RÔLE DE LA MAP3K DLK DANS LA RÉPONSE DES NEURONES À L’ION CALCIUM, UN MÉDIATEUR DE DÉGÉNÉRESCENCE ET RÉGÉNÉRESCENCE
Par :
Mathieu Dalpé-Mainville
Mémoire présenté à la Faculté des Sciences
en vue de l’obtention du grade de maître es science (M.Sc.)
FACULTÉ DES SCIENCES UNIVERSITÉ DE SHERBROOKE
Le 16 août 2016
Le jury a accepté le mémoire de monsieur Mathieu Dalpé-Mainville dans sa version finale
Membres du jury
Professeur Richard Blouin Directeur de recherche Département de biologie
Professeur Nicolas Gévry Conseiller interne Département de biologie
Professeur Nancy Dumais
Conseillère interne et président-rapporteur Département de biologie
iii SOMMAIRE
Le corps humain est composé de plusieurs milliards de cellules neuronales, lesquelles ont un impact primordial sur les systèmes vitaux. En effet, le système nerveux, étant lui-même un système vital du corps humain, effectue de nombreuses fonctions afin de voir au bon fonctionnement des autres systèmes du corps, tels que le système cardiovasculaire, le système digestif, le système musculaire et bien d’autres. Plusieurs maladies neurodégénératives telles que l’Alzheimer et la maladie de Creutzfeldt-Jakob, affectent les cellules nerveuses du corps et occasionnent la perte de différentes fonctions causant une diminution du bien-être et pouvant mener à la mort. La hausse du nombre d’individus atteint de maladies neurodégénératives observée au fils des ans nous montre l’importance de comprendre les phénomènes moléculaires qui se produisent lors des mécanismes de dégénérescence axonale dans les neurones.
Lors de ma maîtrise dans le laboratoire du professeur Richard Blouin, j’ai étudié l’impact d’une augmentation intracellulaire de calcium sur la protéine dual leucine zipper kinase (DLK) en utilisant un modèle cellulaire humain, les SH-SY5Y. J’ai pu démontrer que l’entrée de calcium dans la cellule avait un impact sur la quantité de protéine DLK présente dans celles-ci. En effet, j’ai pu observer une diminution de la quantité de DLK dans les cellules suite à une entrée de calcium, un phénomène exclusivement calcium-dépendant. À l’aide d’inhibiteurs pharmacologiques, j’ai pu montrer l’implication, des calpaïnes, des protéases calcium-dépendantes reconnues pour leur rôle dans la dégénérescence axonale. Les essais in vitro montrent que DLK est une cible spécifique des calpaïnes
iv REMERCIEMENTS
J’aimerais remercier mon directeur de recherche le Dr Richard Blouin. Il fut tout ce qu’un étudiant de maîtrise pouvait s’attendre d’un professeur. Patient, ordonné, et inspirant la confiance. Malgré les difficultés rencontrées, Richard pouvait me remettre sur le droit chemin et je ne le remercierais jamais assez pour cela.
Merci également à mes deux conseillers, Nicolas Gévry et Nancy Dumais, qui ont su donnés de leurs temps et ont apporté leur soutien. Merci au CRSNG et à l’Université de Sherbrooke d’avoir contribué financièrement au projet et pour m’avoir permis d’effectuer ma maîtrise à Sherbrooke. Merci aux membres des laboratoires de Sébastien Rodrigue, Nicolas Gévry et Luc Gaudreau ainsi que mes collègues de baccalauréat. Vous avez agrémenté mes heures de travail avec d’excellents moments, que ce soit à l’université ou ailleurs.
Un énorme merci à ma collègue de travail Andréanne Blondeau. Elle, ainsi que nos deux stagiaires, Laurence Vincent et Lauralyne Dumont, m’ont empêché de devenir fou et elles ont rendu les heures de travail au laboratoire bien plus plaisantes.
Finalement, un immense merci à mes amis, Dominick Matteau, Louis Fréchette, Martin Morin et Mélissa Roy, qui furent bien plus que des amis durant mes quelques années d’université. Nous avons eu un parcours rempli de bons et de moins bons moments, mais je n’oublierais jamais votre optimisme et toutes les folies que nous avons pu faire depuis notre rencontre.
v TABLE DES MATIÈRES
SOMMAIRE . . . iii
REMERCIEMENTS . . . .iv
TABLE DES MATIÈRES . . . v
LISTE DES ABRÉVIATIONS. . . .vii
LISTE DES FIGURES . . . ix
INTRODUCTION . . . 1
1. Système nerveux . . . 1
1.1. Physiologie des cellules nerveuses . . . 3
1.1.1. Caractéristiques structurales des cellules nerveuses . . . 3
1.1.2. Fonctionnement des cellules nerveuses . . . 4
i-) Synapses . . . 5
ii-) Potentiel gradué . . . 7
iii-) Potentiel d’action . . . 8
1.2. Maladies neurodégénératives . . . 9
2. Mécanisme de neurodégénérescence . . . 12
2.1. Dégénérescence wallérienne . . . 13
2.2. Régénérescence axonale . . . 14
3. Signalisation des MAPK . . . 14
3.1. Voies de JNK . . . 18
3.2. Dual leucine zipper kinase . . . 19
3.2.1. Régulation de DLK . . . 21 3.2.2. Rôle de DLK . . . 22 3.2.3. DLK et neurones . . . 23 4. Signalisation calcique . . . 25 4.1. Calcium et neurones . . . 27 4.2. Calpaïnes . . . 28
vi
5. Hypothèses et objectifs du projet de recherche . . . 30
DÉVELOPPEMENT . . . 32
La dual leucine zipper kinase (DLK) est une cible protéolytique des calpaïnes dans les cellules nerveuses humaines . . . 32
Calcium influx triggers calpain-dependent proteolysis of DLK in human neuronal cells . . . 34
ABSTRACT . . . 35
INTRODUCTION . . . . . . 36
MATERIALS AND METHODS . . . 39
RESULTS . . . 45
DISCUSSION . . . 55
AUTHOR CONTRIBUTIONS . . . .58
REFERENCES . . . 56
DISCUSSION GÉNÉRALE . . . 67
1. Impact d’une hausse intracellulaire de calcium sur DLK . . . 67
2. Régulation de l’expression de la MAP3K DLK par les calpaïnes . . . 69
CONCLUSION ET PERSPECTIVES . . . 72
vii LISTE DES ABRÉVIATIONS
Akt: Protéine kinase B
ADN: Acide désoxyribonucléique ARN: Acide ribonucléique ARNm : ARN messager
ASK1: Apoptosis signal-regulation kinase 1
ATF2: Cyclic AMP-dependent transcription factor 2 ATF3: Cyclic AMP-dependent transcription factor 3 Bcl-2: B-cell lymphoma 2
C/EBPα : CCAAT-enhancer-binding proteins α CHIP: Chromatin immunoprecipitation
CREB: C-AMP Response Element-binding protein cTnI Cardiac troponin I
DLK: Dual leucine zipper kinase
ERK: extracellular signal-regulated kinases GABA: l’acide γ-aminobutyrique
GTP: Guanosine triphosphate HSP70: Heat shock protein 70 IRS-2: Insulin receptor substrate 2 JIP: JNK-interacting protein JNK: c-Jun amino-terminal kinases KDa: Kilo Dalton
LZK1: Leucine Zipper Kinase 1 MAP2K: MAP kinase kinase MAP3K: MAP kinase kinase kinase MAP3K12: MAP kinase kinase kinase 12
viii MAPK: Mitogen-activated protein kinase
MEK: MAP/ERK kinase
MEKK: MAPK/ERK kinase kinase
miRNA: micro ARN
MKK3/4/6/7: MAP kinase kinase 3/4/6/7 MLK: Mixed-lineage kinase
MPTP: 1-methyl-4-phenyl-1, 2, 4, 6-tetrahydropyridine MUK: MAPK upstream kinase
mV: Millivolt
NFAT: Nuclear factor of activated T-cell NGF: Nerve growth factor
PDGF: Platelet-derived growth factor PHR1: Pam/Highwire/RPM-1
PKC: Protein kinase C PMK-3: p38 MAP kinase-3
PPARγ2: Peroxisome proliferator-activated receptor gamma PPSE: Potentiel postsynaptique excitateur
PPSI: Potentiel postsynaptique inhibiteur RPM-1 : Regulator of presynaptic morphology 1 SAPK1/2: Stress-activated protein kinases 1/2 SNC : Système nerveux central
SNP : Système nerveux périphérique
USP9X : Ubiquitin specific peptidase 9 X-linked Wnd : Wallenda
ix LISTE DES FIGURES
INTRODUCTION
Figure I.1 Schéma d’un neurone en contact synaptique avec des neurones . . . 4
Figure I.2 Schéma représentatif d’une synapse . . . 6
Figure I.3 Représentation graphique de l’établissement d’un potentiel d’action . . . .8
Figure I.4 Schéma représentatif des différentes voies des MAPK . . . 16
Figure I.5 Modèle d’activation de la MAPKKK DLK et mécanismes neuronaux dans lesquels elle est impliquée . . . 25
Figure I.6 Schéma illustrant l’impact des calpaïnes sur l’activité apoptotique des neurones en réponse à différents stimuli . . . 29
CHAPITRE I Figure 1. DLK is down-regulated in SH-SY5Y cells exposed to the Ca2+ ionophore A23187 . . . 46
Figure 2. A23187-mediated DLK down-regulation in SH-SY5Y cells is calpain-dependent . . . 49
Figure 3. DLK is a direct proteolytic target of calpain . . . 51
1 CHAPITRE 1
INTRODUCTION
1. Système nerveux
C’est en 1888 que Santiago Ramón y Cajal, neurobiologiste espagnol, postula la théorie du neurone. Il apporta des preuves morphologiques indiscutables affirmant que le système nerveux n’est pas un système continu, mais plutôt un système contigu (López-Muñoz et al., 2006). En effet, le système nerveux est constitué de plusieurs cellules nerveuses, communément appelées neurones, lesquelles sont physiologiquement indépendantes les unes des autres. Cet enchainement de cellules qui forment le système nerveux permet le transport d’influx à travers de longues distances et effectue des tâches primordiales à tout moment. Le système nerveux autonome, par exemple, est la partie du système nerveux responsable des fonctions automatiques comme le contrôle des contractions du cœur, la constriction et la dilatation des vaisseaux sanguins, des bronches et bien plus. Un dérèglement du système nerveux autonome, ainsi que toute autre partie du système nerveux, aura des effets néfastes sur l’organisme pouvant mener à la mort.
