DEPARTEMENT DE MICROBIOLOGIE
"PRODUCTION ET CHARACTERISATION DES ANTICORPS MQNOCLONAUX CONTRE"LE CYTOMEGALOVIRUS"
ÜL
THESE
PRESENTEE
A L'ECOLE DES GRADUES DE L'UNIVERSITE LAVAL
POUR L'OBTENTION
DU GRADE DE MAITRE ES SCIENCES (M.Sc. )
PAR
MOHAMED FAWAZ AL-HALABI
BACHELIER ES SCIENCES (ZOOLOGIE) DE L 'UNIVERSITE DE KUWAIT
TO MY PARENTS, WITHOUT WHOM I WOULD NOT HAVE BEEN ABLE TO COMPLETE MY STUDIES.
rra 9^ ni
-I-RESUME
L1 objectif de ce projet a été la production des anticorps monoclonaux dirigés contre le cytomegalovirus (CMV) pour fin d'utilisation potentielle en diagnostic et/ou l'utilisation comme outil pour etudier les aspects fondamentaux du CMV.
Des anticorps monoclonaux ont été obtenus par fusion de lymphocytes de souris (immunisées par quatre injections intrapéritonéales de 500 mg à intervale de deux semaines) et des cellules de mylome de souris (NSO) maintenues en culture exponentielle. Le dépistage des clones sécréteurs a été effectué par ELISA et immunofluorescence (IF). Les hybridomes intéressants ont été clonés par dilution limite puis congelés dans l'azote liquide et injectés par voie intrapéritonéale chez des souris (BALB/c),traitées avec une injection intrapéritonéale de 0.7 ml de pristane (2,4,10,14-Tetramethylpentadecane) dix jours auparavant, pour la production de liquide ascitique. Parmi
-II-les clones sélectionnés, un seul s'est révélé d'un grand intérêt. Le clone CLB-1 s'est révélé d'une grande reactivité et d'une grande spécificité contre le CMV.
Le liquide ascitique produit par les cellules monoclonales CLB-1 a été testé et les aspects suivants des réactions entre le CLB-1 et le CMV ont été vérifiés: (a) Reactivité et spécificité de l'anticorps contre le CMV. Pour étudier ce phénomène, l'anticorps monoclonal (CLB-1) a été testé par IF (en dilution de 1:100) contre des cellules fibroblastiques infectées par le CMV (AD169) ou par l'Herpès simplex type-1 ou type-2. Les résultats ont montré une réaction positive avec les cellules infectées par le CMV (AD169) et négative avec les cellules non-infectées ou infectées par l'Herpès sipmlex type-1 ou type-2. (b) La capacité de cet anticorps (CLB-1) pour la detection des isolées cliniques de CMV a été vérifié. L'anticorps monoclonal a détecté toutes les isolées cliniques trouvés positives au CMV. (c) Pour étudier la cinétique de la synthèse de la protéine contre lequel l'anticorps monoclonal est dirigé, le CLB-1 a été testé en IF avec des cellules fibroblastiques infectées avec le CMV à des temps différents après l'infection. Les résultats ont montré une réaction
-Ill-positive entre 72 et 96 heures après 11 infection prouvant ainsi que la protéine contre laquelle CLB-1 est dirigé est une protéine tardive. (d) La capacité neutralisante de 11 anticorps a été vérifiée et il a été prouvé que ce derniér neutralise le virus à une dilution de 1:100 comparativement au sérum humain ayant une capacité neutralisante à une dilution de 1:5. (e) Finalement le test d'immunodiffusion double sur gel d1 agarose a montré que 1'anticorps monoclonal CLB-1 est de type IgGl.
Plusieurs essais ont été effectués afin de déterminer le poids moléculaire de la (les) protéine(s) contre laquelle 11 anticorps CLB-1 est dirigé. Par immunoblotting, il nous a été impossible d1 identifier cette (ces) protéine (s). La raison probable est la destruction ou la modification de 11 épitope dans le processus de préparation de 11 antigene. Il a été mentionné que la majorité des anticorps monoclonaux ne précipitent pas les antigene contre lesquels ils sont dirigés (Carter and Ter Meulen, In advances in virus research. 1984, 29: 95-130). En conclusion nous avons produit un anticorps monoclonal contre le CMV, cet anticorps peut nous aider pour les diagnostic et probablement pour le traitement de 1'infection virale de CMV.
-IV-ABSTRACT
Fusions of spleen cells obtained from mice immunized with human cytomegalovirus (CMV) and a mouse myeloma cell line were performed.
One monoclonal antibody, CLB-1 was obtained from the four fusions performed. The following aspects of the reaction of the monocolona1 antibody CLB-1, with CMV were investigated: (a) The reactivity and specificity of CLB-1 with CMV; (b) the ability of the CLB-1 monoclonal antibody to detect clinical isolates of CMV; (c ) the kinetics of synthesis of the protein against which the CLB-1 monoclonal antibody reacts ; (d) the ability of the CLB-1 to neutralize CMV; and (e) some characteristics of this monoclonal antibody. The data obtained showed that the CLB-1 monoclonal antibody detected cells infected with CMV A D16 9 , the strain which was used as the immunizing antigen. The CLB-1 antibody showed a high degree cf specificity for the CMV as it did not react with the herpes simplex type-1, herpes simplex type-2 infected cells nor with uninfected human fibroblasts. From the kinetic experiments, the data obtained showed that this monoclonal antibody was directed against a late protein(s) that appeared
V
between 72h and 96h post-infection. The CLB-1 antibody neutralized the virus completely at a dilution of 1:100 of an acitis fluid preparation. Using the double immunodiffusion method, the CLB-1 antibody was found to belong to the IgGl sub-class of immunoglobulins .
-VI-fCKNOWLEDGEMENTS
I am extremely grateful to my thesis supervisor, Dr. Pravin Patel for his guiding, encouragement and sincere support throughout my M.Sc. studies.
To Dr. Robert Letarte, I express my deep appreciation fcr his acceptance to be the co-director of my thesis and for his cheerful guiding and useful discussions.
My deep gratitude to Dr. Gilles Richer and Jean Joncas fcr supplying us with the various clinical isolates used in our research.
I am thankful to Dr. Hans-Wolfgang Ackermann and Dr. Jean Joly for their helpful guiding and useful advices that, helped me throughout, my studies.
Special thanks to my dear friend Brian Lynn for his encouragements and assistance.
Special thanks to Johanne Ouellet, Louise Lizotte, Sylvie Berube, Nathalie Tremblay, and Louis Aubin for their constant help and cheerful encouragement.
I am very grateful to my parents for their financial and moral support and for their faith in me.
I am very grateful to Miss Omnyia M. Qabbara for her helpful assistance in revising this thesis.
-VII-TABLE_p_F_ CONIENI S
Résumé... ... I Abstract... IV Acknowledgements ... VI Table of contents ... VII List of tables ... X List of figures ... XI Diagnosis and clinical source of the viruses used in this
thesis... XII
Introduction ... 1
Isolation of the virus ... 4
The virus ... ... 4
The viral proteins .... ... 6
Immediate early proteins ... 6
CMV early proteins ... 7
CMV late proteins ... 8
Monoclonal antibodies ... 9
Materials and methods ... 17
1.0 Fibroblasts ... 17
1.1 Source of cells ... 17
1.2 Preparation and maintenance of fibroblasts ... 17
1.3 Freezing and thawing the fibroblasts ... 19
2.0 CMV antigen preparation ... 20
-VIII-10.0 Immunofluorescence ... 40
11.0 C'MV titration ... 42
12.0 Neutralization test ... 42
Results ... ... 44
1.0 Standardization of the ELISA test ... 44
2.0 Cell fusions ... 46
3.0 Neutralization of the CMV AD169 by the monoclonal antibody CLB-1 ... 34
4.0 Reactivity of monoclonal antibody CLB-1 with CMV infected cells at different times post infection as revealed by 11F test ... 56
5.0 Reactivity of the monoclonal antibody CLB-1 with CMV clinical isolates and herpes simplex viruses ... 56
6.0 Immunoglobulin typing ... ... ... 61
7.0 Determination of the molecular weight of the viral polypeptide identified by the CLB-1 ... 61
Discussion ... ... 64
-IX-4.0 Enzyme linked immune sorbent, assay (ELISA) ... 23
5.0 Monoclonal antibody production ... 24
5.1 Immunization of mice with CMV antigen ... 25
5.2 Preparation of spleen cells for the fusion ... 25
5.3 Mouse myeloma cells ... 26
5.4 Preparation of 96-well plates containing feeder layers . 27 5.5 Preparation of poly-ethylene-glycol (PEG) ... 27
5.6 Fusion of cells ... 27
5.7 Freezing of hybridomas ... 30
5.8 Cloning ... 30
5.9 Ascetic fluid production ... 31
6.0 Sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE)... 32
6.1 Reagents for SDS-PAGE ... 32
6.2 Preparation of the gel ... 33
6.3 Preparation of the sample ... 34
7.0 Staining the gel ... 35
7.1 Coomasie blue staining ... 35
7.2 The silver staining ... 36
8.0 Western blotting ... 37
8.1 Amido black staining ... 38
8.2 Enzyme Immunoassay for Detecting Proteins on Western Blots ... 38 9.0 Determination of immunoglobulin sub-class 39
-X-Table 1 : Table 2 : Table 3 : Table 4 : Table 5 LIST OF TABLES
Immunization of mice with CMV-infected cell lysate
CMV-specific hybridoma from different fusions performed ... CMV-specific clones derived from M-l fusion ... CMV-specific clones derived from M-5
fusion ... Reactivity arid specificity of ascetic fluids in a CMV-specifie 11F test ....
