numbering SP-A : Surfactant protein A SP-B : Surfactant protein B SP-C : Surfactant protein C SP-D : Surfactant protein D SR : Scavenger receptor
SR-A1 : Scavenger receptor class A member 1 (msr1) SR-A6 : Scavenger receptor class A member 6 (marco) SR-B1 : Scavenger receptor class B member 1 (scarb1) SR-B2 : Scavenger receptor class B member 2 (cd36) TG : Triglycérides
Remerciements
Je tiens d’une part à remercier mes parents qui ont fait de nombreux sacrifices afin que j’accède à la meilleure éducation possible. Ils ont su m’appuyer dans mes démarches, en plus de m’inculquer des valeurs fondamentales telles que la rigueur, la persévérance, l’assiduité, la curiosité, et la valorisation d’un travail de qualité. Ces aptitudes m’ont menée à développer ma débrouillardise, ainsi que ma capacité d’analyse et de résolution de problème face à chacune des difficultés rencontrées. Merci d’avoir été pour moi un exemple de réussite. Je remercie également mes sœurs qui ont su vivre avec mes « rares » angoisses, en plus de rire de mes montagnes d’inquiétude, merci Kaiine et Viiro.
J’aimerais d’autre part remercier Mathieu Morissette, mon directeur de recherche. Merci de m’avoir laissé la chance de te surprendre au cours de ces années au sein de ton équipe. Merci pour ta confiance, ton intérêt et tes conseils. Également, merci pour ta grande « tolérance » envers les cols roulés.
Je remercie aussi mes collègues et amis du Centre de recherche de l’Institut de cardiologie et de pneumologie de Québec. Je tiens particulièrement à remercier Joanie Routhier, Éric Jubinville, Ariane Lechasseur et Maude Talbot pour leur support moral et les nombreux moments mémorables que nous avons partagés. De plus, j’accorde un énorme merci à Marie-Josée Beaulieu, Marie-Ève Paré, Sophie Aubin et Dany Patoine pour votre patience, votre dévouement, votre aide incroyable, et votre épaule. Merci de m’avoir évité la démence.
Un merci tout spécial également à mes deux « tritons » pour leur folie, leur esprit, leurs opinions et leurs conseils. Bref, merci pour cette amitié d’exception ∆. Enfin, merci Karl. Merci pour ton appui, ton écoute, ta compréhension et la motivation que tu m’apportes. Merci pour cette complicité qui ne cesse de croitre à travers les merveilleuses années, et les nombreuses à venir. Merci M. Buhr !
Avant-propos
Un seul article a été inclus dans ce mémoire, présenté dans le chapitre II, contenant la méthodologie et les résultats pertinents à ce mémoire. La participation détaillée des auteurs est décrite à la troisième page du chapitre II.
Chapitre I : Introduction générale et problématique
Le système respiratoire est extrêmement complexe. Il comprend les poumons, mais également les voies aériennes conductrices, les cavités nasales, le nasopharynx, l’oropharynx et le larynx (Scott, 2004). Cette complexité est notamment due au fait que ce système fournit la plus intime et la plus grande superficie corporelle exposée directement à l’environnement extérieur, soit environ 120 m2 (Coxson & Hogg, 2001; Wiebe & Laursen, 1995). Au niveau alvéolaire, où se produisent les échanges gazeux, la barrière biologique se présente comme une interface extrêmement amincie et délicate. En plus de favoriser les échanges, ce mince arrangement cellulaire doit permettre une protection adéquate contre une gamme d’éléments extérieurs d’origine biologique ou non (Burri, 1985; Scott, 2004). Afin d’accomplir cette fonction protectrice, les poumons regroupent divers types cellulaires et structures spécifiques. Les macrophages et le surfactant pulmonaire sont deux de ses joueurs clefs les plus importants.
Impliqué à différents niveaux dans le fonctionnement et la protection des poumons, le surfactant pulmonaire joue un rôle unique dans le maintien de l’homéostasie du système respiratoire. D’une part, cette structure complexe est impliquée dans les fonctions intrinsèques pulmonaires et les capacités fonctionnelles respiratoires (Chretien & Grandordy, 1983; Greenberger, 1997; Mason, Rand, Oulton, MacDonald, & Scott, 1998). D’autre part, le surfactant joue un rôle de barrière protectrice contre une multitude de menaces. En effet, tout organisme ou particule qui pénètre dans le système respiratoire et atteint les alvéoles risque d’entrer en contact avec le surfactant pulmonaire (Phelps, 2001). Ainsi, puisqu’il représente l’une des premières composantes corporelles exposées aux milliers de produits chimiques et toxiques présents dans l’environnement, le surfactant est une structure particulièrement vulnérable. De nombreuses réactions oxydatives découlent directement de ces agents toxiques retrouvés dans de multiples facteurs environnementaux tels que le tabac, et en contexte
2000; Bouhafs et al., 2000; Scott, 2004). En ciblant majoritairement les phospholipides, cette oxydation lipidique altère directement les propriétés fonctionnelles du surfactant et en diminue par le fait même l’efficacité (Calkovska et al., 2008). Lorsque ce dernier est endommagé, des mécanismes de recyclage et de dégradation particuliers sont impliqués. Les macrophages pulmonaires représentent l’un des principaux joueurs dans cette dégradation (Tracy Hussell & Thomas J. Bell, 2014).
Étant responsables d’enclencher une variété de processus biologiques essentiels, les macrophages pulmonaires sont eux aussi nécessaires au maintien de l’homéostasie pulmonaire. Ces macrophages, différenciés spécifiquement dans les poumons, représentent une population cellulaire hautement spécialisée. En jouant le rôle de phagocytes primaires, ils sont impliqués à de nombreuses étapes dans la réponse du système immunitaire inné et adaptatif (Geissmann, Gordon, Hume, Mowat, & Randolph, 2010; Sibille & Reynolds, 1990). Ces cellules sont notamment responsables de l’élimination des bactéries, des virus et des débris cellulaires présents dans les poumons, en plus d’être impliquées dans la présentation d’antigènes et la sécrétion de cytokines (Tracy Hussell & Thomas J. Bell, 2014; Nagy, Szanto, Szatmari, & Szeles, 2012). De surcroît à cette implication dans la défense immunitaire, ils sont également essentiels au maintien de l’homéostasie compositionnelle du surfactant pulmonaire. À maturité, ces cellules acquièrent la capacité de recycler et de dégrader l’excédent de phospholipides, de cholestérol et de protéines de ce dernier, en plus d’être responsables d’en éliminer les débris tels que les lipides oxydés (Fessler & Summer, 2016; Gurel, Ikegami, Chroneos, & Jobe, 2001; Han & Mallampalli, 2015; Ikegami, Na, Korfhagen, & Whitsett, 2005; Lopez-Rodriguez, Gay-Jordi, Mucci, Lachmann, & Serrano-Mollar, 2017; Trapnell & Whitsett, 2002; Wright & Youmans, 1995). Toutefois, les mécanismes utilisés par les macrophages pulmonaires permettant de déceler l’oxydation causée au surfactant pulmonaire ne sont toujours pas compris et caractérisés (Fukuzawa et al., 2013). Des recherches supplémentaires sur le sujet sont nécessaires afin de bien documenter le
comportement cellulaire des macrophages en réponse au surfactant oxydé, et ainsi comprendre la gestion qui en découle.