Le système nerveux peut être séparé en deux sections distinctes, soit le système nerveux central (SNC) et le système nerveux périphérique (SNP). Ils sont tous deux constitués des
2 deux mêmes types cellulaires, les cellules nerveuses et les cellules gliales, mais ont toutefois plusieurs différences au niveau métabolique. Le SNC est constitué de la moelle épinière et de l’encéphale, comprenant le cerveau, le cervelet et le tronc cérébral. Le SNC est très important, car il intègre l’information reçue, coordonne et influence l’activité de toutes les parties du corps. Le SNP, de son côté, est constitué des nerfs crâniens, des nerfs spinaux, des ganglions spinaux et des récepteurs sensoriels et son rôle est de faire circuler les influx nerveux entre les organes et le SNC. La principale différence métabolique entre le SNP et le SNC est que le SNP subit la myélinisation axonale par les cellules de Schwann, alors que dans le SNC, les oligodendrocytes remplissent cette fonction. Les neurones du SNP ont aussi la capacité de régénérer une cellule nerveuse fonctionnelle suite à un dommage cellulaire. Chez les mammifères, cette capacité de régénération n’est pas présente dans les neurones du SNC qui sont alors protégés par une enveloppe osseuse, constituée de la boîte crânienne et de la colonne vertébrale servant à protéger l’encéphale et la moelle épinière respectivement. Les dommages observés au SNC lors de maladies neurodégénératives, étant graves et irréversibles, les recherches sur les mécanismes de mort cellulaire prennent de plus en plus d’importance.
3
1.1.
Physiologie des cellules nerveuses1.1.1. Caractéristiques structurales des cellules nerveuses
Les unités fonctionnelles du système nerveux, les neurones, ont acquis la capacité de transmettre un signal bioélectrique appelé l’influx nerveux. Leur morphologie particulière et leurs composantes protéiques permettent le fonctionnement de la cellule, et du système nerveux (Figure I.1). Le neurone est composé d’un corps cellulaire, aussi appelé péricaryon, qui signifie en grec « autour du noyau » ou soma, dont la membrane se prolonge afin de former les dendrites et un prolongement unique nommé l’axone. Le corps cellulaire est composé des organites ainsi que du noyau, responsable de la synthèse des macromolécules, et il permet de donner une base structurale au neurone. Les dendrites forment plusieurs arborisations dendritiques autour du corps cellulaire. Elles sont nombreuses, courtes et très ramifiées. Les liaisons des dendrites avec des cellules nerveuses en amont, via des synapses neuro-neuronales, ou des cellules sensorielles, via des synapses sensori-neuronales, vont permettre la création d’un potentiel d’action qui sera dirigé sur de courtes distances, vers le cône d’implantation de l’axone. C’est au cône d’implantation que commence l’axone qui conduit le potentiel d’action de manière centrifuge en direction de l’arborisation terminale de l’axone. Les terminaisons axonales pourront faire des contacts synaptiques avec des neurones, généralement via des synapses axo-dendritiques ou des synapses axo-somatiques, qui relient l’axone à la cellule nerveuse en aval sur ses dendrites ou sur son corps cellulaire, respectivement. L’axone peut aussi faire des contacts synaptiques avec des fibres musculaires, via des jonctions neuromusculaires, ou avec des cellules glandulaires, via des synapses neuro-glandulaires. Ainsi, entre une cellule réceptrice et une cellule effectrice, plusieurs neurones forment un réseau qui propage un influx nerveux sous la forme d’un
4 Figure I.1 Schéma d’un neurone en contact synaptique avec des neurones.
potentiel d’action. L’axone est entouré de cellules de Schwann, lesquelles vont produire la gaine de myéline, une couche lipidique importante dans le transport du signal dans l’axone.
1.1.2. Fonctionnement des cellules nerveuses
Les cellules nerveuses, bien qu’étant des unités cellulaires indépendantes, doivent former un réseau capable de transmettre le signal d’une cellule à une autre. Cette communication cellulaire se fait spécifiquement au niveau des synapses et les cellules doivent pouvoir
5 déplacer le signal bioélectrique de part et d’autre de leurs dendrites et de leur axone, sur une distance pouvant aller jusqu’à plus d’un mètre. Cette transmission de signal se fait en plusieurs étapes, soit la réception d’un signal au niveau d’une synapse, la création d’un potentiel gradué local et la création d’un potentiel d’action au niveau du cône d’implantation de l’axone. Ces étapes seront décrites plus en détail dans cette section.
i-) Synapses
Les synapses désignent les zones où une cellule nerveuse fait un contact fonctionnel avec une autre cellule. Tel que vu précédemment, il existe différents types de synapses selon les types cellulaires et les zones du neurone où se produit le contact synaptique. Le concept de synapse fut introduit en 1897 par un physiologiste britannique, Charles Scott Sherrington, affirmant que l’arborescence terminale n’est pas continue, mais plutôt en contact partiel avec la cellule voisine via des connections qu’il nomma « synapse » (López-Muñoz et al., 2006). Une synapse est constituée de trois parties distinctes qui sont illustrées dans la figure I.2, soit l’élément présynaptique, la fente synaptique et finalement, l’élément postsynaptique. L’élément présynaptique provient de la cellule en amont du système, soit une cellule réceptrice ou un neurone. Elle est remplie de vésicules synaptiques contenant les neurotransmetteurs, lesquels sont synthétisés sur place par les nombreuses mitochondries et l’appareil de Golgi présent dans l’élément présynaptique. Les types de neurotransmetteurs sont très variés, les plus communs étant l’acétylcholine, le glutamate, la glycine et l’acide γ-aminobutyrique (GABA). Les neurotransmetteurs sont relâchés dans la fente synaptique lorsque la synapse est activée par un potentiel d’action afférent. Les neurotransmetteurs dans la fente peuvent alors être perçus par les récepteurs présents au niveau de la membrane de l’élément postsynaptique. L’élément postsynaptique, généralement situé au niveau des dendrites ou du corps cellulaire, est celui qui discrimine le signal selon les types de
6 Figure I.2 Schéma représentatif d’une synapse. La figure montre des récepteurs
ionotrope et métabotrope permettant l’entrée d’ions suite à la liaison du neurotransmetteur libéré dans la fente synaptique.
récepteurs présents sur la membrane. Il existe deux types de récepteurs présents sur la membrane postsynaptique, les récepteurs ionotropes et les récepteurs métabotropes. Les premiers sont des protéines membranaires sous forme d’un canal ionique. Ils s’ouvrent suite à la liaison du ligand et permettent l’entrée d’ions tels que Na2+, K+, Ca2+ ou Cl-. Les seconds sont des récepteurs utilisant un second messager intracellulaire, généralement des protéines G trimériques associées à la membrane interne, afin de permettre l’ouverture de canaux ioniques. Les récepteurs ionotropes ont la particularité de convertir un signal extrêmement rapidement contrairement aux récepteurs métabotropes. L’ouverture des canaux ioniques à l’intérieur de l’élément postsynaptique permet la production d’un potentiel gradué.