43
47
49
5 2
-XI-L 1ST OF FIGURES
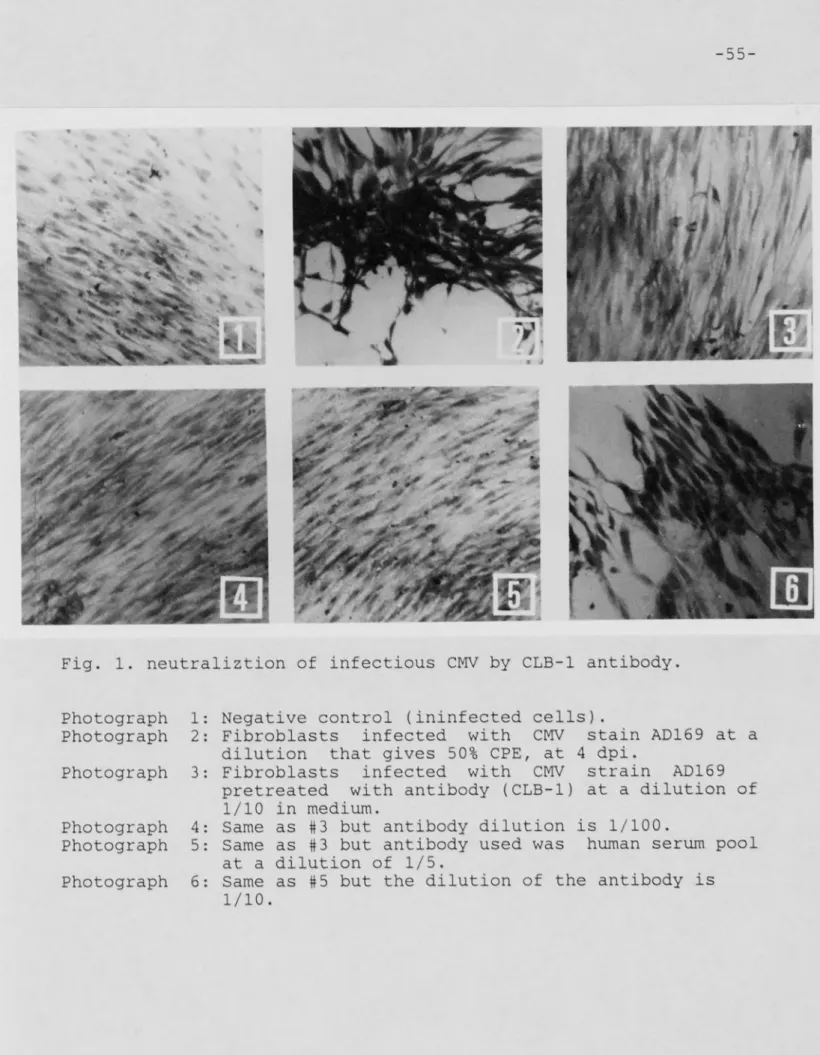
Figure 1: Neutralization of CHV by CLB-1 antibody ... 55
Figure 2: Reaction of CLB-1 antibody with cells
infected with CMV strain AD169 at different
times post infection ... 57
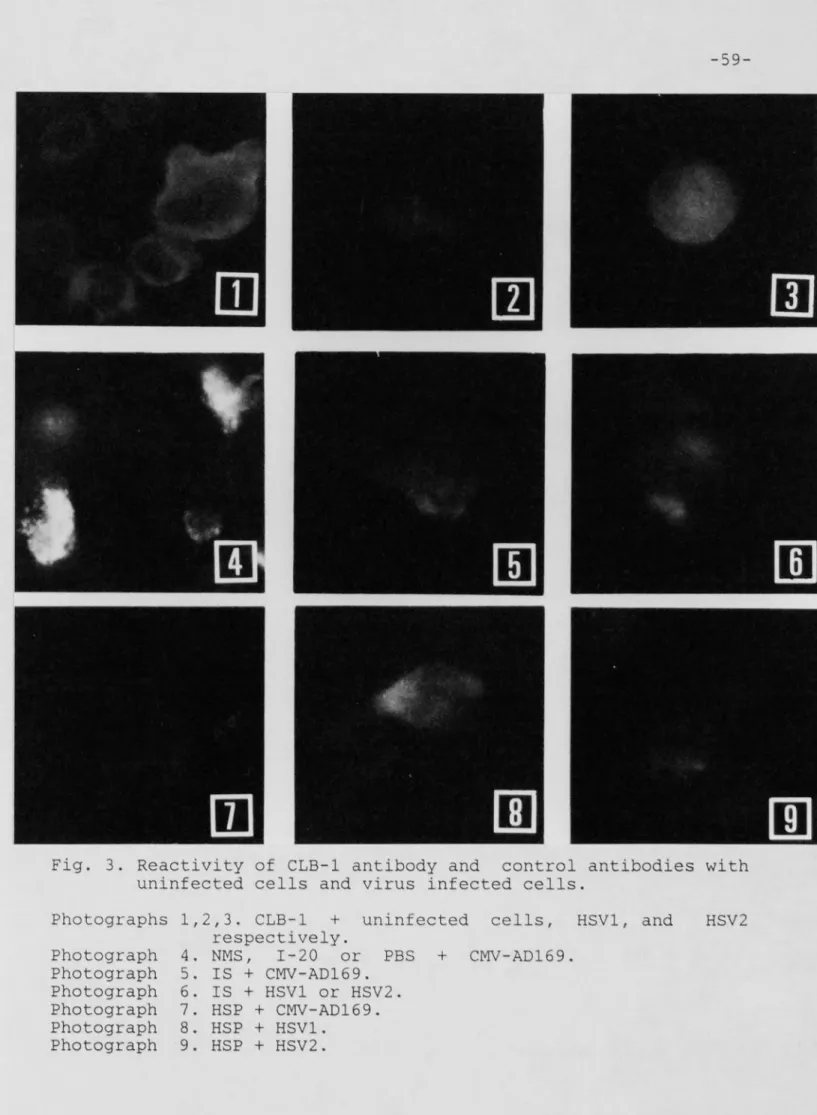
Figure 3: Reaction of CLB-1 antibody and control antibodies with CMV-AD169, HSU-1, HSU-2
infected cells, and uninfected cells ... 59
Figure 4: Reaction of monoclonal antibody CLB-1
with CMU clinical isolates ... 60
-XII-DIAGNOSIS AND CLINICAL SOURCE OF VIRUSES USED IN THIS THESIS:
Isolate Diaqnosis Clinical sample
2467 (Wise) Jaundice urine
5562 Pregnancy urine
8969 (Beaudet) Pericarditis, Pneumopathy
urine
2464 Congenital infection urine
-1-IN1ADDUCTION
Cytomegaloviruses (CMV) comprise a group of agents within the herpesvirus family known for their widespread distribution in humans and in numerous other mammals ( Reynolds et al. 1979 ). One of the important characteristics of the CMV infection is the ability to cause chronic infections of fetus, newborn, and young infants, leading, in some cases, to onset sequelae which is (Stagno et al. 1984). This virus is important as it causes severe congenital anormalities in about 10,000 infants per year in the U.S.A.. CMV can be found in the cervix of up to 10% of health)' women. Thus, it is important to expand our knowledge regarding the basic biology, diagnostic methods as well as treatment of infections caused by this virus. Inspite of the great structural and biological similarities in this group of viruses, it does possess a strong species specificity . This group of viruses can only be permissive in host cells of homologous origin, except simian cytomegalovirus, which can be grown in human fibroblasts and probably causes disease in humans (Huang and Huong, 1960).
-2-Cytomegalovirus can cause severe infection of the fetus. The reasons why some infants are severely affected while others remain symptom-free are not fully understood. Several reasons may contribute to* the severity of the infection, such as the type of maternal infection during pregnancy (primary or recurrent infection) and the fetal age at the time of the transmission of the virus from mother to fetus in utero. Genetic factors that control the mothers immune responses may also be important. The differences in the virulence of the viral strains and the possibility of immunologically mediated injury may operate in utero. Severe infection of the fetus in utero may cause the death of the fetus (Meyers, 19 84) . The exact sequence of events attending intra-utrine transmission remains unclear,but blood-born spread to the placenta and ultimately to the fetus is generally accepted (Reynolds et. al. 1979).