Quels sont les mécanismes employés par le macrophage pulmonaire lui permettant de détecter le surfactant pulmonaire oxydé dans son environnement? Voilà précisément la problématique étudiée lors de ce projet de recherche. Plus précisément, des macrophages ont été stimulés in vitro avec surfactant pulmonaire sain ou oxydé, suite à quoi des analyses transcriptionnelles et fonctionnelles ont été effectuées. Afin de caractériser le comportement de ces cellules, les analyses ont ciblé les aspects principaux du transport lipidique cellulaire, soit la capture, l’accumulation intracellulaire et l’export lipidique. De plus, par l’ajout d’un inhibiteur de la polymérisation des filaments d’actine (Coue, Brenner, Spector, & Korn, 1987; de Oliveira & Mantovani, 1988), le rôle de l’endocytose du surfactant par le macrophage dans la perception du dommage oxydatif a été étudié. Les différents aspects supplémentaires pertinents à la compréhension de ce projet seront ainsi décrits en aval.
1.1 Le surfactant pulmonaire
1.1.1 Les rôles du surfactant pulmonaire
Situé au pourtour de la couche interne alvéolaire, le surfactant pulmonaire procure deux fonctions essentielles au maintien de l’homéostasie pulmonaire. Premièrement, il représente la première barrière protectrice contre les différents agents externes et potentiellement nocifs. Impliqué dans la réponse immunitaire, il est ainsi responsable de l’élimination de divers pathogènes (Han & Mallampalli, 2015). Deuxièmement, le surfactant pulmonaire joue un rôle fonctionnel indispensable au maintien des fonctions respiratoires. Agissant à titre de lubrifiant, il contribue à la diminution de la tension de surface à l’interface air-liquide des alvéoles pulmonaires (Scott, 2004). Sans surfactant pulmonaire, la cage
aériennes lors de l’expiration, ce qui causerait l’affaissement des alvéoles pulmonaires (Creuwels, van Golde, & Haagsman, 1997; Whitsett, Wert, & Weaver, 2010, 2015). Cette force qui résulte de la résistance d’un liquide à prendre de l’expansion, due à l’attraction des molécules les unes envers les autres, est appelée tension de surface (Daniels, Lopatko, & Orgeig, 1998). Ainsi, l’expiration entraine une diminution de la surface pulmonaire et, par conséquent, une diminution de la tension superficielle, alors qu’à l’inverse, une tension superficielle relativement élevée est produite lorsque la surface du poumon est importante comme après l’inhalation (Creuwels et al., 1997). Ces deux fonctions spécifiques prodiguées par le surfactant sont possibles grâce à sa structure unique. En effet, les différentes composantes lipidiques et protéiques de ce dernier sont essentielles au maintien des capacités tensioactives et de la défense de l’hôte contre une infection. La formation et la régulation du surfactant s’effectuent ainsi localement, dans les alvéoles, par des cellules pulmonaires spécifiques.
1.1.2 La formation du surfactant pulmonaire
Représentant environ 5% de la surface alvéolaire totale (Han & Mallampalli, 2015; Herzog, Brody, Colby, Mason, & Williams, 2008), les pneumocytes de type II sont responsables de la production et de la sécrétion du surfactant pulmonaire (Creuwels et al., 1997; Goss, Hunt, & Postle, 2013; Han & Mallampalli, 2015; Scott, 2004). Cette production de novo s’effectue de manière collaborative entre les différents types cellulaires. Notamment, une grande quantité des triglycérides est fournie aux pneumocytes de types II par les lipofibroblastes qui les entreposent (Han & Mallampalli, 2015; McGowan & Torday, 1997; Scott, 2004; Trapnell & Whitsett, 2002). Les composantes du surfactant sont préalablement générées dans le réticulum endoplasmique des pneumocytes de type II puis transportées dans l’appareil de Golgi pour subir de plus amples modifications structurelles (voir figure 1.1) (Goss et al., 2013; Whitsett et al., 2015). Elles sont ensuite emmagasinées sous forme de corps lamellaires, jusqu’à leur sécrétion par
exocytose dans l’hypophase alvéolaire (Goss et al., 2013; Moxley, Jacoby, & Longmore, 1991; Weaver, 1998). Dans cette phase aqueuse, où se trouvent également les protéines du surfactant, les lipides forment une structure en treillis appelée myéline tubulaire (Goss et al., 2013; Nag, Munro, Hearn, et al., 1999). Cette structure est transportée à l’interface air-liquide des alvéoles afin de former une monocouche lipidique continue, le surfactant pulmonaire. Cette couche monomoléculaire de phospholipides dépourvue de protéines représente l’épiphase. Toutefois, lors de l’inhalation, une portion du surfactant est présente dans l’hypophase sous forme de multicouche lipidique transitoire (Scott, 2004). Ainsi, ces vésicules lipidiques, formées par la pression du mouvement respiratoire, sont à leur tour recyclées par les pneumocytes de type II puis dégradées par les lysosomes (voir figure 1.1) (Goss et al., 2013). Ces étapes de recyclage et de synthèse sont hautement régulées, ce qui permet simultanément de fournir une quantité de lipides adéquate aux exigences alvéolaires, tout en empêchant une accumulation excessive (Goss et al., 2013; Lopez-Rodriguez et al., 2017). Également, cette régulation homéostatique permet de maintenir des proportions stables de chacune des composantes du surfactant (Fessler & Summer, 2016; Trapnell & Whitsett, 2002).
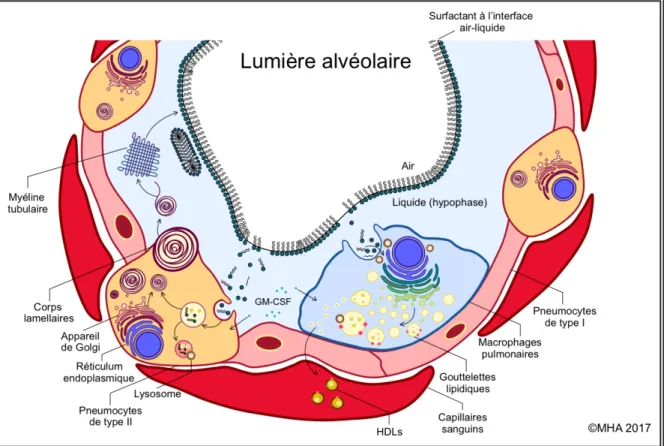
Figure 1.1 Le métabolisme du surfactant pulmonaire.
