Supervised learning to tune simulated annealing for in silico protein structure prediction
Texte intégral
Figure
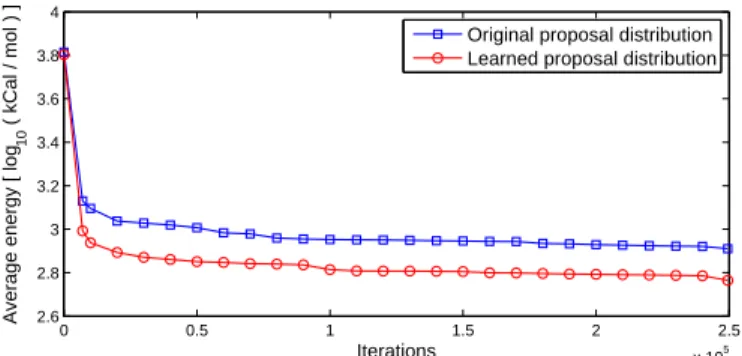
Documents relatifs
We analysed: (i) the risk of major cardiac events (MCEs) for each component of imaging test; (ii) the upper limit for each risk, in order to avoid underestimation of a risk;
Figure 10 shows the expectation value of the very lowest energy observed over the ensemble as a function of the ensemble size M. The total computer effort is calculated
The one-dimensional Ising spin glass is studied to exemplify the procedure and in this case the residual entropy is related to the number of one-spin flip
We show some theorems on strong or uniform strong ergodicity on a nonempty subset of state space at time 0 or at all times, some theorems on weak or strong ergodicity, and a theorem
As for strong ergodicity at time 0, if (P n ) n≥1 is weakly ergodic at time 0 and (ii) from Theorem 2.4 holds, then it does not follow that it is strongly ergodic at time 0..
Chocolate improves over Vanilla by using Iterated F-race instead of F-race as the optimization algorithm and is able to achieve significantly better results, indicating that
To sum up, there is no doubt that application the second fitness function gives us better results but if we compare Cuckoo Search Algorithm and Simulated Annealing it turns out that
A Simulated Annealing Algorithm for the Vehicle Routing Problem with Time Windows and Synchronization Constraints.. 7th International Conference, Learning and Intelligent