In the postnatal period, infection as documented serologically becomes increasingly pre velant with advancing age. The CMV infection is inapparent at birth in about 9 5% of the cases, even though the newborn bears a congenital infection. In diseased infants, death is uncommon, but may develop one or more of the following clinical symptoms: jaundice with hepatcsplenomegaly, thrombocytopenic purpura, spiky fever,
leukopenia, arthralgia, allograft dysfunction, intra-uterine growth retardation, pneumonitis, microcephaly with or without periventricular calcification, chcrioretinitis, optic atrophy, inguinal hernias in males, branchial arch abnormalities, psychomotor retardation and sensorineural hearing loss (Reynolds et. al. 1979, Linhares et al. 1981).
In the normal host and especially in children, infection with CMV is often asymptomatic, although it is the cause of a typical mononucleosis syndrome during adolescence and adulthood (Klemol a and Kaariainen, 1965). The situation is quite different in the immunocompromised host, as severe disseminated CMV disease, including pneumonia, often develops after kidney allografting. Additionally, pneumonia has been a frequent manifestation of CMV infection following bone marrow allografting (Meyers et al. 1983). Both the primary manifestation of CMV infection and the superinfection increases the morbidity and mortality arid decreases the success of organ allografting. An incidence of CMV infection of 50-100°i has been described for renal (Peterson et. al. 1980), cardiac (Pollard et al. 1982), and marrow transplant patients (Meyers et al. 1980). The infection in many of these patients is completely asymptomatic even though viral excretion may be detectable for years after transplantation
4
-Isolation of Human CMV : By the year 1950, the viral etiology of "Salivary gland virus disease" of rodents was demonstrated by passage of filtrates in animals; the virus was host-specific. In humans, morphologic evidence of infection was the presence of enlarged, inclusion-containing cells in the salivary gland of infants. Propagation of this virus was not possible un till the cell culture system was achieved in about 1953. Smith has finally succeeded in cultivating the virus isolated from parotid tissues in cell culture (Smith ; 1957). The strain Davis was isolated from liver biopsy, from a child that, was admitted to a hospital in Boston for having a Toxoplasma infection (Weller et al. 1957). This was the first time that the virus was isolated from a living patient. Two other strains, Kerr and Esp were isolated from the urine of infants (Weller et al. 1957). In 1953 the strain AD169 was isolated by Rowe and co-workers from human adenoid tissue
(Rowe et al. 1953).
The Virus: CMV is morphologically indistinguishable from Herpes simplex or Varicella zoster cr any other herpes virus. The virion consists of a 64 rim core enclosed by a 110 nm icosahedral capsid with 162 capsomeres. The complete particles are surrounded by a double membrane envelope which measures about 180 nm in diameter.
The genome of the CMV is a linear double stranded DNA. The size of the genome is about 150 to 160 million daltons or 230 kb pair. The structural proteins of CMV are produced in abundance late in infection during assembly of progeny virions. As many 30 polypeptides have been associated with CMV virion; a number cf these are glycosylated and comprise the virion envelope (F ia 1 a et al. 1976; Stinski 19 7 6) . Glycoproteins of CMV are inserted into membranes of infected cells and share antigenic determinants with those in the in the virion envelope (Kanich et al. 1972; Spear 1976 ; and Stinski et al. 1979).
The inactivation of the virus can be accomplished by exposing it to 20% ether for 2 hours , to acidic pH less than 5.0, to a temperature of 56° C for 30 minutes, or UV light for 5 minutes. The virus is more stable in media containing 5 to 10% Fetal Bovine Serum (FES) at 3 7 ° C but not at 4° C (Vonka and Benyesh-Melnich, 1966a). Infectivity is preserved by storage of the virus at -90°C with 3% sorbitol (Vonka and Benyesh-Melnich, 1966b) or in liquid nitrogen.
The Viral Proteins : After the entrance of the CMV into the permissive host cell, the virus starts its replicative cycle which is characterized by the appearance of viral proteins in the host cell . Different types of viral proteins start to accumulate in the infected cell at different times post-infection. According to time at which t; h e s e proteins start to appear in the infected cells, they are named immediate early, early, or late proteins.
Immediate Early Proteins: By definition, the immediate early proteins are those viral specific proteins that can be detected in the host cells whithin one hour post-infection, and are transcribed in the absence of prior viral protein synthesis (Stinski, 1984). Obviously, the importance of these proteins lies in the regulation of subsequent viral genome expression, therefore hypothetically, if these proteins are inhibited or inactivated, the whole virus replication cycle can be aborted. The immediate early proteins are expressed from a region between 0.660 to 0.770 map units for Towns strain (Stinski et. al; 1983). The immediate early mRNA originating from these map units remains associated with the nucleus (De Marchi, 1983).
-7-One of the major immediate early proteins is the 72k protein which is detectable within the first, hour post infection (Wat.hen et. al. 1981). However in ether reports , the 6BK immediate early protein was found to be the major protien for the Towne (Cameron and Preston, 1981) , Davis and ADI 69 (Li. n h ares et al. 1981) strains. Other immediate early proteins with molecular weights 14, 31, 35, 47, 58, 60 62, 70, 71, 73, 77, 78, and 85Kd have been reported (Cameron and peterson, 1981; Blanton and Tevethia, 1981). The importance of studying the immediate early proteins is that they may be useful in the early diagnosis of CMV infection.
CMV Eajly Proteins: The early proteins are those viral proteins that are synthesized after the immediate early proteins, but before virai. DNA replication. Human cytomegalovirus has a prolonged phase of early viral gene expression, since it starts four to six hours post infection, and stays unchanged for 24 hours, (i.e. the profile of virai. mRNA and proteins remain the same) . These viral early mRNAs differ in size and in translation from the immediate early mRNAs (Wathen et al. 1981). The early mRN A is transcribed from genes which reside ir. the the large repeated and adjacent sequences in the large region of the viral genome (Stinski, 1984).
-8-Antibodies produced sgainst the early proteins in patients have been tested for their potential use in diagnosis of CMV infections. In the infected neonates, the antibody titer against the early proteins are measurable in cases where the mothers are seropositive, but it usually declines after- birth (Stagno et al, 1975). In the perinatally infected infants, the antibody titer rises diagnostically after the initial fall as measured by immunofluorescence, while the congenitally infected infants do not have an initial fall in the antibody titer against the early proteins, but remains elevated all the time. Although the early proteins appear from 4 to 6 hours post infection, the presence of antibodies against them does not necessarily mean a current infection. The antibodies against the early proteins can remain elevated for as long as 250 days after a proven CMV mononucleosis in a normal patient (Ten Nap el and The , 1980).
CMC Late Proteins: The late proteins are those viral proteins that are synthesized after viral ONA replicaltion. Newly synthesized viral ONA accumulates in the infected cell slowly. At a multiplicity of infection (MOI) of 10 to 20 PF'U per cell, newly synthesized viral DNA is detectable by 12 to 15 hours post-infection, and reaches its maximum level at 72 to 96 hours post-infection (Stinski; 1978). The relative amounts of
-9-accumulated late viral proteins correlate directly with the viral DNA synthesis (Stinski ; 1978) and reache their highest rate of synthesis in about 24 hours post infection and continue at a constant high rate for many days after the infection. The rate of synthesis of the viral specific proteins at this time reaches about 50% to 60% of the total protein synthesis in the host cel] is viral (Stinski; 1977). One of the major viral proteins is the polypeptide VP68, which constitutes about 15% of the proteins associated with the purified virions and dense bodies.(Stinski ; 1984). Other viral proteins such as the VP155, VP120 and VP 83 constitute a significant percentage of the virions and dense bodies but in though concentrations.
The viral envelope is estimated to have at least eight glycoproteins. Their approximate molecular weights are around 145, 132, 120, 115, 90, 70, 64, 55, and 12kd as determined by 505-PAGE. The neutralizing glycoprotein(s) may be one or more of these proteins.
Mo no c 1 ona 1 ant ibodi e_s j_
Since their initial description by Kohler and Milstein in 1975, the monoclonal antibodies have contributed to many advances
10-in the area of biological research. The basic property of a monoclonal antibody is that it combines specifically with one epitope or a family of related epitopes, and that it can provide a wide spectrum of data concerning relatedness, structure, function, synthesis, processing, and intracellular or tissue distribution of a single protein.
Monoclonal antibodies have been applied to several areas of virology such as virus identification where it has been used for diagnosis, taxonomy and epidemiology. Monoclonal antibodies produced against the varicella zoster virus have been successfully used to identify the virus in cells obtained from skin lesions (Forghani. et al. 1982). Monoclonal antibodies have also been used for typing the Epstein-Barr virus (Mueller-Lantzsch et. al, 1981) and Herpes simplex viruses (Richman et. al.1982). The application of the monoclonal antibodies in Hepatitis B virus radioimmunoassay test were found to detect surface antigen in patients who were negative by the conventional testing (Wands et al. 1982). Also four serotypes of the Dengue virus can be distiguished by monoclonal antibodies which were otherwise difficult to distinguish using conventional sera. (Gentry et. al. 1982).