Cette figure illustre la compréhension actuelle de la sécrétion et du recyclage ainsi que des voies cataboliques des phospholipides et des protéines du surfactant pulmonaire. Les phospholipides du surfactant sont synthétisés de novo dans le réticulum endoplasmique (RE) des pneumocytes de type II, suite à quoi ils subissent des modifications dans l’appareil de Golgi et sont emmagasinés sous forme de corps lamellaires avec les protéines du surfactant. Ces corps lamellaires sont ensuite exportés dans l’hypophase alvéolaire, où ils s’assemblent pour former une structure appelée myéline tubulaire. Les phospholipides de ses agrégats se déplacent pour former une monocouche continue à l’interface air-liquide des alvéoles, les têtes polaires étant orientés vers le liquide et les chaines acyles vers l’air. Due entre autres à la forte pression superficielle exercée sur cette interface, cette monocouche est fréquemment endommagée. Ainsi, une portion de ses composantes est dégradée par les macrophages pulmonaires (voir figure 1.3), alors que la majorité est recyclée par les pneumocytes de type II. Une fois internalisés dans les pneumocytes de type II, les phospholipides recyclés sont en fait triés par des lysosomes puis catabolisés ou utilisés en vue d’une éventuelle sécrétion.
1.1.3 Les rôles et les proportions des composantes du surfactant pulmonaire
Représentant la majorité de sa composition, les nombreuses espèces lipidiques du surfactant pulmonaire ont des structures et des fonctions respectives spécifiques (Fessler & Summer, 2016; Trapnell & Whitsett, 2002; Veldhuizen, Nag, Orgeig, & Possmayer, 1998). En effet, les lipides occupent 90% de la totalité des composantes du surfactant, le reste étant composé de protéines. Quatre-vingt-cinq pourcent de ces lipides sont des phospholipides, alors que 5% sont des lipides neutres sous forme de triglycérides, d’acides gras et de cholestérol (voir figure 1.2) (Fessler & Summer, 2016; Orgeig & Daniels, 2001; Tolle, Meier, Rudiger, Hofmann, & Rustow, 2002; Trapnell & Whitsett, 2002; Veldhuizen et al., 1998). Dans la fraction phospholipidique, une espèce domine aux dépens des autres, soit la phosphatidylcholine (PC), qui représente entre 70 et 80% du total, cette fraction contenant aussi 15% de phosphatidylglycérol (PG) et de phosphatidylinositol (PI). En raison de sa structure particulière, la PC est présente en grande quantité à travers le corps. En effet, elle représente la composante principale des membranes cellulaires, où ses chaines acyles s’y trouvent en configuration 1-saturée et 2-insaturée (Hamm, Fabel, & Bartsch, 1992). Par contre, dans le surfactant pulmonaire, la PC adopte une autre configuration qui est exclusive à cette structure lubrifiante.
Présente sous une forme unique dans le surfactant pulmonaire, la PC est impliquée dans les fonctions spécialisées de cette couche lipidique. Contrairement aux PCs qui forment les bicouches membranaires, une grande proportion des PC constituant le surfactant possèdent des chaines acyles de configuration 1,2-saturées. En effet, 60 à 70% des PCs retrouvés dans le surfactant sont bisaturés (Disaturated Phosphatidylcholine; DSPC), ce qui en fait l’espèce lipidique la plus abondante (voir figure 1.2) (Creuwels et al., 1997; Hamm et al., 1992; Lopez-Rodriguez et al., 2017). Ses deux chaines acyles, dont la majorité est de type
palmitate (Dipalmitoylphosphatidylcholine; DPPC), sont donc formées de 16 atomes de carbones et d’aucune insaturation (16:0) (voir figure 1.2). Cette structure unique du DPPC permet aux chaines acyles d’être étroitement serrées les unes contre les autres, ce qui procure une très faible tension superficielle durant la compression de la monocouche du surfactant, lors de l’expiration (Amrein, von Nahmen, & Sieber, 1997; Schurch, Possmayer, Cheng, & Cockshutt, 1992; Veldhuizen et al., 1998; Whitsett et al., 2015). Également, le maintien de sa forme de gel ordonné à température corporelle procure au DPPC un avantage quant à ses capacités d’étalement à la surface alvéolaire (Ross, Krol, Janshoff, & Galla, 2002). Malgré l’importance qu’occupe cette proportion lipidique dans le surfactant, certaines des aptitudes de cette structure sont fournies par une fraction protéique qui, elle aussi, est essentielle.
Le surfactant pulmonaire est composé d’environ 10% de protéines uniques associées aux aspects fonctionnels des lipides et impliqués dans la réponse immunitaire, soient SP-A, SP-B, SP-C et SP-D (voir figure 1.2) (Capote, McCormack, & Possmayer, 2003; Scott, 2004). SP-A et SP-D sont des protéines hydrophiles présentes dans l’hypophase alvéolaire et sont principalement synthétisées par les pneumocytes de types II (Han & Mallampalli, 2015; Lopez-Rodriguez et al., 2017; Scott, 2004). Faisant partie de la famille des collectines, ces protéines jouent un rôle dans l’immunité innée et adaptative (Griese, 1999; Han & Mallampalli, 2015; Lopez-Rodriguez et al., 2017). En effet, en tant qu’opsonine alvéolaire, ces deux protéines ont un impact au niveau de la clairance bactérienne et virale (Fessler & Summer, 2016; Han & Mallampalli, 2015; McCormack & Whitsett, 2002). Agissant souvent de concert, elles sont également impliquées dans la formation de myéline tubulaire (Korfhagen et al., 1996; Williams, Hawgood, & Hamilton, 1991).
SP-A est la plus abondante des protéines du surfactant et remplit un nombre important de fonctions supplémentaires. Notamment, elle est impliquée dans le recyclage et la restauration des propriétés biophysiques du surfactant oxydé, en modulant directement la capture de PC par les pneumocytes de type II (Capote et al., 2003; Scott, 2004; Tsuzuki, Kuroki, & Akino, 1993). Les protéines structurelles SP-B et SP-C, également synthétisées majoritairement par les pneumocytes de type II (Scott, 2004), sont quant à elles hydrophobes. Majoritairement associées à la portion de phospholipides, ces deux protéines procèdent au remodelage des couches de surfactant en favorisant l’incorporation et l’étalement des lipides lors de l’expansion de la monocouche durant l’inhalation (Amrein et al., 1997; Knebel, Sieber, Reichelt, Galla, & Amrein, 2002; Veldhuizen et al., 1998; Whitsett et al., 2015). De surcroît, SP-B est impliquée dans l’adsorption sélective des DSPC par le surfactant et aide à la réorganisation de ces derniers à l’interface air-liquide (Rodriguez-Capote, Nag, Schurch, & Possmayer, 2001; Ross et al., 2002; Takamoto et al., 2001). Ces protéines spécifiques, tout comme les lipides, agissent en collaboration afin de maintenir l’homéostasie fonctionnelle du surfactant pulmonaire. Toutefois, en agissant dans la défense pulmonaire, ces composantes sont en constante interaction avec des molécules potentiellement néfastes qui peuvent en altérer la composition (Putman, van Golde, & Haagsman, 1997; Stenger et al., 2009).
Figure 1.2 Structure et composition du surfactant pulmonaire.
Le surfactant pulmonaire est composé à 90% de lipides et 10% de protéines. La portion lipidique contient principalement des phospholipides, en particulier du dipalmitoylphosphatidylcholine (DPPC), qui est responsable de ses fonctions biophysiques. Les protéines hydrophiles SP-A et SP-D jouent un rôle important dans la défense immunitaire de l’hôte, alors que SP-B et SP-C, deux protéines hydrophobes, participent principalement à la modulation des propriétés fonctionnelles du surfactant. PC = Phosphatidylcholine; PG = Phosphatidylglycérol; PI = Phosphatidylinositol; PE = Phosphatidylethanolamine; PS = Phosphatidylsérine.