7 ii-) Potentiel gradué
Un potentiel gradué est produit lors de l’entrée d’ions au niveau des dendrites ou du soma causé par des canaux ligand-dépendants. Ce changement transitoire de la différence de potentiel électrochimique cause une dépolarisation (augmentation du potentiel membranaire) ou une hyperpolarisation (diminution du potentiel membranaire) de la membrane, en fonction du type d’ions qui a été pompé à l’intérieur de la cellule. Ces signaux pourront se diriger vers le cône d’implantation de l’axone où il y aura une sommation des signaux afin de créer un potentiel d’action. Ces potentiels gradués sont locaux et ne peuvent traverser de longues distances, comme l’axone, puisqu’il y a dilution des ions à l’intérieur du cytoplasme et puisqu’un grand nombre de pompes permettront le rétablissement du potentiel à la normale. Il est alors important de pouvoir créer un potentiel d’action afin de pouvoir voyager à travers l’axone et transmettre le signal vers l’arborisation terminale du neurone. Les potentiels postsynaptiques peuvent être soit excitateur (PPSE), créé par la dépolarisation membranaire suite à l’entrée de cations, soit inhibiteur (PPSI), créé par la polarisation de la membrane par l’entrée d’anions. Afin d’atteindre un potentiel d’action dans l’axone, il doit souvent y avoir sommation des potentiels gradués, créés au niveau de différents membranes postsynaptiques, lorsque ceux-ci ne sont pas suffisamment forts, afin d’atteindre le seuil d’excitabilité du neurone (Figure I.3). L’atteinte du seuil d’excitabilité dans le cône d’implantation permet la production du potentiel d’action dans l’axone.
8 iii-) Potentiel d’action
Lorsque le seuil d’excitabilité de l’axone est atteint, il y aura certainement la création d’un potentiel d’action et donc d’un influx nerveux pouvant transmettre un signal jusqu’à l’arborisation terminale de l’axone. Le potentiel d’action est, comme le potentiel gradué, une dépolarisation de la membrane, mais causé cette fois-ci par l’activation de canaux sodiques voltage-dépendants. L’entrée d’ions sodium (Na2+) causera une dépolarisation de la membrane et les ions présents dans l’axone pourront activer les canaux voisins, créant ainsi une réaction en chaîne qui permet le transport de l’influx. Cette dépolarisation sera rétablie
Figure I.3 Représentation graphique de l’établissement d’un potentiel d’action. A) la dépolarisation causée par le potentiel gradué ne permet pas l’atteinte du seuil d’excitabilité de -50mV. B) La sommation de plusieurs potentiels postsynaptiques excitateurs permet la dépolarisation de la membrane jusqu’à l’atteinte du seuil d’excitabilité permettant la formation d’un potentiel d’action à 30mV. C) La sortie d’ions potassium causée par la dépolarisation va résulter en une hyperpolarisation sous l’état de repos, empêchant la membrane d’être dépolarisée une seconde fois dans un intervalle trop court.
9 par la sortie d’ions potassium (K+) qui, en étant pompées à l’extérieur très rapidement, va créer une période réfractaire durant laquelle la membrane sera hyperpolarisée et ne pourra pas être dépolarisée avant le retour à l’état de repos (Figure I.3). La vitesse du potentiel d’action est aussi grandement amplifiée grâce à la gaine de myéline présente sur la membrane de l’axone. Ces multiples couches lipidiques de sphingomyéline produites par les cellules de Schwann dans les neurones du SNP et par les oligodendrocytes dans les neurones du SNC, empêchent la présence de protéines transmembranaires, dont les canaux ioniques. Les canaux sodiques voltage-dépendants seront présents entre les couches lipidiques, au niveau des nœuds de Ranvier et permettront la propagation de l’influx. En bloquant la présence de canaux à certains endroits de la membrane de l’axone, la gaine de myéline empêche la repolarisation de la membrane par la sortie d’ions sodiques et permet à ceux-ci de voyager d’un nœud à l’autre afin d’activer les canaux voltage-dépendants. Ce signal bioélectrique produit par la dépolarisation membranaire permet, au niveau des éléments présynaptiques, le relâchement de neurotransmetteurs dans la fente synaptique afin de produire un influx en chaîne jusqu’à la cellule effectrice.
1.2.
Maladies neurodégénérativesLes rôles et l’importance des cellules nerveuses et du système nerveux font qu’un dérèglement de l’activité de plusieurs neurones aura des répercussions graves sur l’organisme.En effet, plusieurs maladies neurodégénératives bien connues sont causées par une perte de fonctions, ou la mort de plusieurs neurones. L’Alzheimer, le Parkinson, la maladie de Creutzfeldt-Jakob, la chorée de Huntington, la sclérose en plaques, le glaucome et bien d’autres de ces maladies affectent les cellules neuronales par différents mécanismes de neurodégénérescence qui sont souvent peu compris. Dans le but ultime de développer des
10 outils de prévention ou de guérison, les recherches sur les mécanismes de neurodégénérescence au niveau moléculaire permettront une meilleure compréhension du fonctionnement du système nerveux et des problèmes qui s’y rattachent.
Plusieurs facteurs peuvent causer la perte de fonction ou la mort des cellules neuronales. Les cellules nerveuses peuvent être affectées par des facteurs physiques. Le glaucome, par exemple, est une maladie neurodégénérative du nerf optique qui cause une perte progressive de la vision. Elle est souvent causée par une augmentation de la pression intraoculaire qui comprime le nerf optique jusqu’à détruire les cellules nerveuses qui le composent. Ce symptôme est irréversible et peut mener jusqu’à la cécité (Tezel, 2006; Société canadienne de recherche sur le glaucome).
La sclérose en plaques, quant à elle, est une maladie causée par le système immunitaire ciblant le SNC. Les symptômes observés sont causés par une démyélinisation des fibres nerveuses du système nerveux. Les symptômes sont irréversibles et très variables d’une personne à l’autre, et parfois même, variable d’une journée à l’autre chez une même personne. Les personnes atteintes peuvent souffrir de troubles de l’équilibre, troubles urinaires, troubles intestinaux, troubles cognitifs, dépression, fatigue, névrite optique, troubles sexuels, tremblements et bien d’autres. Les facteurs à l’origine de la sclérose en plaques restent encore inconnus (Société canadienne de la sclérose en plaques).
La maladie de Creutzfeldt-Jakob, plus communément appelée la maladie de la vache folle, est une maladie neurodégénérative du SNC causée par l’accumulation d’un prion. Le prion,
11 protéine naturellement présente dans le cerveau, se voit déformer par la présence d’un autre prion à la forme non naturelle. Les prions vont s’attaquer au cerveau et détruire les cellules, causant des brèches. La maladie peut être transmise à l’homme s’il mange de la viande d’animal infecté et les symptômes vont de troubles de l’équilibre et de la sensibilité jusqu’à la démence et la mort (Société Alzheimer Canada).
L’Alzheimer est la maladie neurodégénérative la plus répandue. Elle représente 64% de tous les cas au Canada. Elle se caractérise par deux types de lésions. Premièrement, l’accumulation de plaques amyloïdes extracellulaires libérant des composés neurotoxiques augmentant l’inflammation et causant la mort cellulaire par apoptose ou par nécrose. Deuxièmement, une agrégation des protéines Tau intracellulaires menant à une déstabilisation des microtubules de la cellule. Ces deux processus causent la mort des cellules nerveuses occasionnant des symptômes graves comme la perte de mémoire, la démence et même la mort (Bossy-Wetzel et al., 2004; Beharry et al.,2014; Société Alzheimer Canada).