-
11-Monoclonal antibodies have also been used in epidemiological studies. Many workers have compared field isolates using monoclonal antibodies directed against the virus glycoproteins and the nucleocapsid proteins. In this way differences have been detected between viruses that were considered identical. The vaccin strains of the yellow fever virus derived from different laboratories have been tested to confirm the required similarities between these vaccines. Similar comparative studies have been done between the Saukett and the Sabin vaccines of polio virus (Monath et. al. 1983; Ferguson et al. 19 8 2 ; a n d Schlesinger et. al. 1983)
Monoclonal antibodies have also been used in identifying the functional properties of viral proteins. For example, the glycoprotein CP-1 of the corona viru s MHV was shown to be responsible for cell attachement and virus spread through cell/cell fusion (Collins et a 1 . 1982) . A modified immunoprécipitation experiment has demonstrated that a subset of the SV4Q-T antigen present in the infected cell is able to bind to the initiation site of virus DMA replication (Scheller et al. 1982). Monoclonal antibodies have been used for identifying the functional epitope or antigenic domain on a protein molecule. By means of function inhibition analysis, a panel of monoclonal
12-antibodies specific for measles virus hemaglutinin was divided into five groups: the first group comprised of antibodies which had no detectable activities in hemagglutination inhi.biton (HI) or neutralization (NT) tests , the second group was that which had HI activity but no NT activity, the third group with equivalent activities in both tests, while the fourth group had a higher HI activity than NT activity and the fifth group was the reverse. Thus a panel of monoclonal antibodies against one type of protein can be used to differentiate between neutralizing and hem agglutinating proteins. (Ter Meulen et. al. 1981).
Once epitopes are identified, it is useful to determine their topographical relationship to each other cn the surface of the molecule. The competitive binding assay has been applied successfully to many viruses (Carter and ter Meulen, 1984) . Monoclonal antibodies specific for the vesicular stomatitis virus (VSV ) G protein gave a detailed antigenic map including the epitopes intstrumentai in virus neutralization and cross-reacting and specific determinants for the strains Indiana or New Jersey (Volk et al. 1982).
Monoclonal antibodies have been useful in studying the biosynthesis and maturation of viral proteins. The maturation of
-13-the early proteins of -13-the HSV-1 was studied by Misra et al. (1982). These workers found that tunicamycin-sensitive glycosylation was necessary for the cell surface expression of these proteins. Glycosylation was not required for insertion into the rough endoplasmic reticulum and intracellular transport of the protein to the plasma membrane. This detailed study also elucidated intermediates formed in the protein modification pathway leading to the mature molecule. A comprehensive immunofluorescence study using monoclonal antobodies has examined the distribution of a number of Moloney murine leukemia protein on the infected cell membrane (Satake et. al., 1981; Satake and Luftig, 1983). In addition the same workers were able to demonstrate by double immunofluorescence that two virus-specific antigens, P15 and P15E, accumulated at the same sites.
Monoclonal antibodies could distinguish readily between native and unfolded forms of the Sindbis virus glycoprotein El ( Rcehri ng et. al.1982) and between native (N) and heated (H) fcrms of the poliovirus capsid (Brioen et. al. 182). More biological information concerning virus assembly has been derived from studies of the poliovirus. Two monoclonal. antibodies were obtained which bound to poliovirion precursor structure. The sites recognized were different, one antibody bound to infectious virions, 80 5 empty capsids in the N form and 14 S precursor subunits, the other bound only to virions and empty capsids
-14-(Ferguson et al. 1981).
Many investigators have prepared monoclonal antibodies against CMV. Goldstein et al. (1982) have prepared monoclonal antibodies that detect an early protein and another that specifically detects a J ate protein. The former antibody showed that the early protein remains localized in the nucleus and has a molecular weight of 72,000 daltons. The later detected a 80,000 dalton protein that remains localized in the cytoplasmic inclusion bodies. Both monoclonal antibodies reacted with laboratory strains of CMV as well as clinical isolates when tested by the indirect immunofluorescence (11F ) test.
Pereira et. al. (1982) have developed anti-CMV monoclonal s, these antibodies were able to detect, the CMV- infected cells by the I IF test, and have been grouped into seven catigories according to their reactivity with the surface proteins of CMV, their ability to precipitate different molecular weight proteins, and their neutralizing activities. Some of these monoclonal antibodies did indeed neutralize the CMV but failed to precipitate the protein against which they were directed. Other types of monoclonal antibodies neutralized the virus and precipitated the proteins against which they were directed. Most of these monoclonais detected both laboratory and clinical isolates of CMV.
-15-Rasmussen e t al (1984) using anti-CMV mcmoclonals, have shown that there are antigen! c similarities between the laboratory strain CMV AD169 and many clinical isolates, as the monoclonal antibody prepared against the CMV ADI69 neutralized not only the CMV ADI69 strain but also the other laboratory strains (Davis and Towne). This monoclonal antibody also neutralized all the clinical isolates tested. The protein against which this antibody was directed has a molecular weight of 86,000 daltons and is localized in the cytoplasm of the infected cells.
Thus monoclonal antibodies in virological research allows us to view the fate of a protein in the infected cell, from synthesis tc virus assembly, in a continueous process with interactions between virus specific gene products and the cellular machinery. Moreover, the use monoclonal antibodies provides a valuable insight into the working of the protein both as an enzyme and as a target for the host immune response. The application of monoclonal antibodies in virology has not only improved information obtained from well established assay procedures, but also has led to processes yielding an entirely new type of information.
Use of monoclonal antibodies for theraputic purposes is still experimental. It is likely that the antibodies would be used in three main areas. In emergency, injection of a
— 16 —
neutralizing antibody can help recovery from disease. Human monoclonal antibodies are now becomings available and are likely to produce fewer problems with allergic reaction. Animal experiments in the mouse have indicated that neutralizing monoclonal antibodies are able to localise HSV infection and prevent spread to the nervous system, promot recovery from ocular infection or protect against a later footpad challenge (Carter et al 1984).
The objectives of the present thesis project were to raise monoclonal antibodies against human CMV and to study their potential use in diagnosis and application as tools in studying the fundamental aspects of the CMV. As mentioned previously, the use of the neutralizing monoclonal antibodies as a theraputic tool against virus infection may prove the importance of the monoclonal antibodies raised against the CMV as will be mentioned later in the discussion.
-17-MATERIALS AMD METHODS
1.0: Fibroblasts
1.1: Source of cells
Fibroblasts were either purchased (F5000, Flow Laboratories # 02-012-83) and passaged as shown later , or prepared in our laboratory from tissues collected from children undergoing circumcision at the Jewish General Hospital in Montréal, or at Le Centre Hospitalier de 1 Université Laval in Quebec City.
1.2: Preparation and maintenance of the fibroblasts
Tissues were sent to our laboratory under sterile conditions in 10 ml tubes containing MEM-M199 medium supplemented with 100 U/ml penicillin, 100 micrograms/ml streptomycin, and 10% inactivated (at 5 60 C for 30 minutes) fetal bovine serum (IFBS). MEM-199 is a composite medium made up of equal parts of Minimum Essential Medium (MEM; GIBC0, cat # 410-1100 ) and medium 199 (CISCO; cat.# 410-1900). Upon reception, the tissues were cut in small pieces (about 1 mm 2 ), washed twice in sterile fresh
-18
medium, and placed in 2 5 cm2 sterile tissue culture flasks with a distance of about 1 cm between each piece of tissue . Then , about 0.5 ml of fresh MEM-M199 was added, supplemented with 100 LI/ml penicillin, 100 ug/ml streptomycin, and 10% 1FBS. This medium will hereafter be refered to as MEM-10. Medium containing 3% ESS will be refered to as MEM-3. The flasks were incubated at 3 7 ° C in a humid 5% carbon dioxide incubator. These incubation conditions were used in all the experiments described henceforth. The next day, about 1 ml of fresh medium was added to the flasks making sure that, the pieces did net float in the medium. The medium was changed every 3 days. After 3 to 4 weeks, the fibroblasts emerged from the tissue pieces and became confluent in the flasks. At this time, the cells were ready for the first passage.
Passage of the fibroblasts was carried out as follows : The medium was discarded and the fibroblasts were washed 3 times with sterile PBS containing 0.05% EOT A (ethylenediaminetetrs acetic acid). This was followed bythe addition of 1 ml of 1% trypsin (Flow Labs # LS00-04454). The flasks were then incubated at 37° C for about 1 to 3 minutes. The flasks were then shaken gently to detach the fibroblasts and 10 ml of fresh medium was added to each flask to stop the action of the trypsin. The contents of each flask were transferred into a 10 ml. sterile centrifuge tubes, the tubes were left to stand for about 5 minutes to allow
-19-the tissue pieces to s e: 111 e to -19-the bottom of -19-the tubes. The supernatant, containing the fibroblasts was transferred into 75 cm culture flasks and incubated at 3 7 0 C. The medium was changed every 2 to 3 days until the fibroblasts monolayer became confluent. They were then passaged by trypsinization into 3 new flasks, each flask containing a final volume of 18 to 20 ml.