1.2 L’oxydation du surfactant pulmonaire
1.2.1 Les causes de l’oxydation du surfactant pulmonaire
Les polluants d’origine atmosphérique représentent une source importante de stress pour les organismes vivants. En effet, lorsque les défenses de l’hôte ne suffisent plus à la protection, un stress oxydatif peut potentiellement affecter les différentes structures exposées, dont le surfactant pulmonaire (Cross et al., 2002). La résultante de ce contact oxydatif se résume souvent à un dommage au niveau protéique et lipidique, suivi de réponses adaptatives, inflammatoires, à la fois nuisibles et réparatrices (Cross et al., 2002). À travers la population de lipides ordonnés du surfactant, s’y trouvent également du cholestérol et des phospholipides insaturés, responsables de sa fluidité (Fessler & Summer, 2016; Glasser & Mallampalli, 2012; Orgeig & Daniels, 2001; Whitsett et al., 2015). Cette portion non négligeable de lipides insaturés comprend divers types de chaines acyles mono ou polyinsaturées. De par leur configuration instable, les phospholipides insaturés du surfactant représentent une cible particulièrement vulnérable à l’oxydation (Calkovska et al., 2008; Cross et al., 2002; Reis & Spickett, 2012).
1.2.2 Les différentes sources d’espèces oxydatives
Les nombreuses espèces oxydantes présentes dans l’air, pouvant affecter ces lipides vulnérables du surfactant, proviennent de multiples provenances. En effet, de nombreux agents oxydants peuvent provenir de sources endogènes, telles que la respiration, le métabolisme et la phagocytose (Droge, 2002), ou de sources exogènes par inhalation de polluants environnementaux tels que la fumée de cigarette et une exposition aux radiations (Ciencewicki, Trivedi, & Kleeberger, 2008; Reis & Spickett, 2012). Les modifications oxydatives qui en résultent dépendent grandement de la nature de ces espèces oxydantes, puisque cela
Un bon nombre des agents oxydants sont générés directement par le corps et peuvent interagir in vivo avec des composantes biologiques, menant à la génération de multiples dommages. Lors d’infections respiratoires, de nombreuses espèces réactives de l’oxygène libérées par les phagocytes activés peuvent être une source importante d’agents oxydants (Bouhafs et al., 2000). Plus particulièrement, beaucoup d’espèces radicalaires peuvent être générées en contexte inflammatoire et lors de processus de signalisation cellulaires (Lambeth, 2002). Notamment, les radicaux hydroxyles (•OH) et superoxydes (O2-•) sont bien connus comme étant des espèces très réactives et peu sélectives (Reis & Spickett, 2012). Malgré tout, les •OH sont plus efficaces et réagissent rapidement avec diverses molécules biologiques telles que les phospholipides, au contraire des O2-• qui eux, ne sont que très peu lipophiles en raison de leur charge négative. Également, du dioxyde d’azote (•NO2) est fréquemment formé en contexte inflammatoire suite à une réaction de catalyse par l’enzyme myéloperoxydase, présente chez différentes cellules phagocytaires du système immunitaire telles que les macrophages (Eiserich et al., 1998). Étroitement lié au •NO2, l’oxyde nitrique (•NO) générée lors de réactions biologiques multiples joue aussi un rôle central dans la pathogenèse de divers troubles infectieux et inflammatoires (Moncada & Higgs, 1993; Wei et al., 1995). Cette espèce radicalaire relativement stable réagit fréquemment in vivo avec des ions superoxydes pour former une espèce oxydante particulièrement dommageable, le peroxynitrite (Beckman & Koppenol, 1996; Beckmann et al., 1994; Koppenol, Moreno, Pryor, Ischiropoulos, & Beckman, 1992; Pryor & Squadrito, 1995; Reis & Spickett, 2012).
Le peroxynitrite (ONOO-) et sa forme protonée (ONOOH) ne sont pas des espèces radicales, mais peuvent être au cœur d’une multitude de réactions. Ces derniers peuvent à la fois générer des réactions oxydantes et nitrantes (Richards, Bommert, Szabo, & Miles, 2007). Plus particulièrement, le peroxynitrite est relativement stable en condition basique et n’a un temps de demi-vie que de
quelques secondes lorsqu’il se trouve à pH 7,4, sous forme de ONOOH (Koppenol et al., 1992). Sous ces conditions, cet agent oxydant se dissocie en différents sous-produits oxydatifs pouvant mener à la génération de •NO, •NO2, O2−• et •OH, qui sont particulièrement réactifs (Pryor & Squadrito, 1995; Reis & Spickett, 2012). Le peroxynitrite est également un agent oxydant puissant bien connu pour son implication dans la peroxydation lipidique, l’oxydation des sulfhydryles et la nitration des résidus aromatiques des protéines (Beckman, Beckman, Chen, Marshall, & Freeman, 1990; Beckmann et al., 1994; Koppenol et al., 1992). En plus du ONOO-, d’autres espèces non radicales telles que le peroxyde d’hydrogène (H2O2), l’acide hypochloreux (HOCl), l’ozone (O3), l’oxygène singlet (O2) et l’oxyde nitrique (NO) provoquent aussi des réactions d’oxydation sélectives in vivo avec diverses biomolécules (Ciencewicki et al., 2008; Ford, 2010; Pacher, Beckman, & Liaudet, 2007; Panasenko, 1997). Ces différentes sources endogènes d’agents oxydants sont toutes, à différents niveaux, respectivement responsables des constantes altérations apportées au surfactant pulmonaire.
Il est aussi important de considérer l’apport exogène d’agents oxydants dommageables puisque leur présence dans l’environnement n’est évidemment pas à négliger. Que ce soit suite à un contact avec un acarien, un allergène, un virus, une bactérie ou tout autre aérosol inhalé (Cross et al., 2002), les différentes composantes du surfactant sont implicitement affectées. En effet, les nombreuses sources de polluants atmosphériques oxydatifs représentent une cause importante de stress pour les organismes vivants. L’une de ces sources oxydatives exogènes les plus documentées est certainement la cigarette de tabac. En effet, l’usage du tabac et l’exposition à la fumée secondaire sont devenus une problématique extrêmement importante et aux conséquences énormes pour la santé pulmonaire au cours des dernières décennies (Bringezu, Pinkerton, & Zasadzinski, 2003; Scott, 2004).
Pouvant contenir jusqu’à 4000 substances distinctes (Robin et al., 2002) et ayant des effets toxiques pour le système respiratoire, la fumée de cigarette de tabac est une source particulièrement importante d’agents oxydants. Le surfactant pulmonaire est la principale victime de cette myriade de composés toxiques. Les répercussions engendrées sur ce dernier varient selon la quantité et le type de particules qu’il rencontre (Bringezu et al., 2003; Nikula, Vallyathan, Green, & Hahn, 2001; Pinkerton et al., 2000). Une bonne portion de ces espèces s’apparentent à celles générées de manière endogène et comprend une grande quantité d’espèces nitrantes et oxydantes (Morse & Rosas, 2014; Papaioannou et al., 2016). La fumée de cigarette active également différents types cellulaires tels que les macrophages pulmonaires, ce qui entraine une production supplémentaire de radicaux oxygénés (Lowry, Rosebrough, Farr, & Randall, 1951; McCormack, 1998; Nag, Munro, Inchley, et al., 1999; Repine, Bast, & Lankhorst, 1997). Ainsi, en induisant un large éventail d’effets biologiques sur les poumons, la fumée de la cigarette de tabac endommage et oxyde entre autres les lipides du surfactant pulmonaire. Ces dommages engendrent diverses répercussions inflammatoires et fonctionnelles (Hohlfeld, Fabel, & Hamm, 1997; Papaioannou et al., 2016; Scott, 2004).