La maladie de Parkinson est une maladie neurologique chronique dégénérative qui affecte le SNC. Elle est la deuxième plus fréquente maladie neurodégénérative après l’Alzheimer et est causée par un dérèglement du système dopaminergique. La dopamine est un neurotransmetteur important dans le cerveau qui joue un rôle modulateur essentiel des sorties motrices. Les individus atteints de la maladie de Parkinson souffrent de tremblements, de rigidité musculaire, de problème d’équilibre et de lenteur (Bossy-Wetzel et al., 2004; Société Parkinson Canada).
12 La maladie de Huntington est une affection génétique causée par un nombre anormal de répétitions des nucléotides CAG sur le gène HD au niveau du chromosome 4p16.3. La maladie fonctionne par un phénomène d’anticipation, c’est-à-dire que les symptômes s’aggravent selon le nombre de répétitions de la séquence observée. C’est une maladie génétique dominante, signifiant qu’elle est transmissible une fois sur deux aux futures générations. Les personnes atteintes souffriront de dégénérescence neurologique menant à d’importants troubles moteurs, cognitifs et psychiatriques. Cependant, des formes plus graves de la maladie causent une perte d’autonomie et la mort (Bossy-Wetzel et al, 2004; Société Huntington du Québec).
2. Mécanismes de neurodégénérescence
La dégénérescence des cellules nerveuses est un évènement qui se produit à différentes occasions, par exemple, lors du développement du système nerveux, lors du dérèglement de l’organisme, caractérisé par une maladie neurodégénérative et lors d’un dommage causé à l’axone d’une cellule. Les mécanismes menant à la dégradation d’un neurone sont variés et complexes. Lors du développement du système nerveux, par exemple, des neurones immatures sont produits en grande quantité, et possèdent tous un grand nombre de neurites pouvant former des synapses avec les cellules voisines. Cependant, plusieurs neurones immatures complets ne s’intègrent pas au réseau, et doivent être retirés par l’activation de la mort cellulaire programmée et plusieurs neurites ne sont pas en contact synaptique et doivent aussi être retirées par un mécanisme de dégénérescence ou de rétraction. Ce principe, appelé l’élagage, permet la formation d’un réseau efficace constituant le système nerveux (Neukomm et Freeman, 2014). La dégénérescence neuronale observée chez les
13 patients atteints de maladies neurodégénératives est variée et souvent mal comprise. Elle est provoquée par une grande variété de stimuli et cause beaucoup de dommage à l’organisme. Finalement, le modèle le mieux compris de dégénérescence observée dans une cellule nerveuse est le modèle de la dégénérescence wallérienne. Ce modèle de dégénérescence est utilisé lors des recherches sur les mécanismes de dégénérescence axonale depuis plusieurs années, car il peut être provoqué par une incision de l’axone.
2.1.
Dégénérescence wallérienneEn 1850, le neuropathologiste britannique, Augustus Waller observa, pour la première fois, qu’après incision de l’axone, la portion distale, soit celle qui n’est plus en contact avec le corps cellulaire, subissait une dégénérescence progressive (Ma et al., 2013). En effet, la dégénérescence wallérienne est un phénomène qui survient lorsqu’il y a un dommage axonal causant la division de l’axone en au moins deux sections : la section proximale et la section distale. Lorsque l’axone est scindé, la section distale subit une décomposition du cytosquelette, la destruction de ses organelles internes et une désintégration granulaire causant l’élimination de cette section (Neukomm et Freeman, 2014). Ces évènements vont activer une série de réponses chez les cellules voisines, les cellules gliales, qui subiront des changements et permettront le recrutement, à l’axone en décomposition, de macrophages, lesquels pourront éliminer, par phagocytose, les débris produits lors de la dégénérescence de l’axone (Luo et O’Leary, 2005).
14
2.2.
Régénérescence axonaleDe son côté, la section proximale, soit la portion de l’axone qui est toujours en liaison avec le corps cellulaire, est soumise à un phénomène plus complexe. Chez les mammifères, lorsqu’un neurone du SNP est coupé au niveau de son axone, la section proximale produite subira un phénomène de régénérescence axonale. Bien que la section distale de l’axone soit éliminée, les cellules de Schwann formeront un tube permettant de diriger l’axone en formation jusqu’à sa cible (Brushart, 1993). Ce phénomène, observé chez les invertébrés, quelques animaux à sang-froid et chez les cellules du SNP de mammifères, permet la formation d’un axone fonctionnel qui reformera des contacts synaptiques avec les cellules voisines (Liu et al., 2011). La régénérescence axonale est un évènement qui n’est pas observée lors de l’incision d’une cellule du SNC. Les dommages engendrés à ces cellules sont permanents et mènent à la perte de leurs fonctions (Lu et al., 2014). Des modèles animaux tels que C. elegans, la drosophile et la souris sont utilisés pour la recherche sur les mécanismes de dégénérescence et régénérescence axonale afin d’approfondir les connaissances sur les facteurs moléculaires qui sont impliqués dans ces mécanismes.
3. Signalisation des MAPK
Les cellules d’un organisme doivent être en mesure de détecter un signal provenant de leur milieu, mais aussi de transmettre adéquatement l’information du récepteur jusqu’à l’effecteur intracellulaire ciblé. Pour ce faire, les cellules ont recours à diverses protéines qui agissent comme messagers. Ces messagers, qui agissent en cascade, vont interagir entre eux,
15 permettant l’amplification d’un signal perçu à un récepteur vers un grand nombre de molécules effectrices. Parmi ces protéines messagères, on retrouve la voie des mitogen-activated protein kinases (MAPK) exclusivement chez les organismes eucaryotes. Cette dernière est composée de plusieurs voies de signalisation différentes, lesquelles fonctionnent généralement de la même manière. Chez les mammifères, cependant, trois voies de MAPK participent activement à la transduction de nombreux signaux, soit les voies de extracellular signal-regulated kinases (ERK), c-Jun amino-terminal kinases (JNK) et p38 (Bost et al., 2005; Philpott et Facci, 2008). Ces voies, par un mécanisme d’assemblage sur une protéine échafaud, forment un complexe contenant une chaîne de trois kinases (Figure I.4). Une première protéine du complexe, une MAP Kinase Kinase Kinase (MAP3K), pourra être activée par un des divers stimuli affectant cette voie. Une fois activée, généralement par la phosphorylation de celle-ci, la MAP3K pourra activer par phosphorylation la seconde protéine, une MAP Kinase Kinase (MAP2K). Cette dernière pourra à son tour phosphoryler et activer la dernière protéine du complexe, une MAPK. La MAPK activée pourra également phosphoryler et/ou interagir avec différentes protéines afin de créer un effet biologique en réponse au stimulus détecté (Bost et al., 2005).
La famille des protéines kinases représente environ 1,7% du génome humain, ce qui en fait la plus grande famille de gènes (Roskoski, 2010). Ces dernières sont des catalyseurs des réactions de phosphorylation permettant l’ajout d’un groupement phosphate à une molécule cible, telles qu’une protéine, un lipide, un carbohydrate, des acides nucléiques ou d’autres macromolécules. Dans le groupe des kinases de protéines, on retrouve trois types de kinases, les tyrosines kinases, les histidines kinases et les sérines/thréonines kinases, lesquelles vont permettre la phosphorylation de leurs cibles au niveau de leurs acides aminés respectifs. Les MAPK présentées plus haut sont des sérines/thréonines kinases (Bost et al., 2005) .Il a été démontré à multiples reprises que les voies des MAPK sont impliquées dans une grande
16 Figure I.4 Schéma représentatif des différentes voies des MAPK. Les trois
voies principales des MAPK : la voie des ERK, des p38 et des JNK sont représentées ici avec les différents médiateurs participant à la transmission d’un signal par les MAPK.
Adapté de (Couture, 2011)
variété de processus métaboliques importants, et ce dans différents types cellulaires de l’organisme.