1.3: Freezing and Thawing the Fibroblasts
Confluent fibroblast monolayers were try p sin i. zed as described above for passaging cells. After stopping the trypsinization reaction, the content of one flask was transferred into a 10 ml sterile centrifuge tube. The cells were centrifuged at 250 X g for 10 minutes in a I EC benchtop centrifuge. After discarding the supernatant, the cell pellet was gently mixed and kept on ice for 10 minutes. Cold medium (1 ml) containing 15% MEM-10, 10% dimethylsulfoxide (DMS0; Fisher # D-128) and 75% FB5 (V : V ) was added. This suspension was then transferred, into s cold freezing vial (NUNC Cryotube, GIBC0 # 366-6 56) arid that was kept on ice and then stored at -80 °C. After 2 4 hours the vials were transferred to a liquid nitrogen freezer.
When needed, the frozen fibrobiasts were thawed b y plaçai the vials in a 37 °C water bath. The vial was thawed in such
-20-that. a small piece of ice was left in the vial. The vial was then immediately removed from the water bath, shaken to dissolve the remaining piece of ice and transferred to a 10 ml sterile centrifuge tube containing cold medium MEM-10. The cells were centrifuged at 250 X g for 10 minutes, the supernatant discarded, pellet mixed and 10 ml of fresh medium was added. The resulting cell suspension was transferred into a 75 cm2 culture flask and incubated under conditions described above. The medium was changed every 3 days until the cells became confluent. On confluency, the cells were passaged into 3 flasks as described above.
2.0: CMV Antigen Preparation
The antigens used in this study were either obtained commercially (Flow Laboratories # 40-613-44 for the CMV antigen AD169 strain, and # 41-613-44 for the control antigen), or prepared in our laboratory as described below : About 18 to 20 flasks (75 cm2) 0f confluent fibroblast monolayers were infected at a high multiplicity of infection (MOI) by adding about 2 ml of a virus preparation over the cells. The flasks were incubated at 37°C for one hour. The flasks were tilted gently every 15 min to insure an equal infection of the cells. At the end of the
-21-incubation period, the supernatant was discarded and about 15 ml of medium MEM-3 was added to each flask. The flasks were then incubated at 3 7 ° C for 5-7 days. To prepare control antigen, the media of another 18 to 20 flasks containing confluent monolayers of fibroblasts were replaced with medium MEM-3 anc the flasks were incubated under the same conditions. The medium was changed every 3 days. When the infected fibroblasts shewed 100% cytopathic effect (CPE) , usually 4 to 5 days post infection (dpi), the monolayers were washed twice with sterile PBS, and then covered with 2 ml of sterile PBS containing 0.05% EOT A (PBS-EDTA). The cells were scraped using a sterile cell scraper (Costar # 3010), pooled and centrifuged at 250 X g for 10 minutes. The cell pellet was then shaken gently and 3 ml of cold sterile PBS was added. The cells were then sonicated, keeping the tube on ice , for 1. minute at 60 Hz (using a Sonic Di smembr atcr ; FISHER ; // 15-338-40) . Large cell fragments were removed by centrifugation at 5 5 0 X g for 20 minutes. The supernatant containing the antigen preparation was removed, aliquots of 100 microliters were frozen at -800C .
3.0: Plotein Determina_t_ionj_
The Lowry method was used for the protein determination which required the following reagents :
-22-Reagent -A- 20% sodium carbonate (SIGMA # 5-2127) and 4% sodium hydroxide (SIGMA # S-55-81), dissolved in distilled water.
Reagent -B- 0.5% cupric sulfate (SIGMA # C-7631), 1% sodium tartrate (SIGMA # 5-0630) dissolved in distilled water.
Reagent -C- 25 ml of reagent (A) plus 0.5 ml of of reagent (B). This mixture should be freshly prepared just before use.
Reagent -D- 2.5ml of Folin phenol reagent 2 N (SIGMA # F -9 2 5 2 ) plus 2.5 ml of distled water.
To generate a standard curve , dilutions of bovine serum albumin (BSA; SIGMA # A-7638) in distilled water were made to give final concentration of 5 micrograms/200 microliters, 10 micrograms/200 microliters, 20 micro grams /200 microliters, 40 micrgrams/200 microliters and 50 micrograms/200 microliters. Distilled water was used as the blank control. Dilutions of the sample to be tested (200 micoliters each ) as 2.5%, 1.25%, and 0.5% were made in distilled water. To each of the dilutions, I. ml of reagent 0 was added while shaking. The tubes were left to stand for 10 minutes and then 100 microliters of reagent
-23-D was added while vcrtexing. The tubes were then left for 30 minutes at room temperature, and then read in the spectrophotometer at 300 nm (Beckman; model 25).
The optical density of each standard was plotted against the concentration of BSA and a standard curve was obtained from which the protein content of the unknown samples was determined.
4.0: Enzyme Linked Immunosorbent Assay (ELISA)
This test was carried out in 96 well plates (Flow it 76-381-04) using the technique described by Voiler et al.1976). The antigen to be coated was diluted to 10 nanograms/microliter in the coating buffer (0.159% sodium carbonate, SIGMA # S-2127, and 0.293% sodium bicarbonate, SIGMA. it 5-8875, pH 9.6). Fifty microliters of the antigen solution was added to each well, except the first. column (A1 to A8) to which only buffer was added. The plate was incubated at 4°C overnight, and then washed three times with PBS-Tween (PBS containing 0.05% Tween-20, SIGMA # P-1379). Fifty microliters of an appropriate dilution of the solution to be tested for the presence of the antibody was added to each well. The plate was incubated at 37°C for three hours, washed 3X for 10 minutes with PBS-Tween. Peroxidase labeled
-24-goat-antimouse IgG and IgM (FLOW # 55-814-00) was diluted to 1:500 in PBS-Tween and 50 microliters of this labeled antibody solution was added to each well, followed by another incubation of 2 hours at 37°C. The plate was washed 3X 10 minutes with PBS-Tween. 0PD (0-phenylenediamine dihydrochloride; SIGMA # P-1526) was used and a fresh solutionof 0PD was prepared by dissolving 10 mg of 0PD in 25 ml of phosphate-citrate buffer (24.355 0.1 N citric acid, BDH ; # 8-10081-34 ; 24.7% sodium phosphate monobasic, SIGMA # S-0786; and 50% distilled water , and 10 microlit.ers of hydrogen peroxide , Fisher # H-237 ) . fifty microliters of the 0PD substrate solution was added to esch well and the plate was then incubated for 30 minutes at 3 7 ° C . The enzyme reaction was stopped by adding 10 microliters/well of 2.5 M sulfuric acid (SIGMA # 680-2). The plate was read at 492 nm using an ELISA plate reader.
5.0: Moneclonal Antibodj Produetion
The general protocols used for monoclonal antibody production were adapted from those described by Galfr and Milstein (1980). The medium used for hybridoma production was Iscove’s medium (Flow # 10-357-22) spplemented with penicillin (100 ll/ml), streptomycin (100 ug/ml), 200 mM glutamine, 200 mM sodium pyruvaten and heat-inactivated FBS at various
-25
concentrations. Iscove's medium containing 0%, 10%, or 20% FBS will be: henceforth referred to as I -0 , 1-10 , and 1-20 respectively. If they contain additional substances such as HAT (hypoxanthine, aminopterin, thymidine) or HT, it will be designated as 1-20 + HAT or 1-10 + HT and so on.
5.1: Jromunizatiojn of Mice with CMV Antigen
Mice received an intraperitoneal injection with 500 ug of CMV antigen mixed 1:1 with Fruend's Complete Adjuvant (DIFC0 // 0638-59). The mice received 3 other injections (500 micrograms of CMV antigen mixed in Fruend's Incomplete Adjuvant) (DIFCC # 0639-59 ) at 2 weeks intervals. Fusions were done usually 7 weeks after the initial immunization with spleen cells obtained from mice showing a good anti-CMV titer as measured by the ELISA technique . On the fourth arid the third day before the fusion, mice received intravenous and intraperitoneal injections of 500 micrograms/mouse of CMV antigen mixed in sterile PBS.
5.2: Preparation of Spleen Cells fcr the Fusion
-26-The spleen was removed aseptica11 y and transferred into a petri dish containing about 2 ml of medium I-0. All the manipulations were carried out under a sterile hood. The spleen cells were removed from the spleen capsule by cutting the organ into small pieces using a sterile scalpel and forceps. The spleen cells usually ooze out into the medium. The cells were then transferred into 50 ml centrifuge tubes, and about 30 ml of medium was added to each tube. The tubes were then spun at 250 X g for 10 minutes. The supernatant was discarded. The cells were shaken gently and 10 ml of medium I-10 was added. One hundred microlit.ers of the cell suspension was added to 90 0 microliters of 1% of glacial acetic acid in distilled water (FISHER # A-38-S). The cells were then counted using the hematocytometer.