1.2.3 L’oxydation lipidique
Différents mécanismes d’oxydation lipidique, ciblant majoritairement les chaines acyles des phospholipides, peuvent se produire dans le surfactant pulmonaire. Entre autres, la peroxydation lipidique est un mécanisme assez fréquent qui permet la génération d’une multitude de composés phospholipidiques. Le profil du phospholipide oxydé généré est évidemment influencé par l’espèce oxydante, le type de liaison de l’acide gras au squelette de glycérol et le type de chaine acyle (Reis & Spickett, 2012). En plus de la peroxydation, les phospholipides insaturés sont également sensibles aux attaques électrophiles par des espèces oxydantes non radicales pouvant générer des espèces lipidiques uniques (Reis & Spickett, 2012). Notamment, des lipides nitrés peuvent être
formés par attaque électrophile d’espèces réactives au nitrogène, telles que le dioxyde d’azote, l’oxyde nitreux et le peroxynitrite (B. A. Freeman et al., 2008; O'Donnell et al., 1999). Les acides gras estérifiés tels que les phospholipides sont des espèces lipidiques particulièrement à risque de subir un tel type de dommage oxydatif.
Représentant presque 50% de la population de phospholipide totale des membranes biologiques (Reis & Spickett, 2012), les PCs sont la population ciblée par la majorité des études s’intéressant à la formation, l’identification et la quantification des phospholipides oxydés (Reis & Spickett, 2012). Tel que précédemment mentionné, le surfactant pulmonaire est composé à majorité d’espèces lipidiques de cette classe, soit des phospholipides comportant un groupement choline en position sn-3 au niveau de leur tête polaire. Ces derniers constituent environ 80% des phospholipides totaux du surfactant, dont 70% sont sous forme de DPPC. De surcroît, plus de 20 espèces de phospholipides insaturés ont également été identifiées dans le surfactant pulmonaire (Almstrand, Voelker, & Murphy, 2015; Fessler & Summer, 2016). Ces espèces insaturées sont particulièrement susceptibles aux modifications en contexte de stress oxydatif, dû à l’instabilité chimique créée par la ou les doubles liaisons présentes sur l’une ou les chaines acyles qui les constituent (Reis & Spickett, 2012). Regroupant plus de 30% de la totalité des phospholipides du surfactant (Agassandian & Mallampalli, 2013; Han & Mallampalli, 2015), ces phospholipides insaturés sont sensibles aux dommages oxydatifs pouvant aussi bien provenir d’agents toxiques oxydants contenus dans la fumée de cigarette ou encore d’espèces oxydatives endogènes. Ainsi l’oxydation de ces différents lipides, insaturés ou non, peut mener à des mélanges complexes d’espèces bioactives de phospholipides oxydés, comprenant des espèces avec des chaines acyles fragmentées (Bochkov et al., 2010; Fessler & Summer, 2016).
Les produits lipidiques générés suite aux modifications oxydatives peuvent grandement varier, selon l’agent oxydant, la longueur de la chaine acyle, le nombre d’insaturations, le groupement de la tête polaire, etc. Ainsi, durant ces modifications chimiques, une multitude de produits d’oxydation secondaire potentiellement toxiques peuvent se produire et interagir avec des protéines et l’ADN, provoquant des dommages biologiques (Frankel, 1984). Certaines de ces espèces lipidiques sont notamment reconnues pour causer divers dommages cellulaires et induire un grand nombre de réponses inflammatoires, telles que l’acide lysophosphatidique qui est entre autres impliqué dans la prolifération cellulaire (Koizumi, Sageshima, Wada, Narita, & Higuchi, 1989; Sacco et al., 2004), la migration (Schneeberger & Karnovsky, 1976; Zhao et al., 2011) et la sécrétion de cytokines et chimiokines (B. A. Freeman, Panus, Matalon, Buckley, & Baker, 1993; Koff, Shao, Kim, Ueki, & Nadel, 2006). Les mécanismes cellulaires permettant la dégradation de ces différentes espèces lipidiques oxydés du surfactant pulmonaire sont donc particulièrement importants. Comme mentionné, les macrophages pulmonaires sont les cellules phagocytaires responsables de la majorité de ces étapes de séquestration (Eguiluz-Gracia et al., 2016; Tracy Hussell & Thomas J. Bell, 2014; Whitsett et al., 2015).
1.2.4 L’impact fonctionnel de l’oxydation du surfactant pulmonaire
Les fonctions mécanistiques et biologiques du surfactant peuvent être affectées de diverses manières, selon les dommages perçus dans sa structure. La majorité de la littérature touchant aux répercussions oxydatives sur le surfactant pulmonaire traite de l’impact du tabagisme dans cette problématique. Toutefois, de nombreux paramètres influencent l’interprétation des effets du tabagisme sur les phospholipides du surfactant pulmonaire, et il serait trop simpliste de n’accuser qu’un seul facteur causal (Morse & Rosas, 2014; Scott, 2004). Malgré tout, l’oxydation lipidique est implicitement incluse lorsqu’il est question de dommage au surfactant induit par le tabagisme (Finley & Ladman, 1972; Morissette et al., 2014;
Morissette, Shen, Thayaparan, & Stampfli, 2015; Rahman et al., 2002; Scott, 2004). Ainsi, l’oxydation lipidique induite par le tabagisme semble causer de nombreux dommages fonctionnels au surfactant pulmonaire.
En altérant la structure des lipides, leur proportion, ainsi que les fonctions protéiques, l’oxydation lipidique engendrée par le tabagisme a de graves conséquences (Honda, Takahashi, Kuroki, Akino, & Abe, 1996; Morissette et al., 2015). À cet effet, il est pertinent de considérer que la cigarette de tabac peut altérer à la fois le contenu du surfactant, mais également l’activité de ce dernier en perturbant son homéostasie lipidique (Bringezu et al., 2003; Honda et al., 1996; Morissette et al., 2015). En altérant sa composition lipidique, la fumée de cigarette semble compromettre directement les capacités de diminution de tension superficielle du surfactant (Hallman, Merritt, Pohjavuori, & Gluck, 1986; Hohlfeld et al., 1997; Stevens, Schadow, Bartholain, Segerer, & Obladen, 1992). En effet, lors d’études d’exposition chronique à la fumée de cigarette, une diminution des quantités de DSPC dans le lavage bronchoalvéolaire a été recensée, suggérant une altération dans la sécrétion du surfactant pulmonaire par les pneumocytes de type II (Bringezu et al., 2003; Subramaniam, Bummer, & Gairola, 1995). Une réduction significative des taux de cholestérol et des ratios cholestérol : phospholipides a aussi été observée chez des fumeurs comparativement aux contrôles non-fumeurs (Hughes & Haslam, 1989, 1990). Puisque le cholestérol est un lipide neutre ayant un impact sur la fluidité du surfactant (Scott, 2004), ces données pourraient en partie expliquer les variations de tension superficielle observées dans un modèle d’exposition chronique à la fumée de cigarette (Subramaniam et al., 1995). Une augmentation de la concentration du surfactant pulmonaire en phosphatidyléthanolamine, phosphatidylglycérol et sphingomyéline a également été mesurée dans le surfactant de fumeurs (Bringezu et al., 2003; Hughes & Haslam, 1989). De plus, les altérations de fonction du surfactant pulmonaire seraient partiellement causées par l’effet des produits sécrétés par les neutrophiles et les macrophages activés sur les protéines du surfactant (Papaioannou et al., 2016). Ces nombreux dommages entrainent ainsi une perte
de la fonction d’abaissement de la tension de surface du surfactant. Cette problématique conduit à une augmentation de gradient de pression à travers la paroi alvéolaire et peut contribuer au développement de diverses maladies pulmonaires (Devendra & Spragg, 2002).