La voie de signalisation de ERK, souvent appelée la voie Ras-Raf-MEK-ERK, fut la première voie des MAPK à être identifiée chez les mammifères (Kyriakis et Avruch, 2012). Elle participe à une grande variété de mécanismes incluant l’adhésion cellulaire, la
17 progression du cycle cellulaire, la migration cellulaire, la survie cellulaire, la différenciation, le métabolisme, la prolifération cellulaire et la transcription (Roskoski, 2010). L’activation de récepteurs membranaires à activité tyrosine kinase suite à la liaison de facteurs de croissance permet l’activation d’une GTPase, la protéine Ras. Par l’action de cette dernière, la MAP3K Raf pourra phosphoryler ses substrats, les MAP2K MEK1 et MEK2, qui ensuite phosphoryleront les MAPK ERK1 et ERK2. Les protéines ERK1/2 activées permettront l’activation de divers facteurs cytoplasmiques ou nucléaires, lesquels contribueront à activer la réponse biologique attendue (Roskoski, 2010).
De son côté, la voie de p38 est principalement activée par le stress ou des facteurs d’inflammation, d’où lui vient son autre nom, la Stress-activated protein kinase 2 (SAPK2). Divisée en quatre isoformes, p38α, p38β, p38γ et p38δ (Bost et al., 2005), dont la présence varie selon les tissus, elle sera activée suite à sa phosphorylation par les MAP2K MKK6, MKK3 ou MKK4. Ces dernières sont elles-mêmes activées par une grande variété de MAP3K dont la MAPK/ERK kinase kinase 3 (MEKK3), la MEKK4, la Mixed-lineage kinase 3 (MLK3), la apoptosis signal-regulating kinase 1 (ASK1) et bien d’autres (Johnson et Nakamura, 2007; Cuadrado et Nebreda, 2010). Une fois activée, la protéine p38 phosphoryle une grande variété de substrats, notamment d’autres protéines kinases, des protéines nucléaires et des facteurs de transcription et de régulation de la chromatine. C’est pourquoi la voie de p38 est impliquée dans plusieurs fonctions physiologiques afin de répondre aux différents stimuli permettant de l’activer (Cuadrado et Nebreda, 2010).
18
3.1.
La voie JNKLa troisième voie principale des MAPK est la voie de JNK. Elle est activée afin de répondre à des situations de stress auxquelles la cellule est exposée comme des expositions au rayonnement ultraviolet, des chocs thermiques et des inhibiteurs de la traduction, par exemple la cycloheximide et l’anisomycine. Elle permet aussi la réponse à des cytokines pro-inflammatoires et à quelques facteurs de croissance (Kyriakis et Avruch, 2012). La protéine JNK, aussi appelé la Stress-activated protein kinase (SAPK), existe sous trois isoformes différentes, JNK1, JNK2 et JNK3, lesquelles sont toutes exprimées sous une forme courte de 46 KDa et une forme longue de 54 KDa. Les isoformes JNK1 et JNK2 sont exprimées de manière ubiquitaire tandis que JNK3 est limitée au cerveau, au cœur et aux testicules. Les trois isoformes vont majoritairement différer au niveau de leur habilité à phosphoryler leurs substrats, ce qui permet de moduler une grande variété de réponses biologiques (Johnson et Nakamura, 2007; Kyriakis et Avruch, 2012). La protéine JNK fonctionne, comme les autres MAPK, en phosphorylant sa cible sur les acides aminées sérines et thréonine. Elle forme un complexe sur l’une des protéines échafauds avec une MAP2K et une MAP3K permettant la cascade d’activation. Les MAP2K activant la protéine JNK sont MKK4 et MKK7, lesquelles peuvent être activées par 14 différentes MAP3K dont la protéine Dual Leucine Zipper kinase (DLK), la protéine d’intérêt dans ce mémoire. Une fois activée, la protéine JNK possède plusieurs substrats qu’elle pourra elle-même activer par phosphorylation. Ses substrats sont variés, allant de facteurs de transcription, comme c-Jun, ATF2, ATF3 et NFAT4, aux facteurs d’apoptose, aux protéines de signalisation, aux microtubules ainsi qu’à une protéine des pores nucléaires. Une fois activés, les substrats de JNK vont contribuer à l’initiation de diverses réponses physiologiques, telles que la prolifération cellulaire, l’apoptose, la mobilité, la réparation de l’ADN, des réactions métaboliques (Johnson et Nakamura, 2007) et aussi possiblement à la suppression de tumeurs (Cazilis et al., 2004).
19 Il a été montré à multiples reprises l’implication de la voie de signalisation de JNK dans le mécanisme de neurodégénérescence. En effet, JNK est un messager important dans les cellules nerveuses, car son activation engendre la mort par apoptose. Des souris JNK3 -/- ou JNK2 -/-, mais pas les JNK1 -/-, ont montré une résistance à la neurotoxine 1-methyl-4-phenyl-1, 2, 4, 6-tetrahydropyridine (MPTP), laquelle cause la mort des cellules nerveuses dopaminergique comme ce qui est observé chez les personnes atteintes de la maladie de Parkinson. De plus, les souris JNK3 -/- sont aussi résistantes à l’apoptose causée par l’accumulation de peptide Aβ42, aussi appelé les plaques amyloïdes, observée chez les patients atteints de l’Alzheimer (Antoniou et al., 2011; Johnson et Nakamura, 2007). L’implication de JNK dans le mécanisme de neurodégénérescence, bien qu’encore peu compris, passe par l’activation de cette dernière par les MAP2K. Il est alors concevable d’affirmer que les MAP2K et les MAP3K de la voie de JNK sont possiblement aussi impliquées dans la neurodégénérescence. Les protéines telles que les MAP2K MKK4 et MKK7 ainsi que plusieurs MAP3K, dont la protéine DLK, sont sujettes à des travaux de recherche visant à comprendre comment la voie JNK est activée et comment cette voie permet d’engager la cellule nerveuse dans le processus de mort cellulaire programmé.
3.2.
Dual Leucine Zipper KinaseLa protéine DLK, aussi appelée MAP Kinase Kinase Kinase 12 (MAP3K12), est une sérine/thréonine kinase de la famille des mixed lineage kinases (MLK), un sous-groupe de MAP3K. Ces protéines se caractérisent sur le plan structural par la présence d’un domaine kinase auquel est juxtaposé du côté C-terminal un domaine leucine-zipper permettant la dimérisation entre les membres de la même famille. La dimérisation de la protéine DLK est essentielle à son autophosphorylation et à son activation (Nihalani et al., 2000; Wang et al.,
20 2004). DLK est connue pour se lier à, et phosphoryler, MKK4 et MKK7, deux MAP2K de la voie JNK. Cependant, la protéine DLK n’a démontré son activité kinase in vitro qu’avec le substrat MKK7 (Hirai et al., 1996; Merritt et a 1999).
Ayant été, pour la première fois, clonée en 1994 à partir des cellules de carcinome embryonnaire humain NTERA-2, une lignée présentant des caractéristiques similaires aux cellules neuronales suite à leur différenciation, la protéine DLK a été initialement appelée Zipper Protein Kinase (ZPK) (Reddy et Pleasure, 1994). Des orthologues chez la souris et le rat ont ensuite été identifiés et nommés DLK et MAPK upstream kinase (MUK) respectivement (Holzman et al., 1994; Hirai et al., 1996). À partir d’un embryon de souris, la plus forte expression du gène DLK observée se trouvait majoritairement au niveau des structures neuronales, comme le cerveau et les ganglions spinaux, mais aussi au niveau de certains organes ou tissus comme la peau, l’intestin, le pancréas et les reins (Blouin et al., 1996; Nadeau et al., 1997). De manière plus précise, la protéine DLK a été localisée, en grande majorité, associée aux microtubules de l’axone des cellules nerveuses et elle a également été observée an niveau de l’appareil du Golgi dans des cellules corticales embryonnaires (Hirai et al., 2002). Par la suite, le gène codant pour la MAP3K12 a été identifié chez la drosophile en 2006 et nommé Wallenda (Wnd) durant une recherche sur la croissance synaptique au niveau des jonctions neuromusculaires (Collins et al., 2006).