5.3: Mo u s_e My e 1 om a Ce lis
The mouse myeloma cell-line NSO was grown in vitro in 1-5 suppleminted with 5% FBS, 0.4 mM/ml Glutamine, 100 LI/ml penicillin, and 100 micro grams/ml of streptomycin. When the cells were in the log phase they were divided into 2 to 3 flasks to have enough cells for the fusion. On the day of the fusion the cells in the log phase were transferred to 50 ml centrifuge tubes and spun down at 250 X g for 10 minutes. The cells were resuspended in I sc eve without FBS and counted using a 1% trypan blue solution.
-27-5.4: Preparation of the 96-well Plates ContainingJeede_r_ _1 a)£rs
Sterile 96-wel 1 plates ( FLOW # 76-032-05 ) were prepared the day before the fusion by distributing 100 microliters of 5 X 10^ cells/ml of normal mouse spleen cells in each well. The normal mouse spleen cells were prepared in the same way as the immune mouse spleen cells and adjusted to 5 X 10 5 cells/ml in 1-20 -t- HAT. The plates were incubated at 3 7 °C in a 5%' CO ^ incubator.
5.5: Preparation o f _ Po_l_y-Ethylene-Glycol (PEG)
A 50% solution (PEG J 500 ; B.D.H. # B-295-75-34) was prepared by taking 6 grams of PEG in a glass tube and melting it in a boiling water bath. Six ml of warm Iscove (40 °C ) was added to the liquified PEG kept at 45°C and was filtered through a 0.22u filter (Millipore # SSWP-022500). The PEG-Iscove solution was kept in a water bath at 37 °C until used.
5.6: Fusion of Cells
-28-the myeloma cells were mixed toge-28-ther in a proportion of 3:1 and centrifuged at 250 X g for 10 minutes. The supernatant was discarded and then the cell pellet was shaken gently and washed three times with Iscove's medium by centrifugation at 250 X g for 10 minutes. After the third centrifugation, the supernatant was discarded, and the tube containing the cells was placed in a water bath at 37 °C for 1 minute. One ml of PEG-Iscove was then slowly added to the cell pellet, and kept at 37°C in the water bath for an additional one minute. Then another 1 ml of medium I-0 was added dropwise over a period of one minute while mixing the cells very gently with the tip of the pipette. Two ml of additional medium I-0 was added three times over a period of one minute each time, finally another 2 ml of I-0 were added and the volume of medium was then made up to 25 ml by adding 15 ml of medium 1-20. The cells were centrifuged at 250 X g for 10 minutes. The supernatant was discarded and the cell pellet was shaken gently (without vortex ) and then resuspended in 1-20 + HAT. The cell concentration was adjusted to 2 X 106 cell s/ml. One hundred microliters of the cell suspension was distributed in each well of 96-well plates containing feeder layers.
The plates were then incubated at 37°C. On the third and the sixth day post fusion, plates were fed with fresh 1-20 + HAT medium by removing about 100 microliters of spent medium from each well and adding fresh medium. Usually 10 days post
-29-infection, small clones would start to appear in the wells. The wells were considered ready for screening the synthesis of antibodies when the clones were large enough to be detected with the naked eye with a simultaneous change i.n the color of the medium (orange to yellow). The ELISA technique described above was used for screening the presence of anti-CMV antibodies. About 110 microliters of the supernatant. was gently taken from the wells to be tested using a sterile pipette tip and transferred into glass tubes. Fifty microliters of each supernatant was added to wells of an ELISA plates which were coated with CMV antigen and control antigen. The ELISA test was carried out as described above. The positive wells were transferred into the wells of a 24 well culture plate (FLOW laboratories // 76-033-05), which contained 0.5 ml of 1-20 + HT medium. The positive wells of the 96-well plate were fed with the same medium and maintained as duplicate cultures. The 24-well cultures were retested again for the presence of anti-CMV antibodies when the cells were 50% confluent. The cells in the positive wells were transferred to 25 cm2 flasks containing about 3 to 4 ml of 1-20 -t- HT. The wells of the 24-well plates were also maintained simultaneously. When the media in the flasks turned orange to yellowish, the cells were fed with another 5 ml of medium. When the medium turned orange to yellowish again, the flasks were retested for the anti-CMV antibodies using the ELISA test. Cells from positive flasks were frozen as described below.
-30-5.7: Freezing Hybridomas
The flasks were shaken gently tc suspend the cells in the medium. The cell suspension was transferred into sterile 15 ml culture tubes and centrifuged at 250 X g for 10 minutes. One ml of complete freezing medium (15% Isecve, 75% FBS and 10% DMSO) was added t.o the cell pellet and kept, on ice for 5-10 minute and then transferred into cold cryotubes. After freezing 2 to 3 vials of cells from each flask, the cells that were producing interesting antibodies were cloned.
5.8 Cloning
Fifty microlit. ers of normal mouse spleen cells at e concentration of 5 X 10^ cells/ml were distributed in the wells of a 96-well plates, to serve as feeder layers. The cloning was done by end-point dilution. One or two ml of the cell suspension from the positive flasks was transferred to a sterile tube and a precise cell count was determined as described above by counting at least 5-10 squares in a hematocytometer. The cell concentration was adjusted to 32, 16, and 8 cells/ml, and 50 microliters of each suspension was distributed to a 96-well plates. The plates were incubated at 3 7 ° 0 for 7-14 days. The medium was surveyed every 2 to 3 days and fresh medium was added
-31-when necessary tc avoid drying out of the wells. When the clones were visible with the naked eye and the cells were about 25-70% confluent, supernatants were tested for anti-CMV antibodies using the ELISA test. The positive cells in the wells were subcultured into 24-well plates and and then into flasks as described above. Interesting clones were frozen as described before.
5.9 Ascitic. Fluid Production
Mice were primed by intraperitoneal injection of 0.7 ml of pristane (2,4,10,14-Tetramethylpentadecane; Aldrich # 12,280-2) 10 days before injecting the cells. The content of 1 flask (7-9 ml) was centrifuged and resuspended into 1 ml of the spent medium. This suspension was injected intrâperitoneally into 1-2 prest.ane-primed mice. Ter. to fifteen days after injecting the cells, the abdomen of the mice became inflated and the ascitis fluid was drawn by inserting a 18G needle into the abdomen of the mouse. The ascitic fluid was centrifuged and the clear super natant frozen at -80 °C .
-32
-6.0: Sodiumdodecylsul_fate Polyacrylamide Gel Electrophoresis (5DS-PAGE)
6.1: The following reagents were prepared to carry out 5DS-PAGE according to the technique of Laemmli (1970):
Reagent -A- : 1.5 M Tris-chloride (SIGMA # T-1530) containing 8 mM EDTA, pH 8.9.
Reagent _-B- : 0.5 M Tris-chloride containing 8 mM of EDTA, pH
6
.
8
.
Reagent -C- : 30% acrylamide (Bio-Rad # 161-0201) in distilled water.
Reapent -D- : 10% SDS (Bio-Rad // 161-0301) in distilled water.
Reagent -E- : 1.5% Ammoniumpersulfate (Bio-Rad # 161- 0700 ) in distilled water.
Reagent -F- : 50 mM Tris-chloride, 0.375 M Glycine (Bio-Rad # 161-0718), 2mM EDTA, 1% SDS; pH 8.3.
Reagent -G- : 1% SDS, 1.2% Tris-Chlori de, 1% Sucrose (SIGMA // 5-9378), 2 mg/100 ml bromophenol blue solution (Bio-Rad # 161- 0404); pH 7.4.
-33-6.2: Preparation of the Gel
The running gel was prepared by mixing the reagents as follows: 18.75 ml of reagent A , 24.75 ml of reagent C, 0.75 ml of reagent D, 75 microliters of TE.MED (Bio-Rad # 161-0601) , 23.25 ml of distilled water ; and 7.5 ml of freshly prepared reagent E. The mixture was added in between the assembled glass plates of the Protean Dual 16 cm slab cel] electrophoresis apparatus (Bio-Rad # 165-1420). About 1 ml of distilled water was added immediatly dropwise on the top of the gel and left to stand on level until it polymerized. The amount prepared above is sufficient to make two 11% gels of 1.5 mm thickness.
As soon as the running gel was polymerized, the spacer gel was prepared by mixing 6.25 ml of reagent. B, 5.6 ml of reagent C, 0.25 ml of reagent D, 25 microliters TEHED, 22.5 ml of distilled water and 7.5 ml of reagent. E. As soon as the mixture was prepared, the water on the top of the running gel was removed and the mixture was added on top of the running gel. A 10 or 15 well comb was placed immediately into the spacer gel in between the glass plates. The gel was left again to stand until the spacer gel polymerized.