Malgré ces nombreuses répercussions, il est difficile d’affirmer si l’oxydation lipidique est explicitement responsable du changement fonctionnel et de la diminution d’efficacité du surfactant induit par le tabagisme. Autrement dit, est-ce le changement structurel du surfactant lui-même qui est responsable de sa diminution fonctionnelle, ou est-ce la variation de ses mécanismes de synthèse et de recyclage, qui eux seraient influencés par la composition lipidique du surfactant? Malgré tout, de récentes données suggèrent fortement que les altérations de l’homéostasie du surfactant seraient responsables de la stimulation des processus inflammatoires déclenchés par le tabagisme (Morissette et al., 2015). Ainsi, l’oxydation des lipides du surfactant pulmonaire aurait pour conséquence d’activer les mécanismes de recyclage et de dégradation des lipides oxydés effectués par les macrophages pulmonaires (Fessler & Summer, 2016; Goss et al., 2013; Han & Mallampalli, 2015; Ikegami et al., 2005; Lopez-Rodriguez et al., 2017; Trapnell & Whitsett, 2002; Wright & Youmans, 1995)
1.3 La biologie lipidique des macrophages pulmonaires
Les macrophages pulmonaires sont bien connus pour leur rôle essentiel dans la défense immunitaire pulmonaire (Tracy Hussell & Thomas J. Bell, 2014; Nagy et al., 2012), en plus de contribuer au maintien de l’homéostasie du surfactant pulmonaire. En effet, stratégiquement positionnés dans l’espace alvéolaire et dans la lumière des voies respiratoires, les macrophages pulmonaires sont les cellules immunologiques prototypiques des poumons(T. Hussell & T. J. Bell, 2014). Principalement originaires des monocytes circulants, qui eux sont produits à partir de cellules progénitrices de la moelle osseuse (T. Hussell & T. J.
Bell, 2014; Nagy et al., 2012; Zhang, Goncalves, & Mosser, 2008), les macrophages pulmonaires se différencient localement dans les poumons et deviennent des macrophages résidents à maturité. Les macrophages pulmonaires en différenciation acquièrent rapidement la machinerie nécessaire pour traiter des quantités élevées de phospholipides, de cholestérol et de protéines. Leur différenciation dépend fortement du facteur de stimulation des colonies de granulocytes-macrophages (GM-CSF), responsable de l’initiation des processus impliqués dans l’absorption et la dégradation du surfactant pulmonaire. Sans ce dernier, les macrophages présentent une déficience dans la dégradation des composantes endommagées du surfactant (Fessler & Summer, 2016; Goss et al., 2013) et dans la défense pulmonaire de l’hôte (Carey & Trapnell, 2010). Ainsi, cette déficience peut engendrer de nombreuses répercussions pulmonaires néfastes, telle qu’une accumulation de lipides anormale dans les alvéoles (Carey & Trapnell, 2010). Ce phénotype particulier est principalement induit par une gestion inefficace des lipides endommagés et n’est pas seulement observé suite à une déficience en GM-CSF. En effet, de nombreux facteurs peuvent enclencher une gestion lipidique inefficace par les macrophages, tel qu’en contexte tabagique où l’on retrouve une grande quantité de lipides oxydés intracellulaires (Morissette et al., 2015). Ainsi, l’activation du transport lipidique pulmonaire est une étape de gestion cruciale au maintien de l’homéostasie respiratoire. De ce fait, la régulation des mécanismes impliqués dans la capture, l’accumulation et l’export lipidique par les macrophages pulmonaires est d’une importance capitale. Toutefois, ces processus complexes ne sont que très peu documentés en contexte oxydatif.
1.3.1 Le transport lipidique par les macrophages pulmonaires
Lors du transport lipidique pulmonaire, les macrophages pulmonaires sont responsables de la capture de différentes espèces lipidiques endommagées et oxydées (Fessler & Summer, 2016). Une fois internalisés, ces lipides endommagés sont dirigés dans des lysosomes spécifiques, ce qui entraine la
libération de cholestérol libre, majoritairement. Ce dernier peut être dirigé dans la membrane plasmique cellulaire pour être directement exporté de la cellule, ou encore être acheminé et accumulé dans la cellule sous forme de vésicules (voir figure 1.3) (Kathryn J. Moore, Frederick J. Sheedy, & Edward A. Fisher, 2013). Ces lipides stockés peuvent ensuite être mobilisés pour permettre leur export.
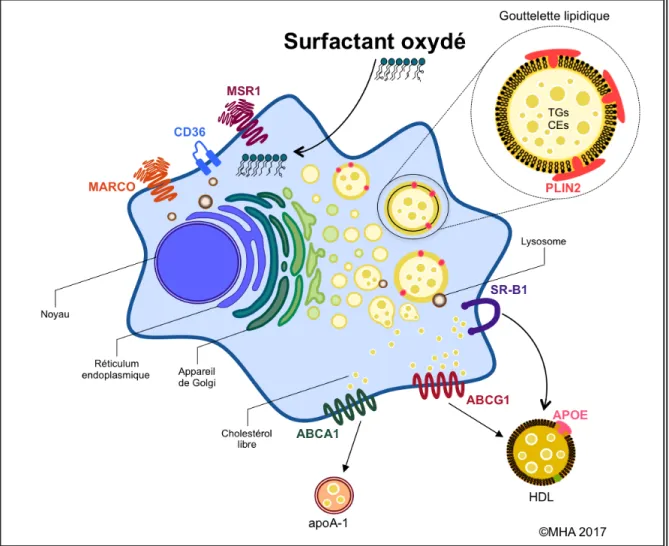
Figure 1.3 Le cycle lipidique dans les macrophages pulmonaires.