21 3.2.1. Régulation de DLK
La régulation de l’activité de DLK est un mécanisme qui est encore peu compris. L’importance de la dimérisation de la protéine a été proposée à plusieurs reprises (Nihalani et al., 2000; Robitaille et al., 2008). Cela est également appuyé par une expérience d’oligomérisation de DLK induite par la transglutaminase, montrant la hausse de phosphorylation de JNK (Robitaille et al., 2004). Plusieurs recherches suggèrent que la quantité intracellulaire de DLK est régulée par ubiquitination. Chez les mammifères, DLK serait ciblée par PHR1, une E3 ubiquitine ligase, afin de la diriger vers le protéasome (Lewcock et al., 2007). Elle serait aussi ciblée et envoyée au protéasome par la protéine HSP70 et sa co-chaperone associé, l’ubiquitine ligase CHIP (Daviau et al., 2006). Ce mécanisme de régulation a aussi été observé chez le vers C. elegans ainsi que la Drosophile, chez lesquels les orthologues de PHR1, soit RPM-1 et Highwire, vont permettre la régulation négative de DLK (Nakata et al., 2005; Collins et al., 2006; Bloom et al., 2007). Des travaux récents ont montré que le niveau de DLK dans l’axone serait régulé par l’action opposée de l’ubiquitine ligase PHR1 et de la désubiquitinase USP9X. Dans ce modèle, le niveau de DLK en condition « normale » est stabilité par les deux protéines. Lorsqu’elle est activée en réponse à un stress, DLK permet l’activation de JNK qui par un mécanisme de rétroactivation phosphorylerait DLK afin de réguler sa stabilité (Huntwork-Rodriguez et al., 2013). Une autre recherche suggère une régulation de DLK par une famille de micro ARN (miRNA), les miR-17, dont l’expression par transfection provoque une baisse des niveaux intracellulaires de DLK (Beveridge et al., 2009). Finalement, en condition normale, la liaison de DLK à la protéine échafaud JNK-interacting protein (JIP) permettrait une régulation négative de l’activité de DLK, empêchant ainsi l’activation des cibles de la protéine en absence de signal. En effet, la liaison à JIP empêcherait la dimérisation de DLK nécessaire à son activation. Une fois JNK associée à la protéine échafaud en réponse à un
22 stimulus, DLK se libère de JIP et s’active en formant un dimère, ce qui lui permet de phosphoryler ses cibles (Nihalani et al., 2001).
3.2.2. Rôles de DLK
Depuis sa découverte, plusieurs groupes de recherche se sont intéressés à la protéine DLK et ont démontré ses implications dans des processus cellulaires différents. En plus de son rôle dans l’activation de la voie JNK, DLK possède la capacité d’activer ERK et p38 dépendamment du contexte cellulaire dans lequel elle est exprimée (Gallo et Johnson, 2002; Daviau et al., 2009). Il a été démontré que DLK participe au processus de différenciation des kératinocytes (Germain et al.2000; Robitaille et al. 2005). En effet, suite à la surexpression de DLK dans une monocouche de kératinocytes humains en culture, surviennent l’arrêt de la croissance et la différenciation des cellules (Germain et al., 2000). Cette observation concorde avec les expériences de localisation, qui ont montré la présence de DLK dans la couche granuleuse de l’épiderme, couche où se produit la différenciation terminale des cellules (Nadeau et al. 1997; Robitaille et al. 2005). Dans un autre modèle, il a été montré par interférence d’ARN que DLK est essentielle pour l’accumulation de lipide et l’expression de deux régulateurs clés de la différentiation des adipocytes : PPARγ2 et C/EBPα (Couture et al., 2009). Parallèlement, la différenciation des préadipocytes 3T3-L1 montre une augmentation graduelle de la quantité de DLK, similaire à l’augmentation des marqueurs adipogéniques PPARγ2, adiponectine et acide gras synthase (Couture et al., 2009). En plus d’être impliquée dans les mécanismes de différenciation, des recherches ont montré que DLK à un rôle dans la régulation de la signalisation par les facteurs de croissance. La déplétion de DLK par interférence à d’ARN bloque l’activation des protéines Akt et ERK, deux kinases impliquées dans la réponse aux facteurs de croissance, après
23 traitement au Platelet-derived growth factor (PDGF) (Daviau et al., 2009). D’autre part, une étude a permis de confirmer que DLK est un médiateur de l’apoptose dans les cellules β du pancréas et des neurones (Plaumann et al., 2008). Effectivement, l’activation de la protéine DLK dans les cellules β du pancréas a permis d’observer une diminution de l’activité de C-AMP Response Element-binding protein (CREB), un facteur de transcription essentiel à la régulation de l’expression des protéines de survie cellulaire IRS-2 et Bcl-2 (Reddy et al., 1999; Jhala et al., 2003; Plaumann et al., 2008).
3.2.3. DLK et neurones
À l’instar de ce mémoire, beaucoup d’études se sont intéressées à l’implication de la protéine DLK dans les cellules neuronales, notamment en raison de son expression préférentielle dans ces cellules au niveau du cerveau et de sa localisation dans les axones. Plus spécifiquement, la délétion de l’ARNm de DLK-1, l’orthologue de DLK chez le vers C. elegans, a permis de mettre en évidence la contribution de cette protéine dans les processus de régénérescence. La régénérescence des neurones suite à une blessure axonale nécessite la protéine DLK et sa capacité à activer la voie JNK ou la voie PMK-3, l’orthologue de p38 chez C. elegans (Hammarlund et al., 2009; Watkins et al., 2013; Stone et al., 2014; Frey et al., 2015). Similairement, des études ont montré que l’orthologue de DLK chez la drosophile, Wnd, stimule l’activation d’une voie de signalisation promouvant la régénérescence axonale (Xiong, et al., 2010; Fang et Bonini, 2012) en réponse à un dommage, mais pas d’implication dans la régénérescence observée suite à l’incision d’un dendrite (Stone et al., 2014). La déplétion de DLK effectuée sur une lignée cellulaire dérivée de phéochromocytome de rat démontre aussi l’importance de la protéine lors de l’élongation et la croissance des axones via la voie JNK (Eto et al., 2010). En parallèle, chez la souris, il
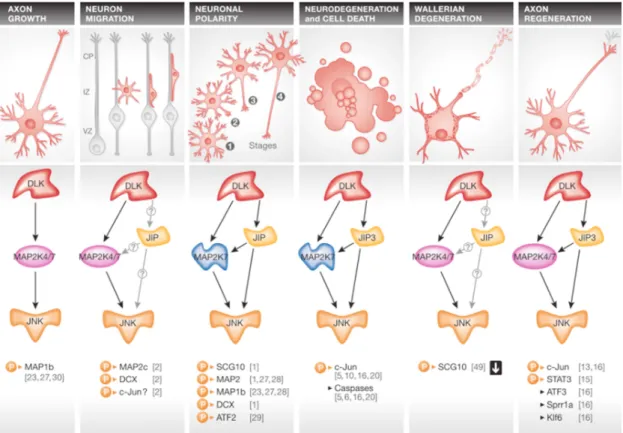
24 a été observé que l’inactivation complète du gène DLK est létale dans les jours qui suivent la naissance et que le développement du cerveau chez ces individus est grandement affecté, suggérant un rôle pour DLK lors de l’embryogenèse et l’organogenèse (Hirai et al., 2006; Bloom et al., 2007). Comme dans les cellules β pancréatiques, il a été montré que la protéine DLK est un médiateur de l’apoptose dans les cellules neuronales. La délétion de DLK dans des ganglions spinaux en culture a permis d’observer la protection des cellules nerveuses contre la mort cellulaire et la dégénérescence axonale causées par la privation de Nerve Growth Factor (NGF) (Ghosh et al., 2011) et a aussi permis d’observer la protection de la dégradation de l’axone après son incision (Miller et al., 2011; Welsbie et al., 2013; Watkins et al., 2013). Parallèlement, dans un modèle in vivo de souris, la délétion du gène DLK a permis d’observer une protection des neurones contre le processus d’apoptose et des processus de mort cellulaire non-apoptotique, tels que l’excitotoxicité (Pozniak et al., 2013). À partir de cellules NIH 3T3 en culture, des fibroblastes murins, il a également été démontré que DLK joue un rôle essentiel dans le processus d’apoptose induit par le calphostin-C, un antibiotique qui inhibe la protéine kinase C (PKC). La délétion de DLK a permis d’observer une diminution de l’activation de la kinase JNK causée par le calphostin-C (Robitaille et al., 2008). Dernièrement, il a été montré que l’activation de la voie JNK via DLK, chez la drosophile ainsi que chez la souris, est impliquée dans le processus de transport axonale (Hirai et al., 2002; Horiuchi et al., 2007). Les multiples rôles de DLK, parfois contradictoires, dépendent du contexte cellulaire et ont besoin d’être éclaircis (Figure I.5).