-34-6.3: Preparation o_f the Sample
Three and a half microliters cf 2-mercspt.oet.hanol (Bio-Rad # 161-0710) was added to 10 ml of reagent G just before use. Then 25 microliters of this reagent was added to 50 microliters of the sample to be electrophoresed. The mixture was boiled in a water bath for one minute , and then left to cool before adding it into the wells in the gel. The comb in the polymerized spacer gel was removed and the wells were washed with distilled water. Fifty microlit.ers of each sample was added in the wells, the total protein content of each sample did not exceed 50 to 70 micrograms. The wells were then filled with reagent. F very slowly so that not to disturb the surface of the sample. The top reservoir cf the electrophoresis apparatus was then assembled with the slab gel glass plates. This reservoir was then filled with reagent F an ci the whole assembly was placed into the bottom reservoir of the apparatus which was filled with about 3.4 L of reagent F. The cover, which contains the electrical connections, was placed on top. The whole apparatus was cooled by a water cooling system. The assembly was then connected to a power supply (Bio-Rad # 165-4700). The voltage was adjusted to 100 volts until the sample reached the running gel, then the voltage was raised to 250 volts and the electrophoresis was carried out. until the blue line of reagent G reached the bottom of the running gel. The power supply was turned off, and the gel was removed from the
-35-bottom reservoir, the plate assemblies removed from the top reservoir and the gel removed from in between the glass plates for either staining or electro-blotting.
7.0: Stain i n cj_ t h e Gel
The gel was stained either by coomasie blue R 250 (Bio-Rad # 161-0400), or by silver nitrate (Accurate Chemicals # APO-4535).
7.1: Coomasie blue staining: The gel was stained with a coomassie blue solution (0.16% coomassie blue in 45.5% methanol , 45.5% distilled water, and 9% acetic acid (FISHER # A-38-S ) ) for 45 minutes on a shaker. The staining solution was then discarded and the gel was washed twice with cold water. The excess of stain and the background was removed from the gel b y placing it in a solution containing 45.5% methanol, 45.5% distilled water, and 9% acetic acid for 1 hour on a shaker apparatus in the presence of pieces of styrofoam kept in the solution. The destaining solution was then discarded and the gel was placed for about 1 hour in a solution containing 20% methanol, 7% acetic acid, 1% glycerin
(FISHER # 6-33-B), and 72% distilled water. The gel was then dried on whatman 3MM filter paper (FISHER # 05-714-5) for 90
-36-minutes with heat in a slab gel dryer model 224 (Bio-Rad # 165-0920) .
7.2: Silver nitrate_staipiny
(a) Fixing : The gel was fixed for 10 minute in a solution containing 50% methanol, 12% acetic acid and 38% distilled water. The gel was then transferred for 10 minutes into a solution consisting of 10% ethanol , 5% acetic acid, and 85% distilled water.
(b) Metal Irnpreynation : The gel was then transferred for 5 minutes into a 0.06% of potassium permanganate (BDH # B-10217-34) solution, retransferred for another 5 minutes into a 0.1% of potassium carbonate (BDH # B-1019-34 ) solution and finally washed with distilled water for 30 minutes with three changes during this period.
(c ) Silver Nitrate Impregnation: After washing the gel , it was transferred into a 0.1% Silver nitrate solution for 10 minutes. The gel was then washed with distilled water for about 3 minutes.
-37-(d) Development : The staining was developed in a solution containing 2 % potassium carbonate and 0.015% formaldehyde (BDH # B-101-13) in distilled water for about 1 to 10 minutes. The reaction was then stopped with distilled water or 1% acetic acid solution. The gel was then dried as described above.
8.0: Western Blotting
The Western blotting was performed on nitrocellulose membrane (Bio-Rad # 162-0115) as described by Burnett (1981). The proteins were transferred from the gel onto the nitrocellulose membrane using a blotting apparatus (Transphor; Hoefer # TE-52). The transfer buffer was composed of 25 mM tris-chloride and 192 mM glycine in of distilled water. One liter of methanol was added to 4 L of the solution and cooled at 4°C. The nitrocellulose membrane (NCM) was always manipulated using disposable gloves (Tru-touch; Becton Dicksom # 2202). An 18 cm X 20 cm piece of NCM was soaked in the transfer buffer and a sandwich (gel holder, scotch-brite pad, Whatman 3MM filter paper, NCM, gel, Whatman 3MM filter paper, scctch-brite pad, and gel holder) was prepared and inserted in the apparatus containing the transfer buffer. The NCM was oriented towards the anode of the apparatus. The blotting was done for 12 hours using a current of
-38-0.3 to 0.5 amperes. At the end of the blotting period, the NCM was taken out of the sandwich and stained with amido black stain (Bio-Rad # 161-0700), or an ELISA was performed as described below.
8.1: Amido Black Staining
The Nitrocellulose membrane was soaked in 0.1% amido black solution (7% Acetic Acid, 30% Ethanol and 63% distilled water) fcr about 4 5 minutes to 1 hour. The NCM was then rinced with distilled water and dried on a filter paper.
8.2: Pr o tein_ Detection_ by Jmmunojblottinc)
The detection of the virai, proteins by monoclonal antibodies was done using an ELISA technique. After the blotting, the NCM was incubated for one hour at room temprature or overnight at 4°C with 1% bovine serum albumin in PBS (PBS-BSA 1%). Next the NCM was washed three times in PBS for 15 minutes. Each washing consists of a 15 minutes period. Henceforth, all washings will be referred to as either "3 X 10 minutes washings" or "3 X 60
-39
-minutes washings". The NCM was then incubated in the solution containing monoclonal antibody or anti-CMV mouse serum at an appropriate dilution for an additional three hours at 37°C. Next the NCM was then washed for 3X 15 minutes in PB5. The NCM was then incubated with a second antibody (goat-antimouse IgC + IgM conjugated with peroxidase; diluted to 1:200 in P8S-BSA 1 % ) for an additional two hours at 37°C. The NCM was then washed for 3X 30 minutes as described above followed by an i.ncotation in a substrate solution for 2 to 10 minutes. The substrate- used was 4-Chloro-l-Naphthol (Aldrich; // C5,780-4 ) and was prepared by dissolving 3 mg of 4-chloro-l-naphthol in 1 ml Methanol. Once dissolved, 5 ml of distilled water was added. Just before use, 40 micro!iters of 3 0% hydrogen peroxide was added to the substrate solution. The enzyme-substrate reaction was stopped by washing the NCM in distilled water, after an adequate color developed.
9.0: Determination of Immunoglobulin Sub-class
The double immune diffusi on kit (Miles Scientific it 42-175) was used to determine the immunoglobulin subclass The gel was prepared by dissolving 1 gram of Ionagar it2 (Oxfoid it L28) in 93 ml Borate Saline (5% borate buffer in PBS). Borate buffer is composed of 0.6184% boric acid, 0.4384% sodium chloride and 0.9536% s o d i u m tetraborate dissolved i.n distilled water. One mg
40-merthiolate (Kodak // 1 1228 ) and 100 microliters of 1 % trypan blue solution were added to the gel solution. The whole mixture was then brought to boil in a boiling water bath. The mixture was aliquoted in 10 ml amounts in glass tubes and stored at 4 ° C until used.
When needed, the agar in the tube was melted in a boiling water bath. The melted agar was transferred to an immunodiffusion plate and left to solidify for 30 minutes at room temprature. The agar was then perforated using the cutter and template provided with the kit. Twelve microliters of the antibody to be tested was then added in the center well , and 12 microliters of the different ant. i mouse-immunoglobulins (goat-antimouse IgC + I g M, rabbit-anti-mouse I g G , rabbit-antimouse I g G , rabbit-antimouse IgG , rabbit-antimouse IgG , and goat-antimouse IgM) were added in the peripheral wells. The anti-mouse immunoglobulins were purchased from Miles Scientific. The plate was then incubated at 3 7 0 0 in a humid atmosphere for 2 à hours to 48 hours. The plate was then examined for precipitation lines.
10.0: Immunofluorescence
Fibroblasts were grown in 25 cm2 flasks to a confluent monolayer were infected with CMV strain ADI 69 and incubated fcr 4
-Al
to 5 days at 37°C. When the cells showed 100% CPE (cytopathic effect) , they were trypsin! zed as discribe cl before. The action of trypsin was stopped with MEM-10 and the cells were transferred into a conical centrifuge tube and washed twice with, PBS. The pellet was resuspended in 0.5 ml sterile PBS. Cont. rol non-infected cells were prepared in the same way. The cells were then distributed on precleaned printed microscope slides (10 microliters/well; Carlson Scientific Inc. II 101006). The slides were dried at room temp rature and fixed for 30 minutes with cold acetone. The slides were stored at -80 °C until used.