Lors du cycle de transport lipidique pulmonaire, les macrophages pulmonaires sont responsables de la capture de différentes espèces lipidiques endommagées et oxydées. Cette étape complexe implique une multitude de mécanismes et de récepteurs spécifiques, dont les récepteurs éboueurs MARCO, MSR1, et CD36. Une fois internalisées, les différentes espèces lipidiques endommagées sont dirigées dans des lysosomes spécifiques. Ces lipides sont dégradés, puis la presque totalité des
composantes lipidiques est acheminée et accumulée dans le réticulum endoplasmique. Dans cette structure cellulaire, ces débris lipidiques dégradés sont transformés en espèces lipidiques neutres telles que le cholestérol-ester et les triglycérides. Ils sont ensuite stockés dans la cellule sous forme de gouttelettes lipidiques cytosoliques. La protéine PLIN2 est la principale responsable de la modulation, la formation et la dégradation de ces gouttelettes. Les lipides neutres emmagasinés sont principalement hydrolysés par des lysosomes. Le cholestérol libre ainsi généré est ensuite exporté de la cellule vers des accepteurs lipidiques lipoprotéiques (HDL et LDL). Cet export s’effectue grâce à des récepteurs spécifiques tels qu’ABCA1, ABCG1, SR-B1 et des apolipoprotéines comme APOA1 et APOE.
1.3.1.1 La capture lipidique
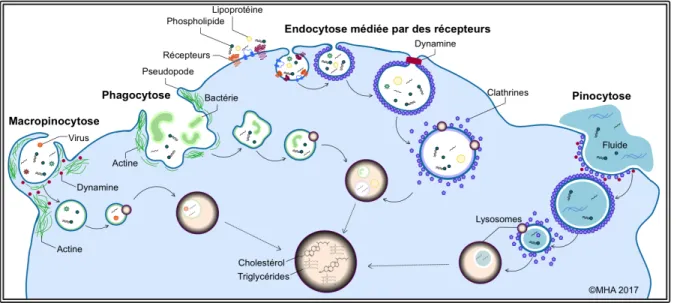
Cette première étape de transport lipidique est particulièrement complexe et implique une multitude de récepteurs et de mécanismes distincts. Une majorité de récepteurs impliqués dans cette capture sont des récepteurs éboueurs (Scavenger Receptor; SR), tels que MSR1, MARCO et CD36. Les mécanismes d’internalisation sont variés et font souvent appel à des étapes d’endocytose médiées par des récepteurs éboueurs, malgré d’autres avenues de captures potentielles (Kathryn J. Moore et al., 2013; Plakkal Ayyappan, Paul, & Goo, 2016). Il est également bien documenté que les macrophages sont au centre du développement de l’athérosclérose, en raison de leur aptitude à internaliser des lipoprotéines modifiées et à libérer des médiateurs inflammatoires (Glass & Witztum, 2001; Lusis, 2000; Zelcer & Tontonoz, 2006). Même si la capture lipidique est en soi un ensemble de processus très complexes, la majorité de la littérature traite précisément de l’implication des macrophages pulmonaires dans la capture et la dégradation des lipoprotéines de faible densité médiée par les récepteurs éboueurs (David & Louise, 2004; Greaves & Gordon, 2009; Kosswig, Rice, Daugherty, & Post, 2003).
Les récepteurs éboueurs font partie d’une superfamille de récepteurs liés à la membrane cellulaire et responsable de l’internalisation d’une variété de ligands tels que les LDL, des protéines et des agents pathogènes endogènes (Zani et al., 2015). En effet, la caractéristique commune de ce groupe disparate de protéines est leur capacité à reconnaitre des ligands communs tels que des ligands polyioniques comprenant des lipoprotéines, des cellules apoptotiques, du cholestérol ester et des phospholipides (David & Louise, 2004; de Winther, van Dijk, Havekes, & Hofker, 2000; Zani et al., 2015). Il existe différentes classes de récepteurs éboueurs, mais les plus importantes pour la capture lipidique par les macrophages sont les classes A et B (Zani et al., 2015).
Les récepteurs éboueurs de classe A (SR-A) incluent notamment les récepteurs SR-A1 (MSR1) et SR-A6 (MARCO) qui sont cruciaux au niveau pulmonaire. Ces derniers sont des glycoprotéines transmembranaires trimériques composées de 6 domaines distincts, dont le domaine « collagene-like » qui représente le site d’interaction avec les lipoprotéines modifiées (Doi et al., 1993; Tanaka et al., 1996). En plus d’être impliqué dans l’adhérence cellulaire (Kosswig et al., 2003), MSR1 est un récepteur associé à l’internalisation des LDLs oxydés et acétylés (de Winther et al., 2000), alors que MARCO présente une plus faible, mais tout de même considérable, affinité pour ces ligands (de Winther et al., 2000; Greaves & Gordon, 2009). De nombreuses études dénotent l’importance particulière de MSR1 dans l’athérosclérose et la formation de cellules spumeuses, soient des cellules présentent à la barrière artérielle qui ingèrent des LDL oxydés et adoptent une apparence spumeuse (de Winther et al., 2000; K. J. Moore, F. J. Sheedy, & E. A. Fisher, 2013; Kathryn J. Moore et al., 2013). Lors d’une exposition tabagique, ce dernier semble jouer un rôle extrêmement important au niveau de la capture du surfactant endommagé (Thakur, Hamilton, & Holian, 2008). Ces deux récepteurs, exprimés par les macrophages pulmonaires, sont connus comme étant des médiateurs de la clairance des lipides oxydés dans le système pulmonaire (Dahl et al., 2007; Zani et al., 2015). Cependant, ils semblent induire des voies de
signalisation intracellulaires distinctes, ce qui génère différents effets sur la réponse immunitaire (Jozefowski, Arredouani, Sulahian, & Kobzik, 2005). Impliqués à différents niveaux, MSR1 et MARCO sont ainsi essentiels dans l’étape de capture lipidique par les macrophages pulmonaires. Malgré tout, ces derniers ne sont pas les seuls récepteurs impliqués dans ces étapes lipidiques, puisqu’ils sont notamment appuyés par les récepteurs éboueurs de classe B.
En incluant de nombreux récepteurs éboueurs ayant des fonctions complémentaires, les récepteurs de classe B (SR-B) sont impliqués dans différents mécanismes importants dans le maintien de l’homéostasie respiratoire. Les récepteurs de cette classe possèdent deux régions transmembranaires situées à proximité des régions N-C terminales qui chevauchent un domaine central glycosylé servant à la reconnaissance du ligand (Zani et al., 2015). D’une part, le récepteur SR-B1 a une grande affinité de liaison avec les HDLs, les virus et les bactéries. Ce récepteur éboueur, exprimé par les macrophages pulmonaires, est particulièrement important dans l’athérosclérose, en jouant un rôle au niveau de la capture de cholestérol par des accepteurs spécifiques, favorisant ainsi l’export lipidique des macrophages (Greaves & Gordon, 2009; Valacchi, Davis, et al., 2011; Zani et al., 2015). D’autre part, SR-B2 (CD36) a de nombreuses fonctions dans la capture lipidique par les macrophages, y compris la capture de LDLs oxydés, favorisant la formation de cellule spumeuse (Kathryn J. Moore et al., 2013; Plakkal Ayyappan et al., 2016; Zani et al., 2015). De surcroît, ses niveaux d’expression augmentent lors d’une inflammation et sous l’effet d’un stress oxydatif (Liani et al., 2012; Nishikawa, Sugimoto, Okada, Sakairi, & Takagi, 2012; R. L. Silverstein, Li, Park, & Rahaman, 2010). CD36 se lie également à une variété d’autres ligands, y compris les phospholipides oxydés, les acides gras à longues chaines, les particules lipidiques modifiées, les cellules apoptotiques ainsi que les pathogènes bactériens et fongiques (R. L. Silverstein & Febbraio, 2009). Une fois internalisées par les macrophages, ces nombreuses espèces lipidiques endommagées subissent alors d’autres étapes de dégradation.