25 4. Signalisation calcique
Le calcium est un métal alcalino-terreux essentiel pour la survie des organismes vivants. Il est important pour la formation des os, des dents et des coquilles et pour le fonctionnement des systèmes vitaux. Cependant, outre son importance pour la matière organique, le calcium
Figure I.5: Modèle d’activation de la MAP3K DLK et mécanismes neuronaux dans lesquels elle est impliquée. La voie de DLK va, selon certaines circonstances, contrôler des réponses qui peuvent être contradictoires dans les neurones.
26 possède un rôle d’une grande importance dans la signalisation cellulaire. Effectivement, cet ion est grandement utilisé comme messager cellulaire, majoritairement via une modification de sa concentration intracellulaire. La concentration intracellulaire de calcium ([Ca2+]i) est hautement régulée via différents mécanismes. L’augmentation de la [Ca2+]i peut être causée par une entrée du calcium extracellulaire via des canaux calciques voltage dépendants ou le relâchement des stocks de calcium à partir du réticulum endoplasmique, des lysosomes ou des mitochondries. Une diminution de la [Ca2+]i sera causée par les mécanismes inverses, soit la séquestration du calcium dans une organelle ou l’expulsion à l’extérieur de la cellule (Burgoyne et Haynes, 2014). L’entrée de calcium aura des effets sur l’expression de gènes via différentes voies de signalisation.
Afin de transmettre un signal qui permettra la régulation de gènes, le calcium intracellulaire interagit avec une panoplie de protéines contenant des domaines de liaison au calcium. L’un de ces domaines, appelé C2 (Gerke et al., 2005) est présent dans la synaptotagmine I, une protéine des vésicules synaptiques possédant deux domaines C2 (les domaines C2A et C2B) dont la conformation en huit feuillets β permet la liaison de plusieurs ions calcium. Cette protéine permet la libération rapide des neurotransmetteurs suite à l’induction d’un signal bioélectrique au niveau des membranes présynaptiques (Fernández-Chacón et al., 2001; McCue et al., 2010). Les annexines sont aussi une famille de protéines pouvant lier le calcium. Reconnues pour leur implication dans les mécanismes d’inflammation et d’apoptose, ces protéines ont la particularité de posséder un site de liaison au calcium, dont la structure en forme de disque permet la liaison au calcium et l’activation de la protéine (Gerke et al., 2005). Enfin, la plus grande des familles de protéines calcium-dépendantes regroupe les protéines à main-EF, un motif hélice-boucle-hélice pouvant lier un ion calcium (Lewit-Bentley et Réty, 2000). La calmoduline, le membre le plus connu et le plus étudié de cette famille, est impliquée dans la régulation de plusieurs processus biologiques tels la mobilité cellulaire, l’exocytose, l’assemblage du cytosquelette et la modulation de la [Ca2+]i
27 (McCue et al., 2010). Les protéines à main EF peuvent être séparées en deux groupes principaux, soit celles dont le motif hélice-boucle-hélice présente quelques variations dans la structure et celles à main EF standard dont font partie la calmoduline, les calpaïnes et plusieurs autres protéines (Zhou et al., 2009; Domínguez et al., 2014).
4.1.
Calcium et neuronesLe calcium est un élément particulièrement important pour les cellules neuronales qui utilisent cet ion afin de produire un potentiel d’action de part et d’autre de l’axone et de libérer des neurotransmetteurs au niveau de la synapse (Augustine et al., 2003). Cependant, un changement de la concentration intracellulaire de calcium des neurones permet de réguler plusieurs autres processus biologiques. Notamment, il a été proposé que le calcium aide la connexion synaptique durant le développement de l’axone (Berglund et Augustine, 2008). Des recherches antérieures montrent des augmentations transitoires de calcium au niveau des cônes de croissance axonale permettant de diriger la croissance dans le sens contraire de la hausse vers le site synaptique (Gomez et al., 2001; Ghosh-Roy et al., 2010). Une étude similaire a pu relater un rôle équivalent au niveau des filopodes dendritiques (Lohmann et Bonhoeffer, 2008). À l’opposé, un rôle du calcium dans la dégénérescence est proposé suite à l’observation d’une plus faible concentration de calcium au niveau d’une incision de l’axone de neurones de rat dont la dégénérescence est grandement ralentie (Adalbert et al., 2012). Il a aussi été décrit que l’activation des calpaïnes suite à l’entrée de calcium causée par une incision de l’axone mène à la décomposition du cytosquelette, une étape du processus de dégénérescence wallérienne (Wang et al., 2012). Cette idée est soutenue par l’observation d’un ralentissement de la dégénérescence lorsqu’il y a utilisation de chélateur de calcium ou d’inhibiteur de calpaïnes lors de l’incision de l’axone (Wang et al., 2012;
28 Yang et al., 2013; Yildiz-Unal et al., 2015), suggérant que la calpaïne est un médiateur de l’activation de la dégénérescence par le calcium.
4.2.
CalpaïnesLes calpaïnes sont des sérine/thréonine protéases calcium-dépendantes de la famille des protéines qui lie le calcium avec un domaine à main EF (Wang et al., 2012; Domínguez et al., 2014). Bien qu’il existe quinze types de calpaïnes, il y a deux isoformes qui sont grandement répandues chez les mammifères, la μ-calpaïne (calpaïne I) et la m-calpaïne (calpaïne-II). Les deux isoformes sont composées de deux sous-unités, soit une de 80kDa, qui est elle-même composée de quatre domaines, et une de 28 kDa, qui est composée de deux domaines. La différence entre les deux isoformes réside dans la concentration nécessaire de calcium intracellulaire pour les activer. Les calpaïnes sont localisées dans le cytoplasme sous une forme inactive, attendant une hausse de concentration de calcium (Camins et al., 2006).
29 L’implication des calpaïnes dans le mécanisme de dégénérescence wallérienne a été démontrée à plusieurs reprises. En effet, l’inhibition de la traduction des calpaïnes de C. elegans, soit CLP-1 et TRA-3, a permis de ralentir la dégénérescence induite chez le vers Figure 6. Schéma illustrant l’impact des calpaïnes sur l’activité apoptotique des
neurones en réponse à différents stimuli. Le rôle de la voie de signalisation des calpaïnes dans le mécanisme de neurodégénérescence est confirmé, mais nécessite encore d’être complété.
30 (Syntichaki et al., 2002). Parallèlement, l’inhibition des calpaïnes chez la souris, grâce à la surexpression de son inhibiteur naturel, la calpastatine, a permis d’observer une protection contre la dégénérescence wallérienne causée par l’incision de l’axone (Ma et al., 2013; Yang et al., 2013), suggérant une importance des calpaïnes dans le processus de neurodégénérescence. Parallèlement, des recherches effectuées sur des neurones de mollusque du genre aplysia ont montré grâce à l’utilisation d’un inhibiteur des calpaïnes, la calpeptine, que l’inhibition des calpaïnes empêche la formation du cône de croissance après axotomie (Gitler et Spira, 2002). Ceci suggère que les calpaïnes possèdent un rôle important dans la régénérescence axonale, en contraste avec son rôle dans le mécanisme de dégénérescence.
5. Hypothèse et objectifs du projet de recherche
Le laboratoire du professeur Blouin s’intéresse, depuis ses débuts, aux fonctions et aux mécanismes de régulation de la protéine DLK, que ce soit au niveau des neurones, des adipocytes, des kératinocytes, des cellules cancéreuses ou des fibroblastes indifférenciés. Comme mentionné plus haut, DLK est une kinase impliquée dans divers processus biologiques importants, notamment au niveau des cellules nerveuses où son expression est considérable. Son importance dans le mécanisme de dégénérescence des neurones en fait une cible intéressante pour des études visant la compréhension moléculaire des maladies neurodégénératives. Sur la base des observations récentes démontrant que le calcium est un régulateur reconnu des processus de régénérescence et de dégénérescence (Wang et al., 2012), et que la protéine DLK est un élément clé de la voie de signalisation du calcium dans les neurones (Yan et Jin, 2012; Ghosh-Roy et al., 2010), l’hypothèse générale de ce mémoire est que l’augmentation de la concentration de calcium intracellulaire va exercer un
31 effet sur l’expression, l’activité et la fonction de DLK. Afin de tester cette hypothèse, les cellules de la lignée neuroblastique humaine SH-SY5Y ont été utilisées. Cette lignée est considérée comme un bon modèle neuronal, car suite à sa différenciation avec de l’acide rétinoïque, elle possède des caractéristiques similaires aux cellules nerveuses (da Rocha et al., 2015). Avec l’aide d’un ionophore de calcium, le A23187, il a été possible d’examiner les conséquences de l’augmentation de la concentration intracellulaire de calcium dans les cellules SH-SY5 sur a) l’expression, b) l’activité et c) la fonction de la protéine DLK. Les résultats de ces travaux, présentés sous la forme d’un manuscrit dans la section suivante, indiquent que le calcium régule de façon négative les niveaux de DLK dans la cellule en favorisant sa dégradation de manière calpaïne-dépendante.