The indirect immunofluorescence (Ilf) test was performed as follows: Ten microliters of an appropriate dilution of the antibody preparation to be tested was added to the wells. The slides were then incubated for 3 hours at 3 7 0 C in a humid atmosphere, followed by 3X 10 minutes washings in PBS. Ten microliters of an appropriate dilution of fluorescein isothiocyanate labeled goat-antimouse IgG + IgM ( H 4- L ) antibody (KPL # 021809) was then added to each well, followed by another incubation for 2 hours at 37 °C in a humid atmosphere. The slides were then washed 3 X 10 minutes in PBS and mounted using PBS-glycer in (50%) (Fisher // 6-33-B). The slides were then examined with a fluorescent microscope and photographed at a magnification cf 4 0 0 X using a 2 minutes exposure. A Kodak Ektachrome EL ] 35-20 film (400 ASA, Kodak # 160-1814) was used.
-42-11.0: CM\/ Titration
Fibroblasts were grown to conflueney in sterile 96-well plates. Several dilutions (tenfold, up to 1 : 10^) of the virus were made in MEM-3. Fifty microliters cf each virus dilution was added in triplicate tc the 96-well plate. Medium alone was used as a negative control. The plate was then incubated for 1 hour at 3 7 0 C in humid 5% 00^ incubator. The virus supernatants ever the cells were then replaced with fresh medium MEM-3. The plate was then incubated for an additional 3 to 8 days at 3 7 0 C. The medium was changed every 3 days. At the end of the incubation period when satisfactory CPE was noted, the plate was washed 3X 3 minutes with PBS and stained with 0.3% giemsa stain (prepared i.n ethanol) for 20 minutes by adding 50 microliters of the stain per well . The plate was then washed twice with PBS and examined with a light microscope. Per neutralization studies three viral dilutions were chosen. The first dilution was one which showed a 50% (CPE); the second was ten fold less concentrated as the first and the third was ten fold mere concentrated than the first dilution .
12.0: Neutralization Test
Fibroblasts were grown to a confluent, monolayer in 96 well plate as described before. Dilutions (undiluted, 1:2, 1:5, 1:10,
-43-and 1:100) of the monoclonal antibody to be tested were made in MEM-199 medium. A human serum pool (undiluted, 1:2, 1:5, and 1:10) was used as a positive control. Three hundred microliters of each of these antibody dilutions were mixed with 500 micro!iters of each of the viral dilutions chosen in sterile tubes. The tubes were incubated for 1 hour at 37 °C. As an antibody negative control , three hundred microliters of each of the virus dilutions chosen in the titration test were incubated for 1 hour under the same conditions with 300 microliters of MEM-199 medium. At the end of the 1 hour incubation period, the medium over the fibroblast monolayer was replaced with 100 microliters/well (in triplicate) of the virus : antibody or virus : medium mixture. The plate was then incubated for 1 hour at 37 °C , washed once with fresh ME M-3 ( 200 microliters/well) and incubated with 200 microliters/well of fresh ME'M-3 for 5-7 days at 3 7 °C. The medium was changed every 3 days and at the end of the incubation period, the plate was washed 3X 5 minutes with PBS, stained for 20 minutes with 3% giemsa stain, examined and photographed using a earner a mounted on a light microscope.
1. Standardization o_f the ELISA technique.
The ELISA technique was used to test for the presence of the anti-CMV antibodies in this project. Different dilutions of the immune and non-immune sera obtained from mice were made. Using dilutions less than 1:100 gave optic density (0.D. ) readings (for the immune mouse serum) over 2.00, which were not useful to give an accurate calculation. Therefore, 2-fold dilutions were made from 1:200 up to 1:25600. The results shown in (Table 1.) enabled us to choose the mice with the highest anti-CMV antibody titers. The mice B - 4 and B-5 were chosen for the first fusion (M-1). At a later date, the mouse A-3 still had a high titer of anti-CMV antibodies, therefore, it was used for the the second fusion ( M - 3 ) . Similarly, mouse B-3 was used for the third fusion (M-4). For fusion M-5 , two mice immunized with CMV-ant.igen prepared in our laboratory were used. The ELISA readings were 1.629, 0.839 and 1.796, 1.177 when tested with the CMV an'tigen and control antigen respectively. The first mouse was chosen as it gave lower reactivity with the control antigen. An ELISA test was done tc
a Table. 1. Immunization of mice with CMV-infected cell-lysate. :
Serum dilution Serum A-1 Serum A-2 Serum A-3 Serum A-4 Serum B-l Serum B-2 Serum B-3 Serum B-4 Serum B-5 1 : 200 0.184 0.077 . 0.657 „ 0.191 . . 0.112 . . 0.260 . . 0.608 . . 1.062 . . 1.249 1 : 400 0.194 0.061 0.452 0.096 0.071 0.199 0.367 0.296 0.565 1 : 800 0.096 0.033 0.298 0.061 0.040 0.106 0.184 0.162 0.270 1 : 1600 0.028 0.023 0.159 0.059 0.020 0.106 0.089 0.243 0.194 1 : 3200 0.021 0.029 0.082 0.018 0.016 0.020 0.039 0.077 0.059 1 : 6400 0.005 0.004 0.036 0.008 0.018 0.024 0.031 0.083 0.055 1 : 12800 0.014 0.006 0.039 0.017 0.015 0.021 0.038 0.069 0.077 1 : 25600 0.006 0.018 0.014 0.008 0.010 0.013 0.017 0.042 0.039
The plates were coated with CMV-antigen at a concentration of 50 ng/well. The plates were read at a wave-length of 492 nm.
The normal mouse serum at a dilution of 1:200 gave a reading od 0.046.
I
-46-test the effect of the detergent Tween-20 in PBS (RBS-Tween) when used as s diluent for the anti-mouse peroxidase-labeled antibody and/or when used for washing the ELISA plates in between the different steps of the technique. No significant differences were noticed therefore RBS-Tween was used in all the subsequent experiments. The amount, of antigen needed to coat the well was determined by titrating the CMV-antigen and the control antigen against a fixed concentration of anti-CMV antibody. Satisfactory optical density readings were obtained when 50 nanograms protein/well was used. This concentration was therefore used in all the ELISA tests described. The appropriate dilution of the peroxidase-labeled anti-mouse antibody was also determined by checkerboard titration using different dilutions. Normally dilutions of 1/500 to 1/10GO were used in the ELISA tests.
2. Cell fusions.
The ELISA results obtained from four fusions between mouse spleen cells (immunized with CMV) and mouse NSO myeloma cells are summarized in (Table 2). The total number cf wells tested in the 9 6 - well plates were 2267, from which 903 were positive. The positive cultures were compared to a CMV-immune serum using the following formula:
TABLE 2. CMV-specific hybridoma from the different fusions performed.
Fusion # # of wells | tested in ] 96-WP (a) ! _ ! # of +ve wells M-l . . 142 ! 1 22 M-3 268 1 47 M-4 257 | 1 243 M-5 1600 j 591 ! # of wells | transferred |to 24-WP (b) # of | +ve j wells ! 1 # of wells transferred to flasks ' # of j hybridomas| frozen • | 22 1 7 ; 7 7 47 21 | 1 21 15 (c)| 243 62 | i 62 26 (c) 1 64 64 ! i i 64 64 a. 96-WP = 96 well plates. b. 24-WP = 24 well plates.c. The number of hybridoma frozen from the flasks were sometimes less than the number of the hybridoma transfereed transfered as the cells either turned negative or died before freezing.
-48-(0.D. of test samp]e - O.D. of negative cont. ) ?o of positivity r
---(O.D. of positive cont.- O.D. of negative cont.)
Where O.D. is the optical density , negative control is medium I — 10 -*• HT without antibody and positive control is s serum obtained from mice immunized with C M V .
From the 903 positive wells, 376 anti-CMV antibody producing bybridomas displaying high reactivities were transferred to wells in 24-we 11 plates. When the cells in the 24-weel plates showed 50% or more con fluency, they were retested by the ELISA technique. 154 wells were positive and were subcultured in 25 cm2 flasks. The cultures in the flasks were tested again when they showed good growth. A total of 112 cultures were frozen as they continued antibody production. The non-productive were discarded.
A few interesting hybridomas from the M-l fusion that showed high anti-CMV antibody titers were cloned (Table 3). The hybridomes that gave positive clones were CLA-2, CLA-6, CLC-5, and CLB-1. A total of 29 clones were obtained and frozen for Later use. When the clones were thawed out arid retested by the ELISA technique, they were either negative or gave nonspecific reactions. When tested by the- CMV-speci fie 11F test the CLB-1

![TABLE 2. CMV-specific hybridoma from the different fusions performed. Fusion # # of wells | tested in ] 96-WP (a) ! _ ! # of+ve wells M-l](https://thumb-eu.123doks.com/thumbv2/123doknet/3700705.109995/61.1170.166.1053.316.547/table-specific-hybridoma-different-fusions-performed-fusion-tested.webp)