1.3.1.2 L’accumulation intracellulaire lipidique
Lors du processus de transport lipidique, une portion des différentes espèces lipidiques du surfactant endommagées et captées par les macrophages doit subir des étapes de dégradation et de séquestration intracellulaire cruciales à la survie cellulaire (Plakkal Ayyappan et al., 2016; Walther & Farese, 2012). Les systèmes vivants sont maintenus par un flux constant d’énergie métabolique. Les lipides, riches en hydrocarbures, constituent une source d’énergie élevée pour tous les organismes eucaryotes (Sletten, Seline, Rudd, Logsdon, & Listenberger, 2014; Thiam, Farese, & Walther, 2013). Ainsi, la capacité d’emmagasiner les lipides dans les cellules et les tissus est souvent une avenue cruciale pour la survie (Sletten et al., 2014; Walther & Farese, 2012). De nombreux types cellulaires, dont les macrophages pulmonaires, sont aptes à emmagasiner une grande quantité de lipides dans des réservoirs distincts (Thiam et al., 2013). Afin de compacter efficacement ces différentes espèces lipidiques, les macrophages les convertissent en lipides neutres, tels que les triglycérides (TG), les esters de stérol et le cholestérol (Thiam et al., 2013; Walther & Farese, 2012). Une fois emmagasinés et séparés du cytosol par une monocouche de lipides amphiphiles (Walther & Farese, 2012), ces réservoirs lipidiques intracellulaires se nomment gouttelettes lipidiques. Compte tenu de leurs diverses fonctions, les gouttelettes lipidiques se situent au centre de la biologie des membranes et du métabolisme énergétique et sont des organites fondamentaux dans le maintien de l’homéostasie cellulaire pulmonaire (Sletten et al., 2014).
Les cellules traitent les lipides endommagés, comme les acides gras, afin de se protéger contre leur lipotoxicité. Par exemple, certaines espèces lipidiques oxydées sont responsables d’enclencher des voies cellulaires apoptotiques (Listenberger et al., 2003; Sletten et al., 2014; Tabas, 2002). Ainsi, la plupart des espèces lipidiques possiblement nocives sont estérifiées et transformées en huiles neutres, dans les bicouches membranaires ou par émulsion (Thiam et al., 2013). Malgré le manque d’informations reliées à leurs processus de formation, les
gouttelettes lipidiques seraient formées par le bourgeonnement des phospholipides du réticulum endoplasmique (RE) chez les eucaryotes (Thiam et al., 2013; Walther & Farese, 2012). Toutefois, la composition en phospholipides diffère entre les bicouches du RE et les monocouches des gouttelettes lipidiques (Thiam et al., 2013). Il est établi que la plupart des enzymes impliquées dans la synthèse de triglycérides et d’ester de stérol sont directement localisées dans le RE, lieu où s’effectuerait donc le clivage et le remaniement des différentes espèces lipidiques endommagées suite à leur capture (Thiam et al., 2013). En plus des phospholipides, la surface des gouttelettes lipidiques est recouverte de protéines spécifiques pouvant agir sur leur régulation, leur croissance et leur utilisation.
Selon le type cellulaire, certaines protéines uniques sont présentes sur les gouttelettes lipidiques et sont responsables de réguler diverses fonctions (Thiam et al., 2013; Walther & Farese, 2012; Yang et al., 2012). Beaucoup de ces protéines ont des rôles dans le métabolisme des lipides et dans le trafic des PCs, des stérols et des triglycérides (Thiam et al., 2013), en plus de jouer un rôle dans l’hydrolyse de ces derniers (Sletten et al., 2014). Certaines de ces protéines, sans être implicitement impliquées dans le métabolisme des lipides, ont un rôle dans le contrôle des propriétés de surface des gouttelettes. Les périlipines, par exemple, sont une famille de protéine impliquée dans la protection des gouttelettes lipidiques contre la lipolyse (Brasaemle et al., 2000), en plus d’être responsable de leur formation par la modulation de leur processus de bourgeonnement (Thiam et al., 2013). La périlipine 2 (PLIN2) est une protéine particulièrement importante recouvrant la surface de ces gouttelettes dans toutes les cellules de mammifères, à l’exception des adipocytes blancs matures, où l’on retrouve la périlipine 1. PLIN2 empêche spécifiquement l’association des lipases avec la surface des gouttelettes et ralentit la dégradation des triglycérides (Plakkal Ayyappan et al., 2016; Sletten et al., 2014). Par conséquent, le niveau d’expression de PLIN2 est intimement lié à l’accumulation des gouttelettes lipidiques dans les macrophages (Becker et al., 2010; Lu et al., 2001; Paul, Chang, Li, Yechoor, & Chan, 2008; Plakkal Ayyappan et al., 2016). Suite à la séquestration lipidique et afin d’en permettre l’export, deux
voies d’hydrolyse des gouttelettes lipidiques sont possibles. Dans la première voie, le cholestérol ester contenu est hydrolysé en cholestérol libre par des hydrolases spécifiques (nCEHs), qui s’associent aux gouttelettes. La deuxième voie implique l’engorgement autophagocytaire des gouttelettes, suivie d’une fusion avec des lysosomes et d’une hydrolyse des cholestérols-ester (Ouimet et al., 2011). Le cholestérol libre ainsi généré est ensuite exporté via des mécanismes d’export lipidique spécifiques.
1.3.1.3 L’export lipidique
L’export lipidique, aussi appelé transport inverse de lipides, représente le transfert des lipides de la cellule vers des accepteurs spécifiques extracellulaires. Ce processus impliquant une variété de protéines et de récepteurs particuliers est la dernière étape dans le transport lipidique pulmonaire. Cette étape est particulièrement importante pour les cellules pulmonaires telles que les macrophages et les pneumocytes de type II qui traitent une charge importante de lipide provenant du surfactant pulmonaire endommagé ou oxydé (Carey & Trapnell, 2010). En contexte tabagique, cette étape semble être limitante, tel qu’en témoigne l’accumulation de lipides, la taille des macrophages et la libération de nombreux médiateurs inflammatoires (Morissette et al., 2015). L’export lipidique des macrophages par ABCA1 et ABCG1 s’effectue, entre autres, vers les lipoprotéines à haute densité (high density lipoprotein; HDL) (Gelissen et al., 2006; Kennedy et al., 2005). Les lipoprotéines sont de grands complexes constitués d'une membrane externe de protéines (apolipoprotéines) et de lipides comprenant
des phospholipides et du cholestérol, ainsi qu’un noyau hydrophobe de
cholestérol-ester (CE) et de triglycéride (TG) (Rhainds & Brissette, 2004). Chacune des protéines qui composent les lipoprotéines assure leur maintien structurel et favorise un transport inverse de cholestérol adéquat à travers le corps.