32 CHAPITRE II
DÉVELOPPEMENT
La dual leucine zipper kinase (DLK) est une cible protéolytique des calpaïnes dans les cellules nerveuses humaines
Les neurones sont des cellules polarisées conduisant un signal bioélectrique sur de considérables distances. En réponse à certains dommages, elles entrent dans un processus d’autodestruction, menant à la dégénérescence axonale et la mort cellulaire. Plusieurs recherches visant à élucider les mécanismes moléculaires de la neurodégénérescence ont identifié un membre de la famille des mixed-lineage kinase (MLK), la Dual Leucine Zipper Kinase (DLK), comme un médiateur essentiel de ce processus. Cependant, comment et quand DLK répond au signal d’un dommage à l’axone est une importante question qui doit être posée. Des observations montrant une hausse rapide de la concentration intracellulaire de Ca2+ en réponse à des dommages axonaux nous ont menés à tester une hypothèse proposant que cet ion régule l’expression et l’activité de DLK. En utilisant la lignée cellulaire neuroblastique humaine, les SH-SY5Y comme modèle, des traitements avec l’ionophore de Ca2+, le A23187, ont montré une baisse du niveau de DLK au niveau protéique par la méthode d’immunobuvardage de type western. La baisse de l’expression de DLK causé par le A23187 fut inhibée par des prétraitements avec soit des chélateurs de Ca2+, ainsi que l’inhibiteur de calpaïne, l’ALLN, suggérant un rôle pour la calpaïne, une protéase dépendante du Ca2+. En support avec cette idée, nous avons montré que DLK est co-localisé avec les calpaïnes dans les cellules SH-SY5Y et que DLK est une cible de
33 l’activité des calpaïnes in vitro. Finalement, une analyse par spectrométrie de masse sur les produits de dégradation de DLK par les calpaïnes a révélé des sites de clivage possible des calpaïnes sur la protéine DLK entre les résidus 18 et 393. Pris ensemble, ces résultats identifient le Ca2+ comme un régulateur de l’expression de DLK dans les neurones humains par un mécanisme impliquant une lyse par les calpaïnes.
Cette section du mémoire est présentée sous forme de manuscrit. Comme premier auteur, j’ai réalisé la majorité des expériences, sous la supervision du professeur Richard Blouin. Daniel Garneau, coordonnateur en instrumentation, a réalisé l’analyse des résultats d’immunocytochimie alors que l’analyse de spectrométrie de masse des produits de dégradation de DLK fut effectuée par M. Denis Faubert, de l’Institut de Recherches Cliniques de Montréal. Ce manuscrit sera soumis pour publication dans les meilleurs délais.
34 Calcium influx triggers calpain-dependent proteolysis of
DLK in human neuronal cells
Mathieu Dalpé-Mainville and Richard Blouin
Département de biologie, Faculté des sciences, Université de Sherbrooke, Sherbrooke, Quebec, JIK 2R1, Canada
To whom correspondence should be addressed: Richard Blouin, Département de biologie, Faculté des sciences, Université de Sherbrooke, 2500 Boul. de l’Université, Sherbrooke, Québec, Canada J1K 2R1
Running head: DLK undergoes calpain-mediated cleavage in response to Ca2+ influx
Key words: DLK, Ca2+, calpain, neurodegeneration
35 ABSTRACT
It is believed that DLK acts as a sensor of injury in neurons to mediate neurodegeneration and axon regeneration. However, how DLK responds to neuronal insults remains enigmatic. Here, we show that DLK level is down-regulated in SH-SY5Y neurons by A23187, an ionophore that increases intracellular calcium (Ca2+) concentration. A23187’s effect on DLK was prevented either by Ca2+ chelators or a calpain inhibitor, suggesting that reduction of DLK abundance is calpain-dependent. This prediction is supported by the findings that DLK co-localizes with calpain in cells and is cleaved in vitro by purified calpain, mainly in the N-terminal region. Collectively, these data identify Ca2+ as a regulator of DLK protein levels in human neuronal cells by a mechanism involving calpain-mediated proteolysis.
36 INTRODUCTION
Neurons are highly specialized cells that transmit electrical and chemical signals to other neurons or output cells, such as muscle. In response to diverse insults, they undergo a self-destruction process that results in axon degeneration and cell death (wang et al., 2012). Apart from its role during development, axon degeneration is central to the pathology of many traumatic and neurodegenerative disorder (Lingor et al., 2012). Another major event occurring after nerve injury or in parallel in many invertebrate neurons and in a number of vertebrate neurons of the peripheral nervous system (PNS) is the induction of axon regeneration (Liu et al., 2011; Bradke et al., 2012). The ability of a neuron to undergo regeneration after injury highly depends on its growth capability and the local extracellular environment (Liu et al., 2011; Bradke et al., 2012). Although the morphological and temporal modalities of axon degeneration and regeneration have been well described, little is known about the molecular pathways that regulate theses processes.
Interestingly, recent findings have brought attention to a conserved protein kinase, designated DLK, as a candidate regulator of axonal damage signaling. Support for this idea is provided by the observations that expression of DLK, which localizes to axons, growth cones and synapses (Mata et al., 1996; Hira et al., 2005; Eto et al., 2010), is up-regulated in response to neuronal stress (Xiong et al., 2012; Welsbie et al., 2013; Watkins et al., 2013). Loss of DLK has been shown to attenuate apoptosis in neurons exposed to multiple insults (Chen et al., 2008; Ghosh et al., 2011; Pozniak et al., 2013; Welsbie et al., 2013; Watkins et al., 2013) and to protect distal axons from Wallerian degeneration Miller et al., 2009). Additionally, the absence of DLK in both C. elegans and mice impairs regeneration of proximal axons after axotomy (Hammarlund et al., 2009; Watkins et al., 2013) and
37 abrogates retrograde transport of injury signals, a mechanism potentially contributing to the regenerative response (Shin et al., 2012). Thus, DLK functions in both the degeneration and regeneration of neurons in response to axonal injury, most probably by controlling a variety of downstream effectors. To date, the mechanisms by which DLK is activated and regulated in neurons after axon injury have not been clearly defined, but current evidence suggests the involvement of phosphorylation (Huntwork-Rodriguez et al., 2013; Hao et al., 2016), interaction with the scaffolding protein JIP3 (Ghosh et al., 2011) and ubiquitin-mediated degradation. (Collins et al., 2006; Xiong et al., 2010; Yan & Jin, 2012) Recent studies have also established that DLK-1, the C. elegans homolog of DLK, is regulated by a Ca2+ -dependent mechanism that switches DLK-1 activity from off to on state in neurons following dissociation of an inhibitory shorter isoform (Yan & Jin, 2012).
Ca2+ is a very important intracellular messenger in the brain, being essential for neuronal development, synaptic transmission and plasticity, memory formation, and the regulation of various metabolic pathways (Kawamoto et al., 2012). Deregulation of Ca2+ signaling has been implicated in the pathogenesis of many neurodegenerative disorders, including Alzheimer’s, Parkinson’s and Huntington’s diseases (Marambaud et al., 2009; Bezprozvanny, 2010; Woods & Padmanabhan, 2012). Studies in various experimental models have shown that one of the earliest responses to axon injury in neurons is a large increase in intracellular Ca2+ (Ziv & Spira, 1993; Wang et al., 2012). This Ca2+ likely comes from a combination of sources, including the damaged membrane, activation of voltage-gated Ca2+ channels, and release of Ca2+ from intracellular stores (Matthews & Dietrich, 2015). Ca2+ influx triggers activation of many different Ca2+-dependent elements, among which the Ca2+-dependent protease calpains are of particular interest in the context of neurodegeneration (Syntichaki et al., 2002; Coleman, 2005; Ma et al., 2013; Yang et al., 2013). Although calpains are known to play a role in axon maturation and maintenance during central nervous system development (Sedarous et al., 2003; Touma et al., 2007), their